Glycerol Oxidation over Supported Gold Catalysts: The Combined Effect of Au Particle Size and Basicity of Support

 ,
, 
 ,
,  ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
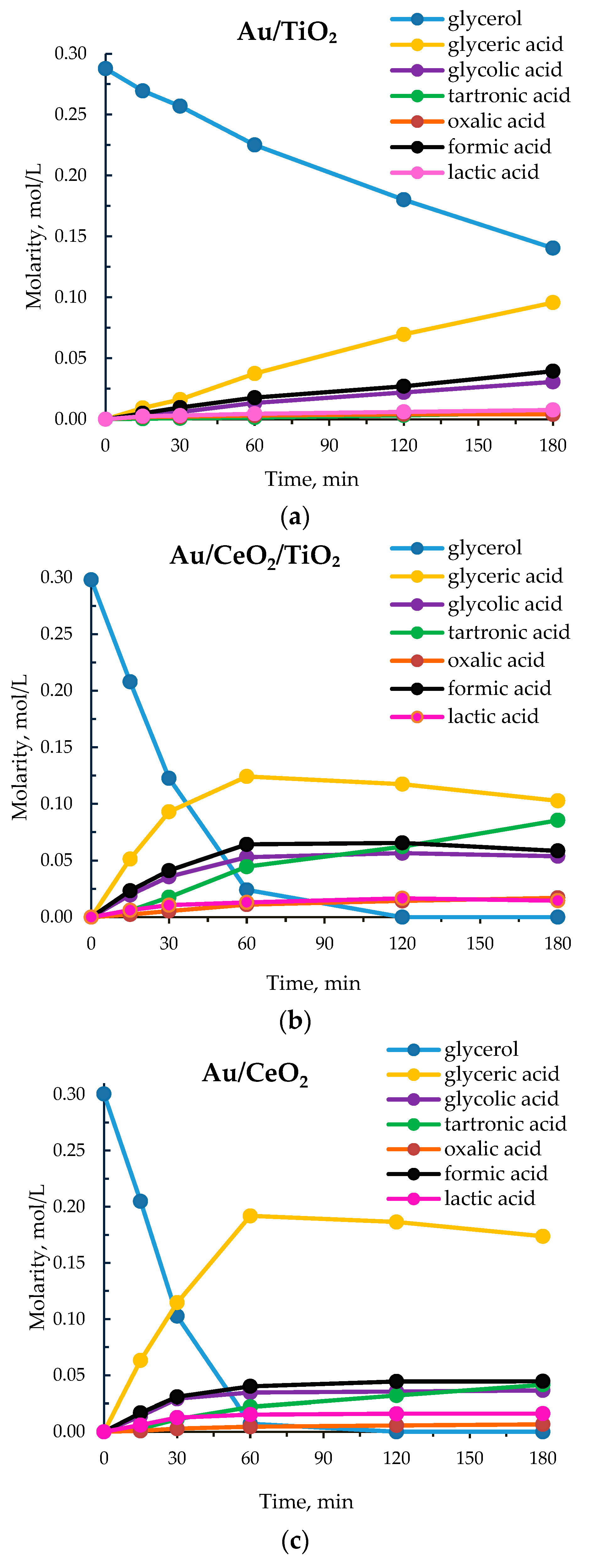
3.1. Catalytic Results
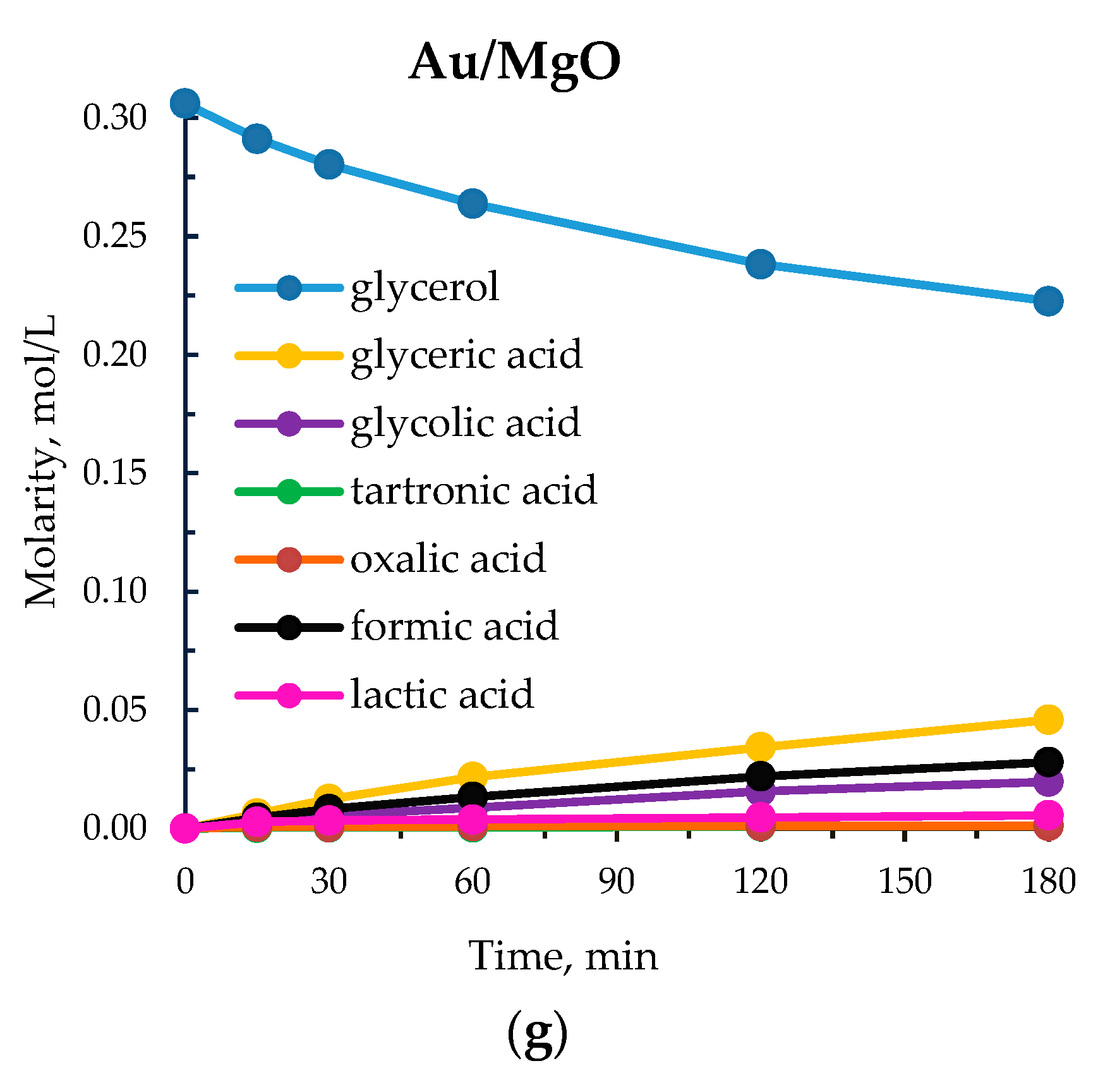
3.2. Study of the Influence of Alkaline Earth Base Additives
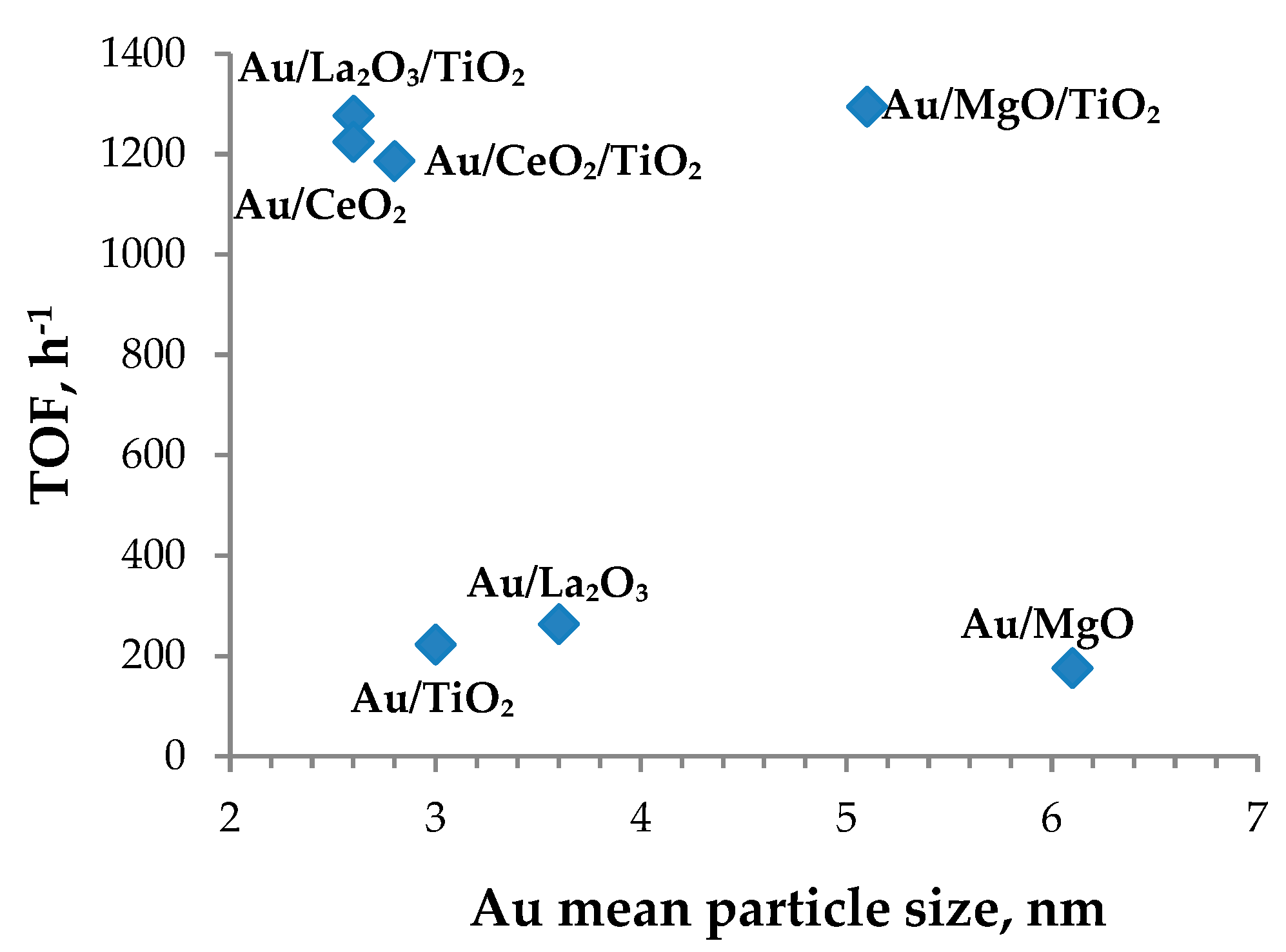
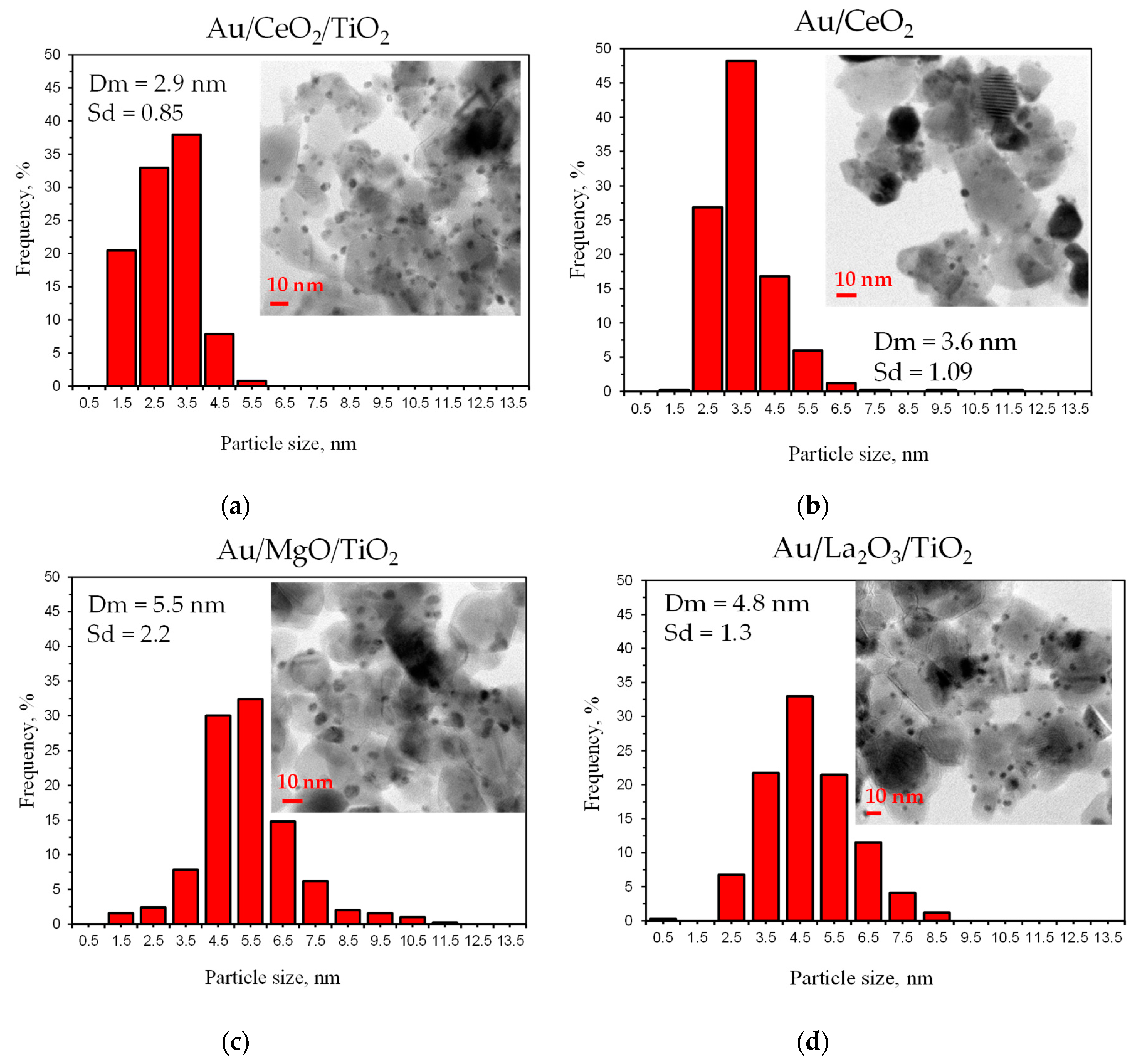
3.3. Catalyst Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass: Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas. Available online: https://www.osti.gov/biblio/15008859-top-value-added-chemicals-from-biomass-volume-results-screening-potpotent-candidates-from-sugars-synthesis-gas (accessed on 16 June 2020).
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Christoph, R.; Schmidt, B.; Steinberner, U.; Dilla, W.; Karinen, R. Glycerol, Ullmann’s Encyclopedia of Industrial Chemistry. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/14356007.a12_477.pub2 (accessed on 16 June 2020).
- Glycerine: An Overview. Available online: https://www.aciscience.org/docs/Glycerine_-_an_overview.pdf (accessed on 16 June 2020).
- Esposito, R.; Raucci, U.; Cucciolito, M.E.; Di Guida, R.; Scamardella, C.; Rega, N.; Ruffo, F. Iron(III) complexes for highly efficient and sustainable ketalization of glycerol: A combined experimental and theoretical study. ACS Omega 2019, 4, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Quispe, C.A.G.; Coronado, C.J.R.; Carvalho, J.A. Glycerol: Production, consumption, prices, characterization and new trends in combustion. Renew. Sustain. Energy Rev. 2013, 27, 475–493. [Google Scholar] [CrossRef]
- Villa, A.; Dimitratos, N.; Chan-Thaw, C.E.; Hammond, C.; Prati, L.; Hutchings, G.J. Glycerol oxidation using gold-containing catalysts. Acc. Chem. Res. 2015, 48, 1403−1412. [Google Scholar] [CrossRef] [PubMed]
- Dodekatos, G.; Schünemann, S.; Tüysüz, H. Recent advances in thermo-, photo-, and electrocatalytic glycerol oxidation. ACS Catal. 2018, 8, 6301–6333. [Google Scholar] [CrossRef]
- The EU Biodiesel Industry. Available online: https://www.ebb-eu.org/stats.php (accessed on 16 June 2020).
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on platinum metal catalysts in aqueous solutions. Catal. Today 1994, 19, 247–283. [Google Scholar] [CrossRef]
- Garcia, R.; Besson, M.; Gallezot, P. Chemoselective catalytic oxidation of glycerol with air on platinum metals. Appl. Catal. A Gen. 1995, 127, 165–176. [Google Scholar] [CrossRef]
- Kimura, H.; Tsuto, K.; Wakisaka, T.; Kazumi, Y.; Inaya, Y. Selective oxidation of glycerol on a platinum-bismuth catalyst. Appl. Catal. A Gen. 1993, 96, 217–228. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef]
- Abad, A.; Almela, C.; Corma, A.; Garcia, H. Unique gold chemoselectivity for the aerobic oxidation of allylic alcohols. Chem. Commun. 2006, 3178–3180. [Google Scholar] [CrossRef]
- Villa, A.; Veith, G.M.; Prati, L. Selective oxidation of glycerol under acidic conditions using gold catalysts. Angew. Chem. Int. Ed. 2010, 49, 4499–4502. [Google Scholar] [CrossRef] [PubMed]
- Carrettin, S.; Mcmorn, P.; Johnston, P.; Griffin, K.; Kiely, C.J.; Hutchings, G.J. Oxidation of glycerol using supported Pt, Pd and Au catalysts. Phys. Chem. Chem. Phys. 2003, 5, 1329–1336. [Google Scholar] [CrossRef]
- Dimitratos, N.; Villa, A.; Bianchi, C.L.; Prati, L.; Makkee, M. Gold on titania: Effect of preparation method in the liquid phase oxidation. Appl. Catal. A Gen. 2006, 311, 185–192. [Google Scholar] [CrossRef]
- Habe, H.; Fukuoka, T.; Kitamoto, D.; Sakaki, K. Biotechnological production of d-glyceric acid and its application. Appl. Microbiol. Biotechnol. 2009, 84, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Habe, H.; Fukuoka, T.; Kitamoto, D.; Sakaki, K. Application of electrodialysis to glycerate recovery from a glycerol containing model solution and culture broth. J. Biosci. Bioeng. 2009, 107, 425–428. [Google Scholar] [CrossRef]
- Habe, H.; Fukuoka, T.; Kitamoto, D.; Sakaki, K. Biotransformation of glycerol to d-glyceric acid by Acetobacter tropicalis. Appl. Microbiol. Biotechnol. 2009, 81, 1033–1039. [Google Scholar] [CrossRef]
- Yunhai, S.; Houyong, S.; Deming, L.; Qinghua, L.; Dexing, C.; Yongchuan, Z. Separation of glycolic acid from glycolonitrile hydrolysate by reactive extraction with tri-n-octylamine. Sep. Purif. Technol. 2006, 49, 20–26. [Google Scholar] [CrossRef]
- Santiago, L.G.; Soccol, C.R.; Castro, G.R. Emerging technologies for bioactive applications in foods. In Food Bioactives: Extraction and Biotechnology Applications; Puri, M., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 205–226. ISBN 978-3-319-51637-0. [Google Scholar]
- Katryniok, B.; Kimura, H.; Skrzyńska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Bizot, P.M.; Bailey, B.R.; Hicks, P.D. Use of Tartronic Acid as an Oxygen Scavenger. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO1998016475 (accessed on 16 June 2020).
- Pagliaro, M.; Rossi, M. The Future of Glycerol: New Usages for a Versatile Raw Material. Available online: https://pubs.rsc.org/en/content/ebook/978-0-85404-124-4 (accessed on 16 June 2020).
- Caselli, G.; Monatovanini, M.; Gandolfi, C.A.; Allegretti, M.; Fiorentino, S.; Pellegrini, L.; Melillo, G.; Bertini, R.; Sabbatini, W.; Anacardio, R.; et al. Tartronates: A new generation of drugs affecting bone metabolism. J. Bone Miner. Res. 1997, 12, 972–981. [Google Scholar] [CrossRef]
- Carrettin, S.; McMorn, P.; Johnston, P.; Griffin, K.; Hutchings, G.J. Selective oxidation of glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide. Chem. Commun. 2002, 696–697. [Google Scholar] [CrossRef]
- Porta, F.; Prati, L. Selective oxidation of glycerol to sodium glycerate with gold-on-carbon catalyst: An insight into reaction selectivity. J. Catal. 2004, 224, 397–403. [Google Scholar] [CrossRef]
- Cai, J.; Ma, H.; Zhang, J.; Du, Z.; Huang, Y.; Gao, J.; Xu, J. Catalytic oxidation of glycerol to tartronic acid over Au/HY catalyst under mild conditions. Chin. J. Catal. 2014, 35, 1653–1660. [Google Scholar] [CrossRef]
- Villa, A.; Veith, G.M.; Ferri, D.; Weidenkaff, A.; Perry, K.A.; Campisi, S.; Prati, L. NiO as a peculiar support for metal nanoparticles in polyols oxidation. Catal. Sci. Technol. 2013, 3, 394–399. [Google Scholar] [CrossRef]
- Wang, D.; Villa, A.; Su, D.; Prati, L.; Schlogl, R. Carbon-supported gold nanocatalysts: Shape effect in the selective glycerol oxidation. ChemCatChem 2013, 5, 2717–2723. [Google Scholar] [CrossRef]
- Sobczak, I.; Jagodzinska, K.; Ziolek, M. Glycerol oxidation on gold catalysts supported on group five metal oxides—A comparative study with other metal oxides and carbon based catalyst. Catal. Today 2010, 158, 121–129. [Google Scholar] [CrossRef]
- Wolski, L. Factors affecting the activity and selectivity of niobia-based gold catalysts in liquid phase glycerol oxidation. Catal. Today 2020, 354, 36–43. [Google Scholar] [CrossRef]
- Murthy, P.R.; Selvam, P. The enhanced catalytic performance and stability of ordered mesoporous carbon supported nano-gold with high structural integrity for glycerol oxidation. Chem. Rec. 2019, 19, 1913–1925. [Google Scholar] [CrossRef]
- Zope, B.N.; Davis, S.E.; Davis, R.J. Influence of reaction conditions on diacid formation during Au-catalyzed oxidation of glycerol and hydroxymethylfurfural. Top. Catal. 2012, 55, 24–32. [Google Scholar] [CrossRef]
- Pakrieva, E.; Ribeiro, A.P.C.; Kolobova, E.; Martins, L.M.D.R.S.; Carabineiro, S.A.C.; German, D.; Pichugina, D.; Jiang, C.; Pombeiro, A.J.L.; Bogdanchikova, N.; et al. Supported gold nanoparticles as catalysts in peroxidative and aerobic oxidation of 1-phenylethanol under mild conditions. Nanomaterials 2020, 10, 151. [Google Scholar] [CrossRef]
- Pakrieva, E.; Kolobova, E.; Kotolevich, Y.; Pascual, L.; Carabineiro, S.A.C.; Kharlanov, A.N.; Pichugina, D.; Nikitina, N.; German, D.; Zepeda Partida, T.A.; et al. Effect of gold electronic state on the catalytic performance of nano gold catalysts in n-octanol oxidation. Nanomaterials 2020, 10, 880. [Google Scholar] [CrossRef]
- Kolobova, E.; Maki-Arvela, P.; Pestryakov, A.; Pakrieva, E.; Pascual, L.; Smeds, A.; Rahkila, J.; Sandberg, T.; Peltonen, J.; Murzin, D.Y. Reductive amination of ketones with benzylamine over gold supported on different oxides. Catal. Lett. 2019, 149, 3432–3446. [Google Scholar] [CrossRef]
- Kolobova, E.; Kotolevich, Y.; Pakrieva, E.G.; Mamontov, G.; Farias, M.H.; Bogdanchikova, N.; Pestryakov, A. Causes of activation and deactivation of modified nanogold catalysts during prolonged storage and redox treatments. Molecules 2016, 21, 486. [Google Scholar] [CrossRef]
- Pakrieva, E.; Kolobova, E.; Mamontov, G.; Bogdanchikova, N.; Farias, M.H.; Pascual, L.; Cortés Corberán, V.; Martinez Gonzalez, S.; Carabineiro, S.A.C.; Pestryakov, A. Green oxidation of n-octanol on supported nanogold catalysts: Formation of gold active sites under combined effect of gold content, additive nature and redox pretreatment. ChemCatChem 2019, 11, 1615–1624. [Google Scholar] [CrossRef]
- Kolobova, E.N.; Pakrieva, E.G.; Carabineiro, S.; Bogdanchikova, N.; Kharlanov, A.; Kazantsev, S.O.; Hemming, J.; Mäki-Arvela, P.; Pestryakov, A.N.; Murzin, D. Oxidation of a wood extractive betulin to biologically active oxo-derivatives using supported gold catalysts. Green Chem. 2019, 21, 3370–3382. [Google Scholar] [CrossRef]
- Kolobova, E.; Mäki-Arvela, P.; Grigoreva, A.; Pakrieva, E.; Carabineiro, S.A.C.; Peltonen, J.; Kazantsev, S.; Bogdanchikova, N.; Pestryakov, A.; Murzin, D.Y. Catalytic oxidative transformation of betulin to its valuable oxo-derivatives over gold supported catalysts: Effect of support nature. Cattod 2020. Accepted. [Google Scholar] [CrossRef]
- Yang, J.; Guan, Y.; Verhoeven, T.; Santen, R.; Li, С.; Hensen, E.J.M. Basic metal carbonate supported gold nanoparticles: Enhanced performance in aerobic alcohol oxidation. Green Chem. 2009, 11, 322–325. [Google Scholar] [CrossRef]
- Fang, W.; Chen, J.; Zhang, Q.; Deng, W.; Wang, Y. Hydrotalcite-supported gold catalyst for the oxidant-free dehydrogenation of benzyl alcohol: Studies on support and gold size effects. Chem. Eur. J. 2011, 17, 1247–1256. [Google Scholar] [CrossRef]
- Ketchie, W.C.; Fang, Y.-L.; Wong, M.S.; Murayama, M.; Davis, R.J. Influence of gold particle size on the aqueous-phase oxidation of carbon monoxide and glycerol. J. Catal. 2007, 250, 94–101. [Google Scholar] [CrossRef]
- Sankar, M.; Dimitratos, N.; Knight, D.W.; Carley, A.F.; Tiruvalam, R.; Kiely, C.J.; Thomas, D.; Hutchings, G.J. Oxidation of glycerol to glycolate by using supported gold and palladium nanoparticles. ChemSusChem 2009, 2, 1145–1151. [Google Scholar] [CrossRef]
- Ketchie, W.C.; Murayama, M.; Davis, R.J. Selective oxidation of glycerol over carbon-supported AuPd catalysts. J. Catal. 2007, 250, 264–273. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
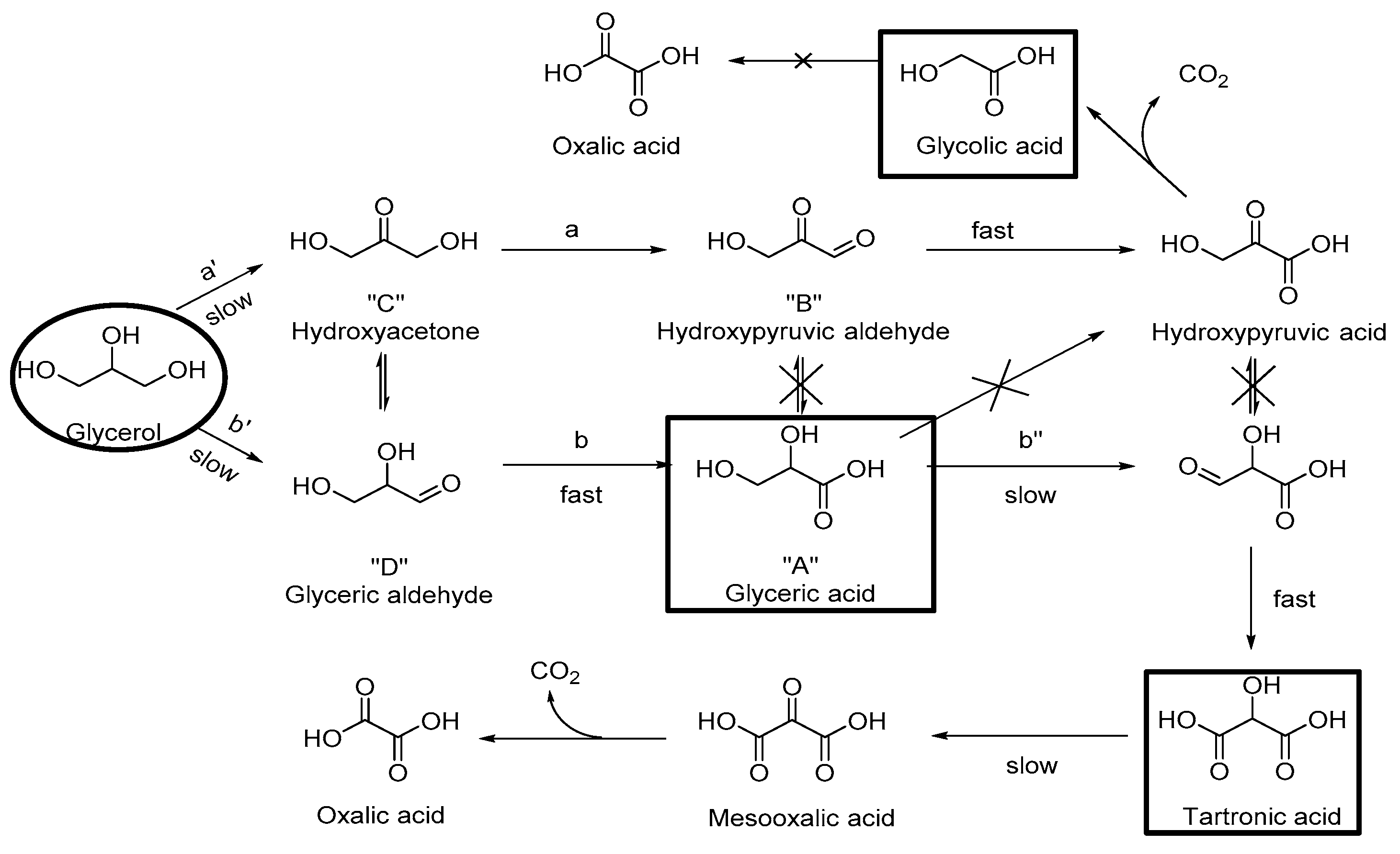{kind=link}
| Catalyst | pO2, atm | T, °C | NaOH eqv. | Reaction Time, h | R a | Conversion, % | Selectivity, % | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Glyceric Acid | Glycolic Acid | Tartronic Acid | ||||||||
| 1% Au/Aсtive carbon (AC) | 3 | 50 | 1 | 3 | 538 | 56 | 100 | 0 | 0 | [27] |
| 1% Au/Graphite | 3 | 60 | 2 | 3 | 538 | 54 | 100 | 0 | 0 | [27] |
| 1% Au/Graphite | 3 | 60 | 1 | 3 | 540 | 43 | 80 | 0 | 20 | [27] |
| 1% Au/Graphite | 6 | 60 | 2 | 3 | 214 | 91 | 92 | 0 | 6 | [27] |
| 1% Au/AC Cit-calc b | 3 | 30 | 4 | 6 | 500 | 90 | 92 | No info | No info | [28] |
| 1% Au/AC Cit-calc | 3 | 60 | 1 | 20 | 500 | 99 | 89 | No info | No info | [28] |
| 1% Au/AC PVAс | 3 | 60 | 1 | 6 | 500 | 99 | 45 | No info | No info | [28] |
| 1.5% Au/AC | 3 | 60 | 4 | 9 | 150 | 90 | 89 | - | 7 | [29] |
| 1.5% Au/CeO2 | 3 | 60 | 4 | 9 | 150 | 98 | 2 | - | 25 | [29] |
| 1.5% Au/NaY d | 3 | 60 | 4 | 9 | 150 | 98 | 27 | - | 44 | [29] |
| 1.5% Au/REY d | 3 | 60 | 4 | 9 | 150 | 99 | 5 | - | 70 | [29] |
| 1.5% Au/HY d | 3 | 60 | 4 | 9 | 150 | 98 | 3 | - | 82 | [29] |
| 1% Au/TiO2 | 3 | 50 | 4 | 4 | 1000 | 68 | 81 | 3 | 5 | [30] |
| 1% Au/NiO | 3 | 50 | 4 | 4 | 1000 | 99 | 55 | 11 | 9 | [30] |
| 1% Au/CNF-PS e | 3 | 50 | 4 | 4 | 1000 | 64 | 55 | 22 | - | [31] |
| 1% Au/CNF-HT f | 3 | 50 | 4 | 4 | 1000 | 70 | 22 | 35 | - | [31] |
| 1% Au-Nb2O5 | 6 | 60 | 2 | 15 | 980 | 94 | 43 | 7 | 5 | [32] |
| 1% Au-Nb2O5 | 6 | 60 | 2 | 5 | 980 | 67 | 47 | 5 | 5 | [32] |
| 2% Au-Nb2O5 | 6 | 60 | 2 | 5 | 500 | 51 | 87 | 3 | 3 | [33] |
| 1% Au/CMK-3 g | 7 | 60 | 4 | 5 | 500 | 82 | 71 | 15 | 8 | [34] |
| 1% Au/NCCR-56 g | 7 | 60 | 4 | 5 | 500 | 84 | 70 | 17 | 7 | [34] |
| 1.6% Au/TiO2 | 11 | 60 | 2 | 3 | 8000 | 83 | 61 | 34 | 3 | [35] |
| 1.6% Au/TiO2 | 11 | 60 | 2 | 3 | 2000 | 91 | 63 | 14 | 13 | [35] |
| 1.6% Au/TiO2 | 11 | 60 | 2 | 3 | 350 | 100 | 50 | 7 | 25 | [35] |
| Entry | Catalyst | TOF b | Conv. % | Selectivity at the Highest Conversion, % | |||||
|---|---|---|---|---|---|---|---|---|---|
| Glyceric Acid | Glycolic Acid | Tartronic Acid | Formic Acid | Lactic Acid | Oxalic Acid | ||||
| 1 | Au/TiO₂ | 223 | 51 | 66 | 14 | 4 | 9 | 5 | 2 |
| 2 | Au/CeO₂/TiO₂ c | 1186 | 100 | 44 | 14 | 24 | 8 | 6 | 4 |
| 3 | Au/CeO₂/TiO₂ d | 1186 | 100 | 38 | 13 | 32 | 7 | 5 | 4 |
| 4 | Au/CeO₂ | 1225 | 100 | 67 | 9 | 12 | 5 | 6 | 1 |
| 5 | Au/La₂O₃/TiO₂ | 1277 | 100 | 68 | 11 | 8 | 4 | 8 | 1 |
| 6 | Au/La₂O₃ | 264 | 56 | 71 | 11 | 3 | 8 | 6 | 1 |
| 7 | Au/MgO/TiO₂ | 1295 | 100 | 56 | 12 | 15 | 7 | 8 | 2 |
| 8 | Au/MgO | 176 | 27 | 60 | 17 | 3 | 12 | 7 | 1 |
| Entry | Alkaline Earth Base | T, °C | Gly/Base | Conv. % | Selectivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| Glyceric Acid | Glycolic Acid | Tartronic Acid | Formic Acid | Oxalic Acid | |||||
| 1 | SrO | 50 | 12 | 7 | 48 | 22 | 4 | 26 | 0 |
| 2 | MgO | 50 | 12 | 10 | 48 | 27 | 8 | 11 | 6 |
| 3 | CaO | 50 | 12 | 8 | 48 | 23 | 4 | 25 | 0 |
| 4 | SrO | 80 | 12 | 9 | 53 | 23 | 4 | 20 | 1 |
| 5 | MgO | 80 | 12 | 14 | 53 | 27 | 5 | 9 | 6 |
| 6 | CaO | 80 | 12 | 13 | 40 | 29 | 3 | 28 | 0 |
| 7 | MgO | 80 | 4 | 16 | 56 | 27 | 10 | 0 | 7 |
| 8 | MgO | 95 | 4 | 20 | 52 | 29 | 12 | 0 | 7 |
| 9 | MgO b | 95 | 4 | 0.4 | 100 | 0 | 0 | 0 | 0 |
| 10 | No base | 95 | 0 | 2 | 54 | 0 | 0 | 46 | 0 |
| Entry | Sample | SBET, m2/g | EDX Au Content, wt.% | Au average Particle Size, nm | Au Relative Content, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Support | Au0 | Au1+ | Au3+ | Aund− | ||||
| 1 | Au/TiO2 | 50 | 55 | 4.0 | 3.0 | 75 | 14 | 11 | 0 |
| 2 | Au/CeO2/TiO2 | 46 | 48 | 4.1 | 2.8 | 68 | 20 | 12 | 0 |
| 3 | Au/CeO2 | 38 | 37 | 4.3 | 2.6 | 57 | 21 | 0 | 22 |
| 4 | Au/La2O3/TiO2 | 43 | 48 | 3.3 | 2.6 | 83 | 17 | 0 | 0 |
| 5 | Au/La2O3 | 10 | 9 | 4.5 | 3.6 | 50 | 12 | 0 | 38 |
| 6 | Au/MgO/TiO2 | 43 | 48 | 4.0 | 5.1 | 51 | 29 | 20 | 0 |
| 7 | Au/MgO | 312 | 141 | 3.9 | 6.1 | 100 | 0 | 0 | 0 |
| Entry | Sample | Concentration of Base Sites, µmol/g, % | TOF, h−1 | Dmean, nm | |
|---|---|---|---|---|---|
| Support | Catalyst | ||||
| 1 | Au/TiO2 | 88 | 73 | 223 | 3.0 |
| 2 | Au/CeO2/TiO2 | 70 | 153 | 1186 | 2.8 |
| 3 | Au/CeO2 | 122 | 230 | 1225 | 2.6 |
| 4 | Au/La2O3/TiO2 | 119 | 161 | 1277 | 2.6 |
| 5 | Au/La2O3 | 84 | 1523 | 264 | 3.6 |
| 6 | Au/MgO/TiO2 | 191 | 236 | 1295 | 5.1 |
| 7 | Au/MgO | 409 | 665 | 176 | 6.1 |
| Entry | Sample | Average Particle Size | Selectivity (%) towards Tartronic Acid after 3 h | |
|---|---|---|---|---|
| Before a Reaction | After Glycerol Oxidation | |||
| 1 | Au/CeO2/TiO2 | 2.8 | 2.9 | 32 |
| 2 | Au/MgO/TiO2 | 5.1 | 5.5 | 25 |
| 3 | Au/CeO2 | 2.6 | 3.6 | 19 |
| 4 | Au/La2O3/TiO2 | 2.6 | 4.8 | 15 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakrieva, E.; Kolobova, E.; German, D.; Stucchi, M.; Villa, A.; Prati, L.; Carabineiro, S.A.C.; Bogdanchikova, N.; Cortés Corberán, V.; Pestryakov, A. Glycerol Oxidation over Supported Gold Catalysts: The Combined Effect of Au Particle Size and Basicity of Support. Processes 2020, 8, 1016. https://doi.org/10.3390/pr8091016
Pakrieva E, Kolobova E, German D, Stucchi M, Villa A, Prati L, Carabineiro SAC, Bogdanchikova N, Cortés Corberán V, Pestryakov A. Glycerol Oxidation over Supported Gold Catalysts: The Combined Effect of Au Particle Size and Basicity of Support. Processes. 2020; 8(9):1016. https://doi.org/10.3390/pr8091016
Chicago/Turabian StylePakrieva, Ekaterina, Ekaterina Kolobova, Dmitrii German, Marta Stucchi, Alberto Villa, Laura Prati, Sónia. A.C. Carabineiro, Nina Bogdanchikova, Vicente Cortés Corberán, and Alexey Pestryakov. 2020. "Glycerol Oxidation over Supported Gold Catalysts: The Combined Effect of Au Particle Size and Basicity of Support" Processes 8, no. 9: 1016. https://doi.org/10.3390/pr8091016
APA StylePakrieva, E., Kolobova, E., German, D., Stucchi, M., Villa, A., Prati, L., Carabineiro, S. A. C., Bogdanchikova, N., Cortés Corberán, V., & Pestryakov, A. (2020). Glycerol Oxidation over Supported Gold Catalysts: The Combined Effect of Au Particle Size and Basicity of Support. Processes, 8(9), 1016. https://doi.org/10.3390/pr8091016