A Newly Designed EGFP-2A Peptide Monocistronic Baculoviral Vector for Concatenating the Expression of Recombinant Proteins in Insect Cells

 ,
,  ,
,
Abstract
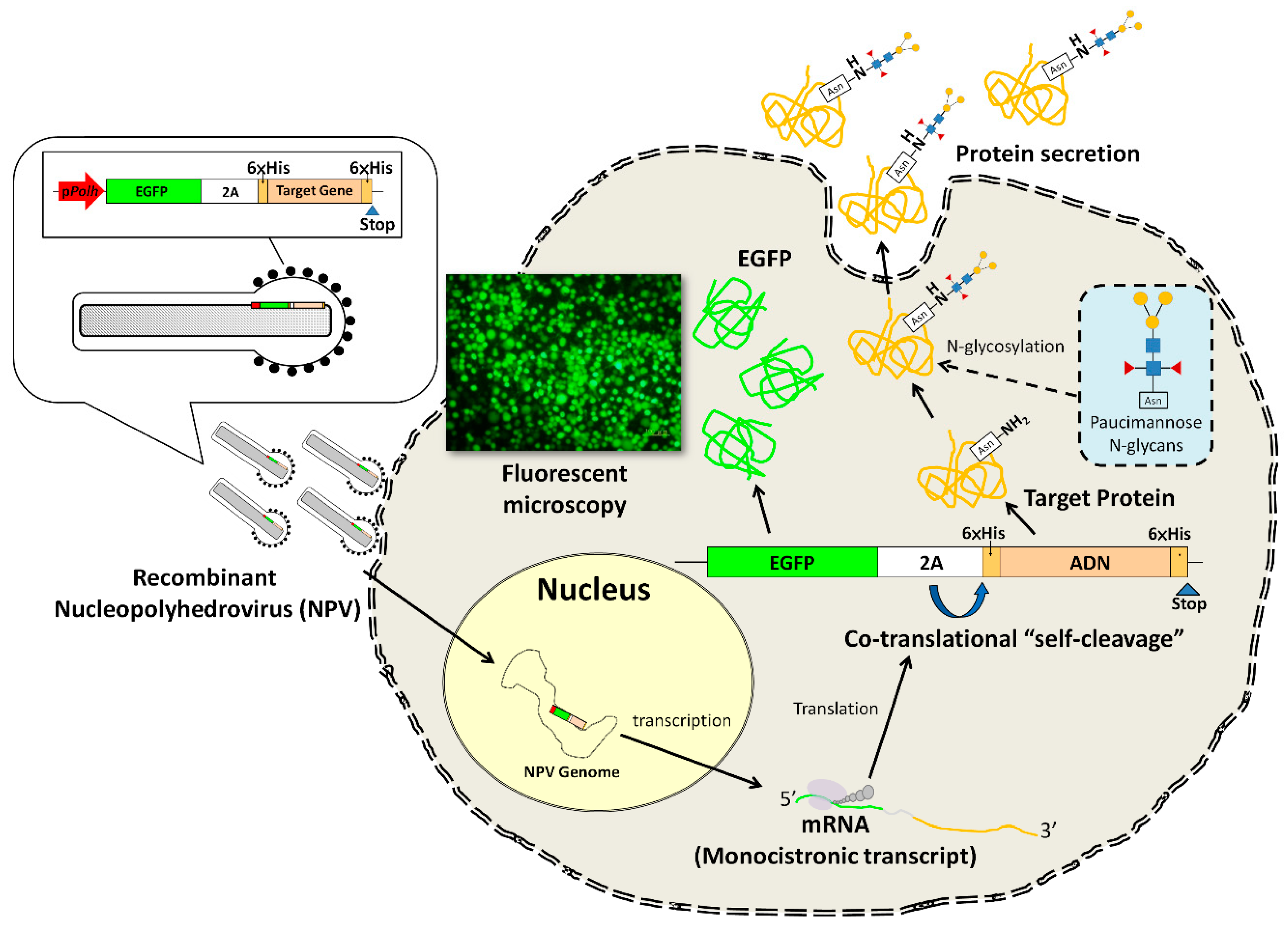
:1. Introduction
2. Materials and Methods
2.1. Cell lines and Virus Growth
2.2. Construction of Transfer Vectors
2.3. Recombinant Virus Generation and Cell Viability Analysis
2.4. Expression and Purification of Recombinant Adiponectin
2.5. Phosphorylation of AMPK and ACC by Adiponectin
3. Results
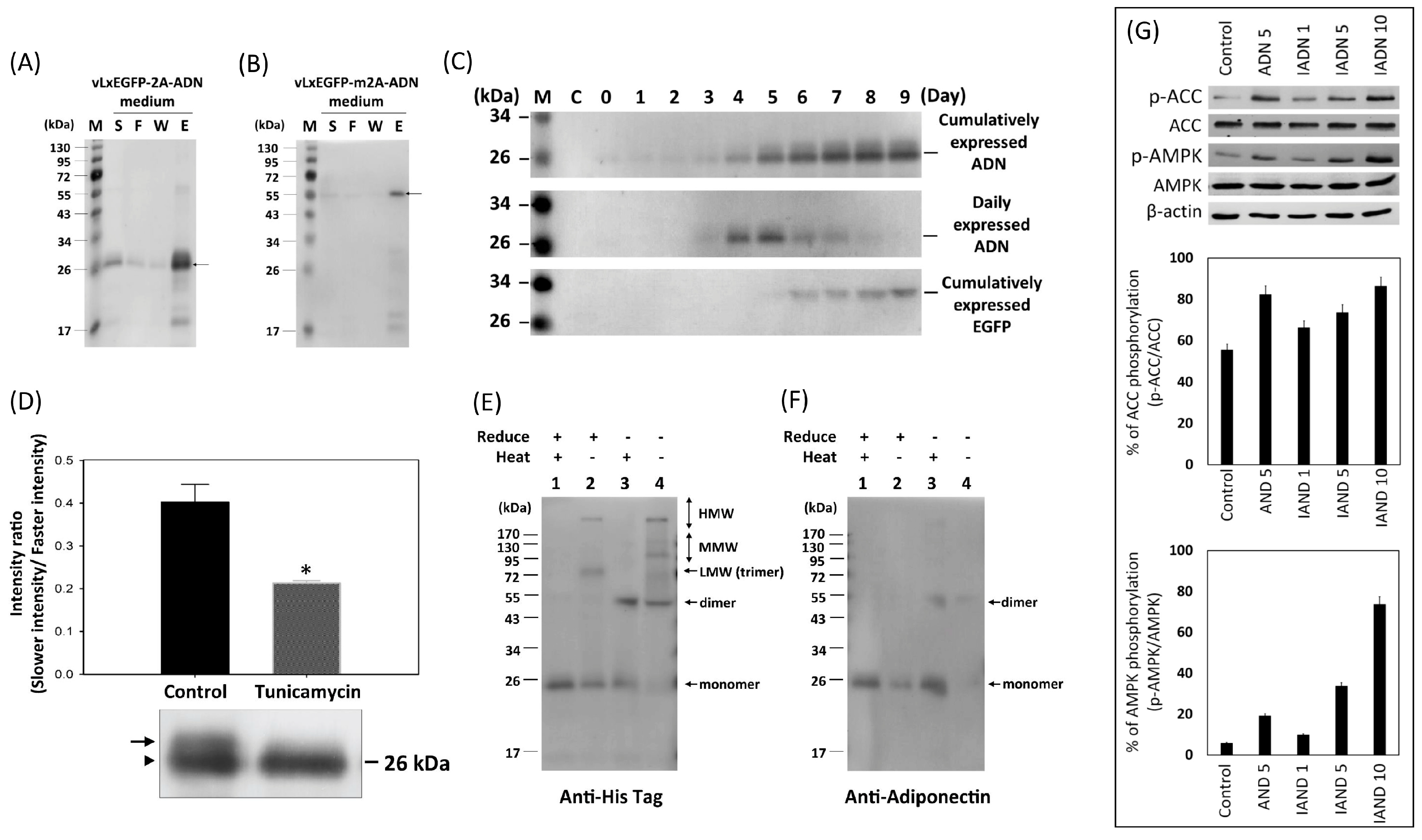
3.1. Recombinant Virus Production and Cell Viability Analysis
3.2. Verification of 2A Peptide Function
3.3. Purification of Recombinant Proteins from Culture Medium
3.4. Glycosylation of Recombinant Proteins
3.5. Multiuser Separation Patterns of Porcine Adiponectin by SDS-PAGE under Various Conditions
3.6. Effect of Recombinant Porcine Adiponectin on the Phosphorylation of AMPK and ACC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Compliance with Ethical Standards
References
- Murhammer, D.W. Baculovirus and Insect Cell Expression Protocols; Methods in Moleculer Biology; Humana Press: New York, NY, USA, 2007. [Google Scholar]
- Smith, G.E.; Summers, M.D.; Fraser, M.J. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol. Cell. Biol. 1983, 3, 2156–2165. [Google Scholar] [CrossRef]
- Miller, L.K. Baculoviruses as gene expression vectors. Annu. Rev. Microbiol. 1988, 42, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Luckow, V.A.; Summers, M.D. Trends in the development of baculovirus expression vectors. Nat. Biotechnol. 1988, 6, 47–55. [Google Scholar] [CrossRef]
- Steele, K.H.; Stone, B.J.; Franklin, K.M.; Fath-Goodin, A.; Zhang, X.; Jiang, H.; Webb, B.A.; Geisler, C. Improving the baculovirus expression vector system with vankyrin-enhanced technology. Biotechnol. Prog. 2017, 33, 1496–1507. [Google Scholar] [CrossRef]
- Wang, J.; Hou, D.; Wang, Q.; Kuang, W.; Zhang, L.; Li, J.; Shen, S.; Deng, F.; Wang, H.; Hu, Z.; et al. Genome analysis of a novel Group I alphabaculovirus obtained from Oxyplax ochracea. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Sussman, D.J. 24-hour assay for estimating the titer of beta-galactosidase-expressing baculovirus. Biotechniques 1995, 18, 50–51. [Google Scholar]
- de Felipe, P.; Ryan, M.D. Targeting of proteins derived from self-processing polyproteins containing multiple signal sequences. Traffic 2004, 5, 616–626. [Google Scholar] [CrossRef] [PubMed]
- de Felipe, P.; Luke, G.A.; Hughes, L.E.; Gani, D.; Halpin, C.; Ryan, M.D. E unum pluribus: Multiple proteins from a self-processing polyprotein. Trends Biotechnol. 2006, 24, 68–75. [Google Scholar] [CrossRef]
- Mansouri, M.; Bellon-Echeverria, I.; Rizk, A.; Ehsaei, Z.; Cianciolo Cosentino, C.; Silva, C.S.; Xie, Y.; Boyce, F.M.; Davis, M.W.; Neuhauss, S.C.; et al. Highly efficient baculovirus-mediated multigene delivery in primary cells. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Ruth Vaseghi, H.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Wang, R.; Zhao, P.; Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Sci. Rep. 2015, 5, 16273. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, F.; Xu, S.; Wang, R.; Chen, W.; Hou, K.; Tian, C.; Wang, F.; Zhao, P.; Xia, Q. Optimization of a 2A self-cleaving peptide-based multigene expression system for efficient expression of upstream and downstream genes in silkworm. Mol. Genet. Genom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Wang, C.H. New cell lines from Lymantria xylina (Lepidoptera: Lymantriidae): Characterization and susceptibility to baculoviruses. J. Invertebr. Pathol. 2006, 93, 186–191. [Google Scholar] [CrossRef]
- Hink, W.; Strauss, E. Growth of the Trichoplusia ni (TN-368) cell line in suspension culture. Int. Conf. Invertebr. Tissue Culture Appl. Med. Biol. Agric. 1976. [Google Scholar] [CrossRef]
- Nai, Y.S.; Wu, C.Y.; Wang, T.C.; Chen, Y.R.; Lau, W.H.; Lo, C.F.; Tsai, M.F.; Wang, C.H. Genomic sequencing and analyses of Lymantria xylina multiple nucleopolyhedrovirus. BMC Genomics 2010, 11. [Google Scholar] [CrossRef]
- Waki, H.; Yamauchi, T.; Kamon, J.; Ito, Y.; Uchida, S.; Kita, S.; Hara, K.; Hada, Y.; Vasseur, F.; Froguel, P.; et al. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J. Biol. Chem. 2003, 278, 40352–40363. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods. 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.E.; Scherer, P.E. Adiponectin--journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J. Intern. Med. 2005, 257, 167–175. [Google Scholar] [CrossRef]
- Richards, A.A.; Stephens, T.; Charlton, H.K.; Jones, A.; Macdonald, G.A.; Prins, J.B.; Whitehead, J.P. Adiponectin multimerization is dependent on conserved lysines in the collagenous domain: evidence for regulation of multimerization by alterations in posttranslational modifications. Mol. Endocrinol. 2006, 20, 1673–1687. [Google Scholar] [CrossRef]
- Osborn, M.J.; Panoskaltsis-Mortari, A.; McElmurry, R.T.; Bell, S.K.; Vignali, D.A.; Ryan, M.D.; Wilber, A.C.; McIvor, R.S.; Tolar, J.; Blazar, B.R. A picornaviral 2A-like sequence-based tricistronic vector allowing for high-level therapeutic gene expression coupled to a dual-reporter system. Mol. Ther. 2005, 12, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Minskaia, E.; Nicholson, J.; Ryan, M.D. Optimisation of the foot-and-mouth disease virus 2A co-expression system for biomedical applications. BMC Biotechnol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, A.; Vande Velde, G.; Reumers, V.; Toelen, J.; Thiry, I.; Vandeputte, C.; Vets, S.; Deroose, C.; Bormans, G.; Baekelandt, V.; et al. Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Hum. Gene. Ther. 2009, 20, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.B.; Zhang, Y.J.; Zhang, H.T.; Yang, S.L.; Gong, Y. Cloning and expression of adiponectin and its globular domain, and measurement of the biological activity in vivo. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 2003, 35, 1023–1028. [Google Scholar]
- Peake, P.W.; Hughes, J.T.; Shen, Y.; Charlesworth, J.A. Glycosylation of human adiponectin affects its conformation and stability. J. Mol. Endocrinol. 2007, 39, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Fukuhara, A.; Shimomura, I. N-linked glycosylation of mouse adiponectin. Horm. Metab. Res. 2011, 43, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Tomas, E.; Murrey, H.E.; Hug, C.; Lee, D.H.; Ruderman, N.B.; Heuser, J.E.; Lodish, H.F. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. J. Biol. Chem. 2003, 278, 50810–50817. [Google Scholar] [CrossRef]
- Yuh, I.S.; Sheffield, L.G. Adiponectin gene cloning and its expression in insect cell expression system. Reprod. Dev. Biol. 2012, 36, 193–198. [Google Scholar]
- Neumeier, M.; Weigert, J.; Schaffler, A.; Wehrwein, G.; Muller-Ladner, U.; Scholmerich, J.; Wrede, C.; Buechler, C. Different effects of adiponectin isoforms in human monocytic cells. J. Leukoc. Biol. 2006, 79, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, A.; Knight, C.; Xu, L.Y.; Cooper, G.J. Hydroxylation and glycosylation of the four conserved lysine residues in the collagenous domain of adiponectin. Potential role in the modulation of its insulin-sensitizing activity. J. Biol. Chem. 2002, 277, 19521–19529. [Google Scholar] [CrossRef]
- Gomez-Sebastian, S.; Lopez-Vidal, J.; Escribano, J.M. Significant productivity improvement of the baculovirus expression vector system by engineering a novel expression cassette. PLoS ONE 2014, 9, e96562. [Google Scholar] [CrossRef]
- Wang, Y.; Lam, K.S.; Yau, M.H.; Xu, A. Post-translational modifications of adiponectin: Mechanisms and functional implications. Biochem. J. 2008, 409, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, M.E.; Gabler, N.K. The development of porcine models of obesity and the metabolic syndrome. J. Nutr. 2008, 138, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.H.; Lin, Y.Y.; Wang, Y.C.; Huang, C.W.; Chen, C.C.; Wu, S.C.; Mersmann, H.J.; Cheng, W.T.; Ding, S.T. Porcine adiponectin receptor 1 transgene resists high-fat/sucrose diet-induced weight gain, hepatosteatosis and insulin resistance in mice. Exp. Anim. 2013, 62, 347–360. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Note |
|---|---|---|
| M13-F | 5′-GTTGTAAAACGACGGCCAGT-3′ | - |
| M13-R | 5′-CAGGAAACAGCTATGACC-3′ | - |
| polh-up(R) | 5′-CCATGATATCTTTACGGTGTATTTGATTTTTCCAAAATCTTTACG-3′ | corresponding to 1977–2011 nt in the LyxyMNPV EcoRV-K fragment; the EcoR V site is underlined |
| polh-down(F) | 5′-GGTTAAGCTTACGATTACGCCTCGGATTCC-3′ | corresponding to 2753–2772 nt in the LyxyMNPV EcoR V-K fragment; the HindIII site is underlined |
| MCS-F | 5′-ATCGGTACCGCTAGCTCTAGACCCGGGCTGCAGCCGCGGA-3′ | - |
| MCS-R | 5′-AGCTTCCGCGGCTGCAGCCCGGGTCTAGAGCTAGCGGTACCGAT-3′ | - |
| GFP-forward primer | 5′-GCGCTCTAGAATGGTGAGCAAGGGCGAGGAG-3′ | the XbaI site is underlined |
| GFP-reverse primer_1 | 5′-GCGCGAATTCGGGCCCAGGATTCGA CTCGATGTCACCATCTTGTGTCAAATCCTTCTGCCCACCACCAATAATATTCTTGTACAGCTCGTCCATGCC-3′ | encoding the normal 2A peptide from Perina nuda virus; the EcoRI site is underlined |
| GFP-reverse primer_2 | 5′-GCGCGAATTCAGGATTCGACTCGAT GTCACCATCTTGTGTCAAATCCTTCTG CCCACCACCAATAATATTCTTGTACAGCTCGTCCATGCC-3′ | encoding a noncleavable mutant 2A peptide; the EcoRI site is underlined |
| EcoRI/His-ADN_F | 5′-GCGCGAATTC CATCACCATCACCAT CACATGCTGTTGTTGGGAGCTGTTC-3′ | the sequences for the EcoRI site and 6× Histidine tag are underlined |
| SacII/His-ADN_R | 5′-CGCGCCGCGGTCAGTGATGGTGATG GTGATGTTCAATGTTGTGGTAGAGAAG GAAG-3′ | the sequences for the SacII site and 6x Histidine tag are underlined |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-Y.; Huang, C.-W.; Nai, Y.-S.; Chu, P.-Y.; Wang, C.-H.; Ding, S.-T. A Newly Designed EGFP-2A Peptide Monocistronic Baculoviral Vector for Concatenating the Expression of Recombinant Proteins in Insect Cells. Processes 2019, 7, 291. https://doi.org/10.3390/pr7050291
Wu C-Y, Huang C-W, Nai Y-S, Chu P-Y, Wang C-H, Ding S-T. A Newly Designed EGFP-2A Peptide Monocistronic Baculoviral Vector for Concatenating the Expression of Recombinant Proteins in Insect Cells. Processes. 2019; 7(5):291. https://doi.org/10.3390/pr7050291
Chicago/Turabian StyleWu, Chih-Yu, Chao-Wei Huang, Yu-Shin Nai, Pei-Yu Chu, Chung-Hsiung Wang, and Shih-Torng Ding. 2019. "A Newly Designed EGFP-2A Peptide Monocistronic Baculoviral Vector for Concatenating the Expression of Recombinant Proteins in Insect Cells" Processes 7, no. 5: 291. https://doi.org/10.3390/pr7050291
APA StyleWu, C.-Y., Huang, C.-W., Nai, Y.-S., Chu, P.-Y., Wang, C.-H., & Ding, S.-T. (2019). A Newly Designed EGFP-2A Peptide Monocistronic Baculoviral Vector for Concatenating the Expression of Recombinant Proteins in Insect Cells. Processes, 7(5), 291. https://doi.org/10.3390/pr7050291