
Toward the Identification of Potential α-Ketoamide Covalent Inhibitors for SARS-CoV-2 Main Protease: Fragment-Based Drug Design and MM-PBSA Calculations

 , , and
, , and 
Abstract
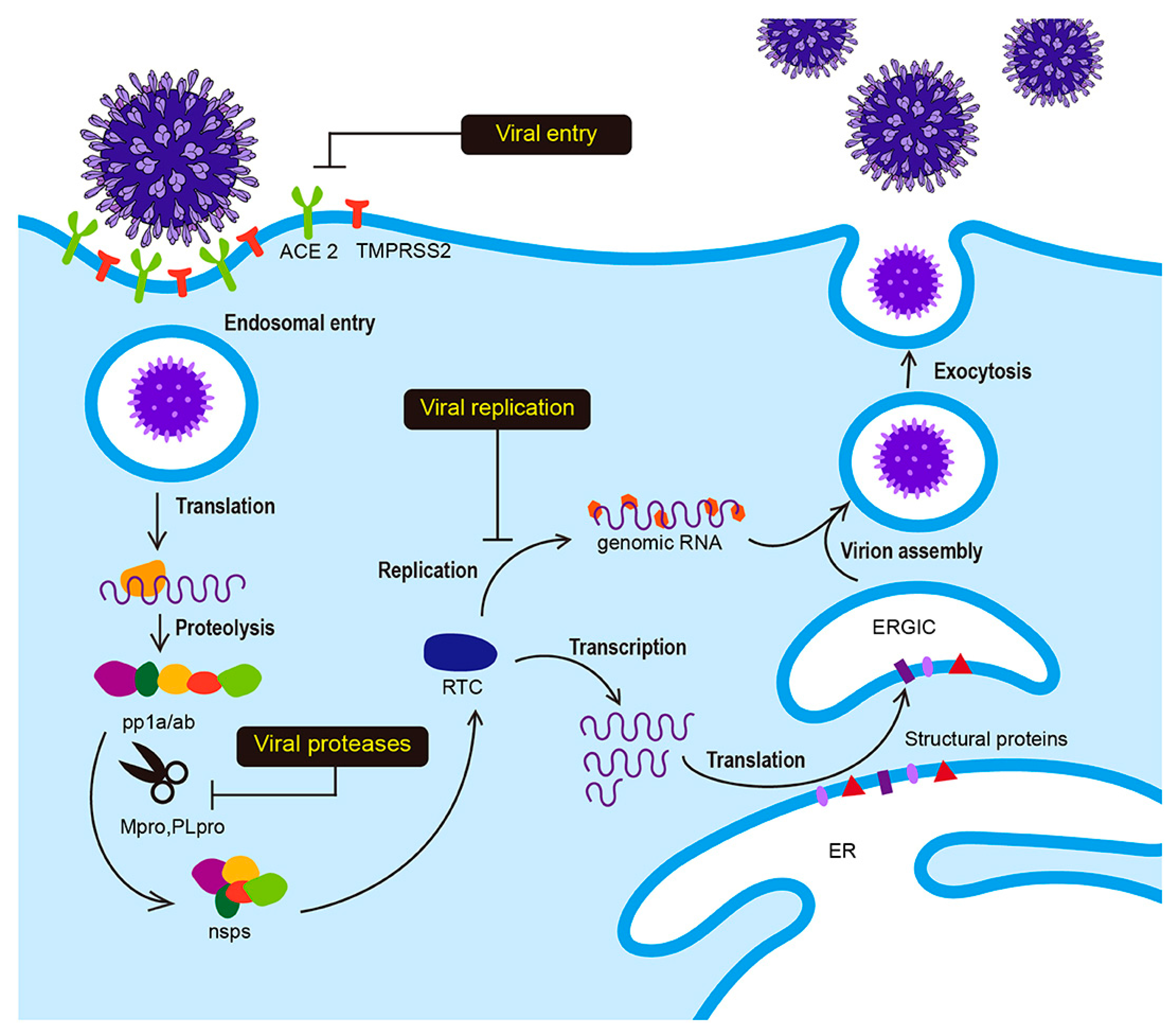
1. Introduction
2. Results
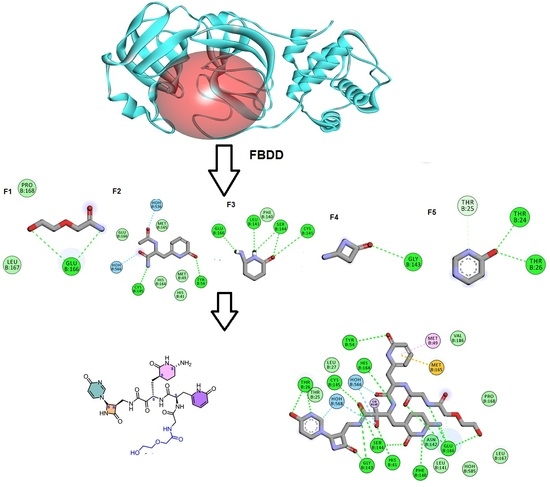
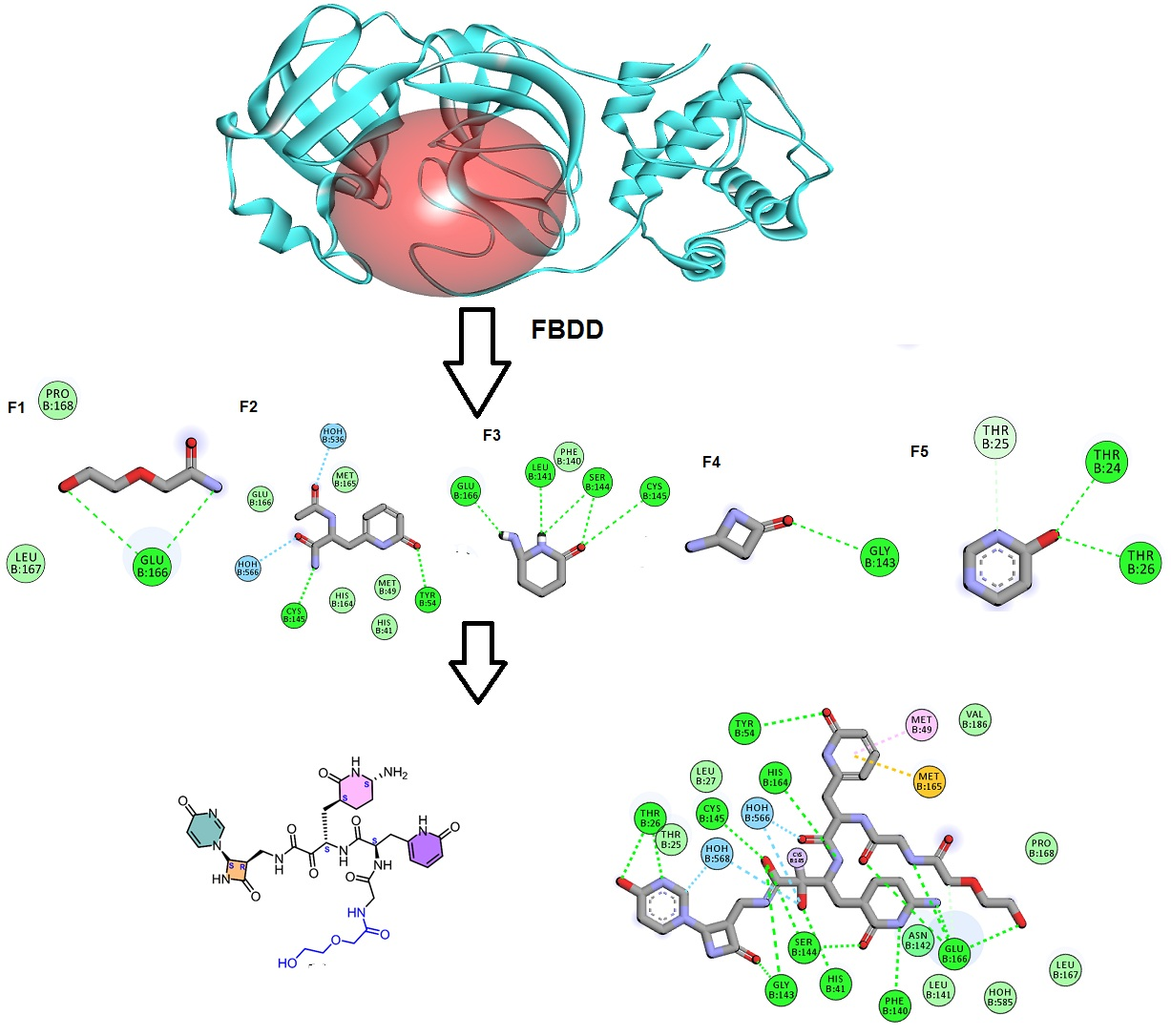
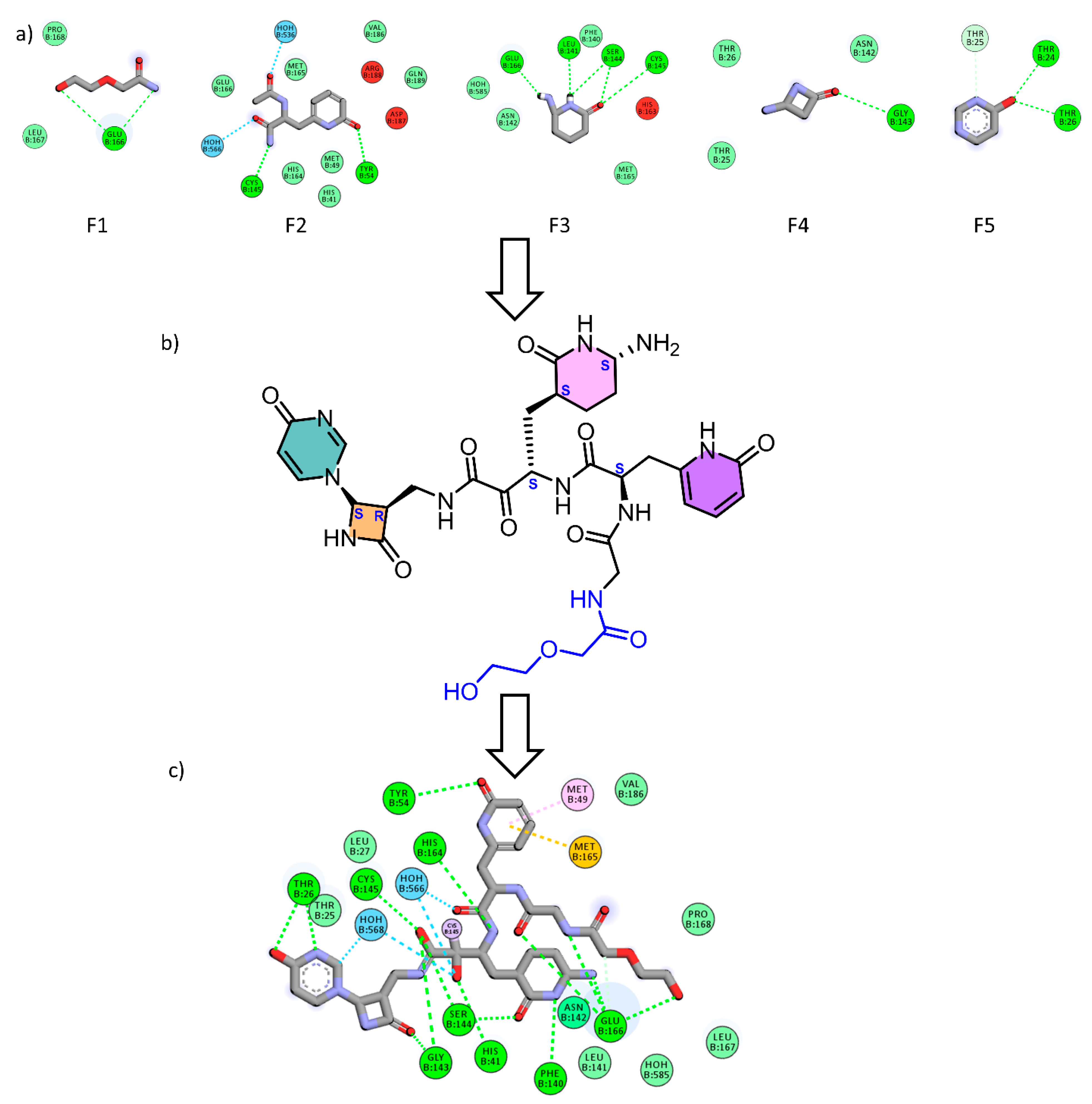
2.1. Fragment-Based Drug Design (FBDD)
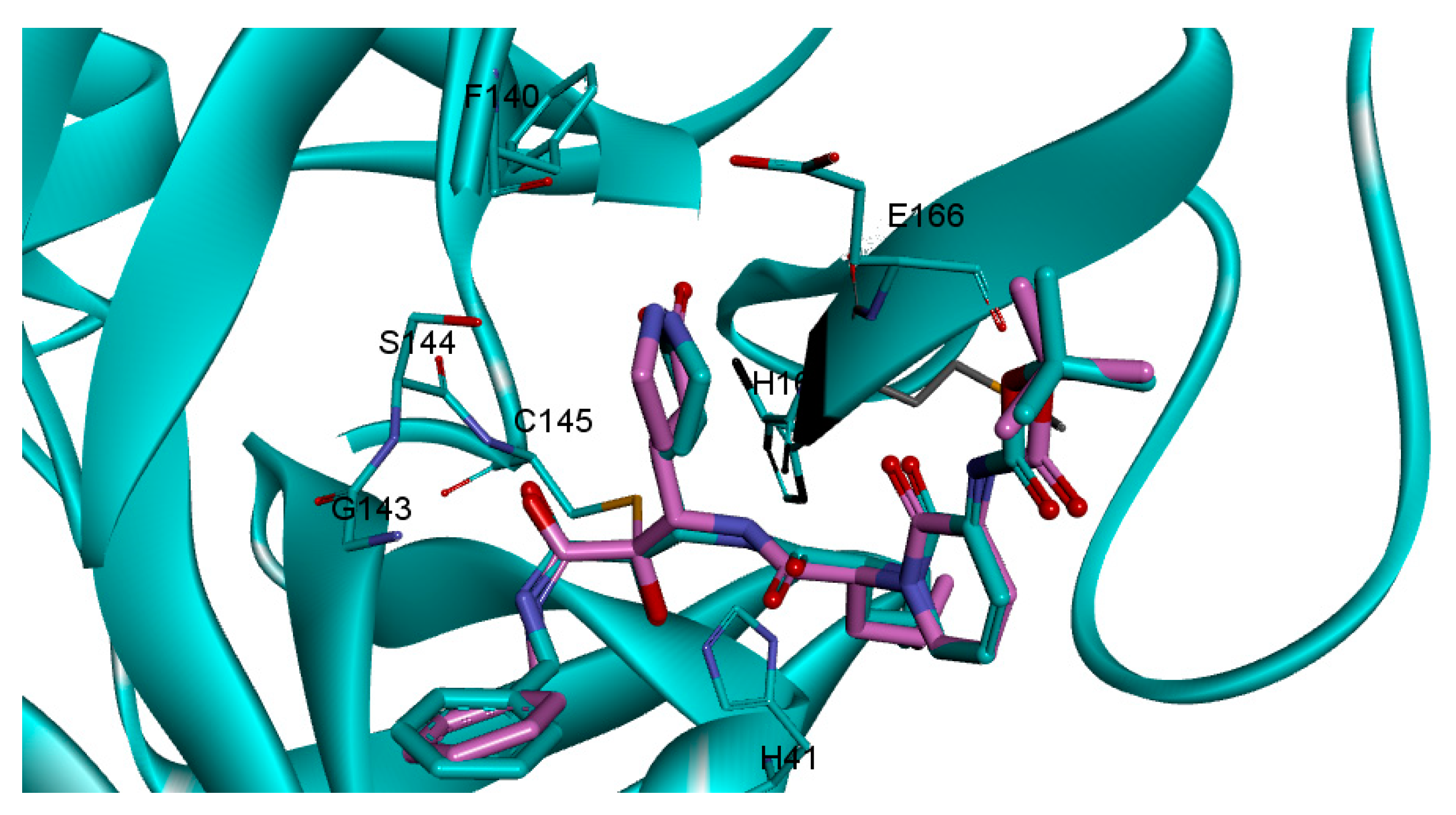
2.2. Covalent Docking
2.3. In Silico ADME and Toxicity Calculations
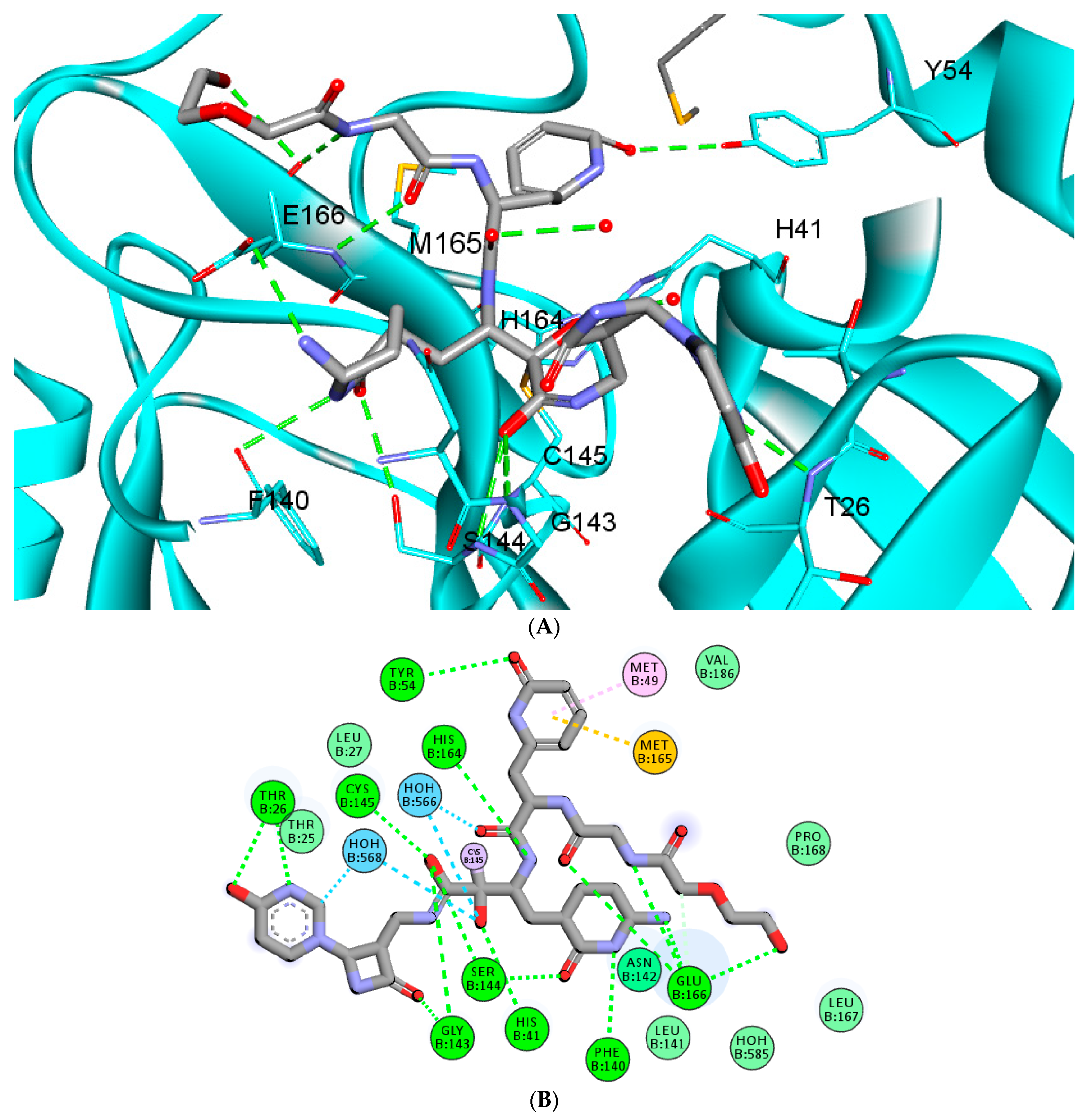
2.4. Molecular Dynamics (MD) Simulations
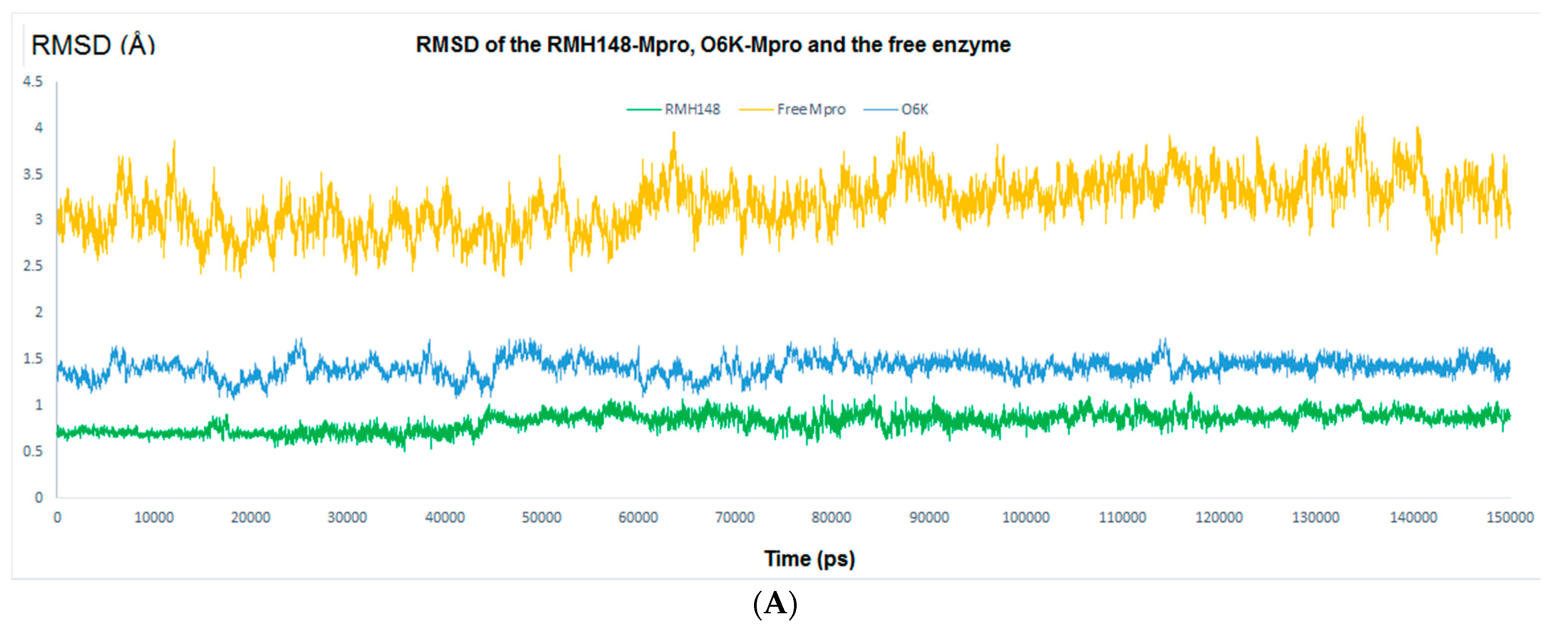
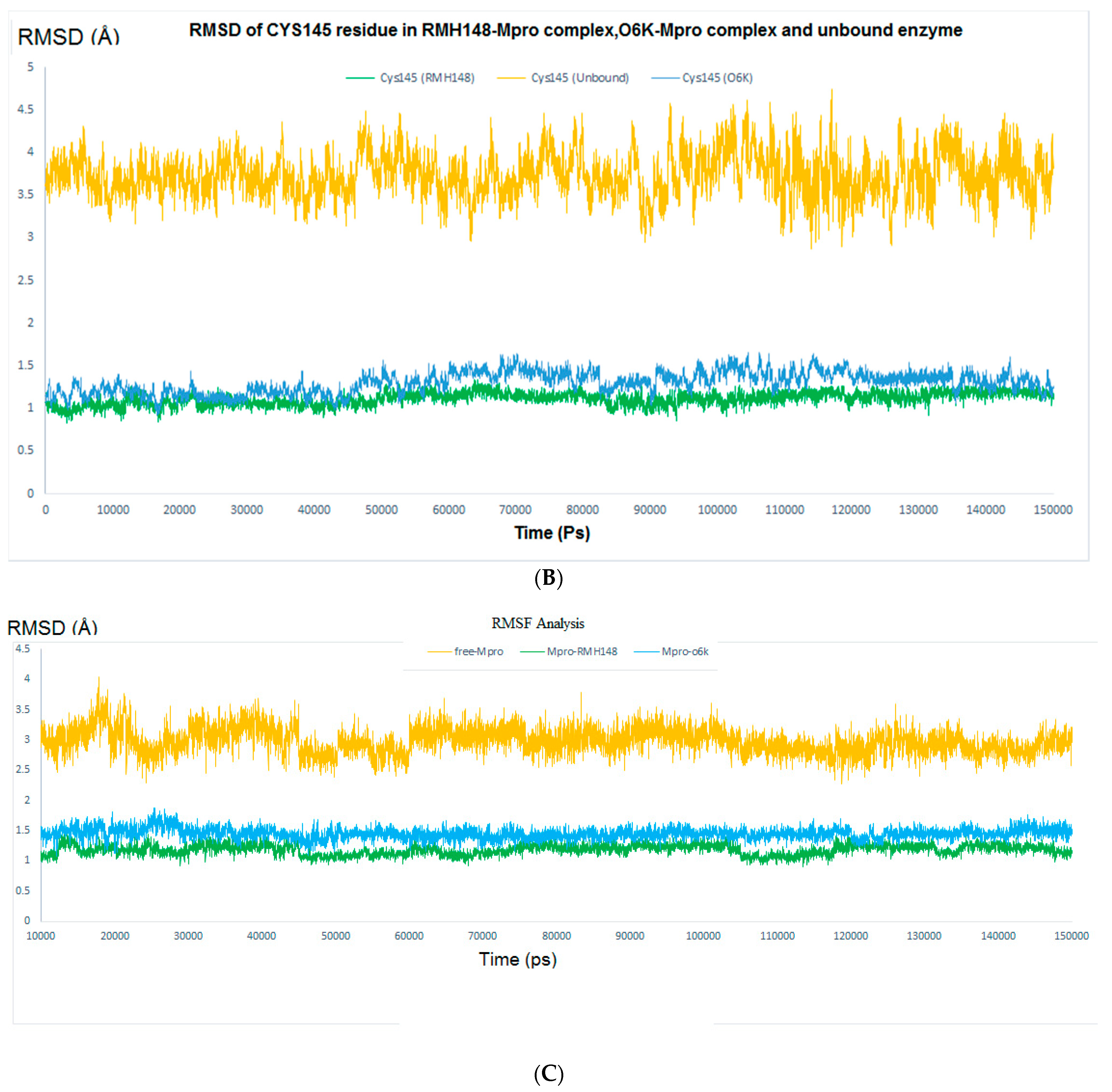
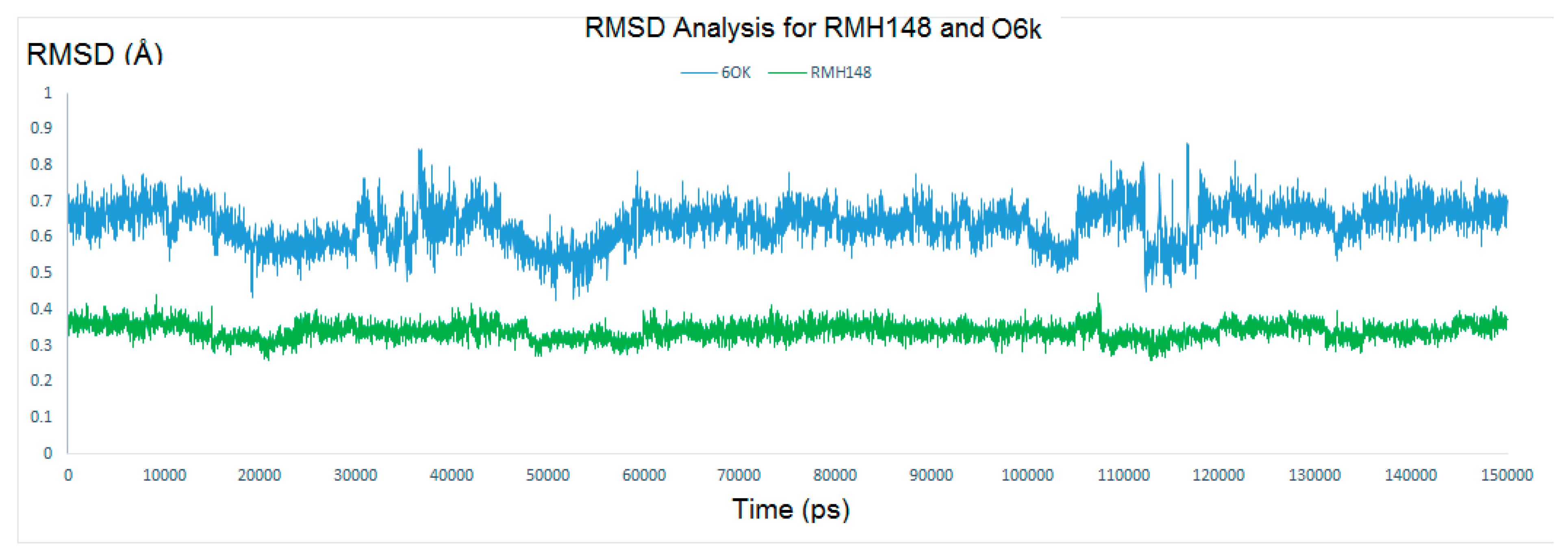
2.4.1. RMSD and RMSF Analysis
2.4.2. Evaluating the Stability of RMH148–Mpro
2.5. MM–PBSA (Molecular Mechanics Poisson-Boltzmann Surface Area) Binding Free Energy Calculations
3. Material and Methods
3.1. Fragment-Based Drug Design (FBDD)
3.2. Covalent Docking
3.3. In Silico ADME and Toxicity Calculations
3.4. Molecular Dynamics (MD)
3.5. MM-PBSA Calculation
4. Future Prospects for RMH148
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.-Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef]
- Borgio, J.F.; Alsuwat, H.S.; Al Otaibi, W.M.; Ibrahim, A.M.; Almandil, N.B.; Al Asoom, L.I.; Salahuddin, M.; Kamaraj, B.; AbdulAzeez, S. State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Arch. Med Sci. 2020, 16, 508–518. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef]
- Jeong, G.U.; Song, H.; Yoon, G.Y.; Kim, D.; Kwon, Y.-C. Therapeutic Strategies Against COVID-19 and Structural Characterization of SARS-CoV-2: A Review. Front. Microbiol. 2020, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Castro, C. The mechanism of action of ribavirin: Lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase. Curr. Opin. Infect. Dis. 2001, 14, 757–764. [Google Scholar] [CrossRef]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Ziebuhr, J.; Gorbalenya, A.; Snijder, E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, A.; Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Du, Q.-S.; Wang, S.-Q.; Zhu, Y.; Wei, D.-Q.; Guo, H.; Sirois, S.; Chou, K.-C. Polyprotein cleavage mechanism of SARS CoV Mpro and chemical modification of the octapeptide. Peptides 2004, 25, 1857–1864. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-like Proteinase from SARS Coronavirus Catalyzes Substrate Hydrolysis by a General Base Mechanism. Biochemistry 2004, 43, 4568–4574. [Google Scholar] [CrossRef] [PubMed]
- De Leuw, P.; Stephan, C. Protease inhibitors for the treatment of hepatitis C virus infection. GMS Infect. Dis. 2017, 5, 8. [Google Scholar]
- Wang, Y.; Lv, Z.; Chu, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV/AIDS Res. Palliat. Care 2015, 7, 95–104. [Google Scholar] [CrossRef]
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018, 38, 1295–1331. [Google Scholar] [CrossRef]
- De Kloe, G.E.; Bailey, D.; Leurs, R.; de Esch, I.J. Transforming fragments into candidates: Small becomes big in medicinal chemistry. Drug Discov. Today 2009, 14, 630–646. [Google Scholar] [CrossRef]
- Blum, L.C.; Reymond, J.-L. 970 Million Druglike Small Molecules for Virtual Screening in the Chemical Universe Database GDB-13. J. Am. Chem. Soc. 2009, 131, 8732–8733. [Google Scholar] [CrossRef]
- Roughley, S.D.; Hubbard, R.E. How Well Can Fragments Explore Accessed Chemical Space? A Case Study from Heat Shock Protein 90: Miniperspective. J. Med. Chem. 2011, 54, 3989–4005. [Google Scholar] [CrossRef]
- Mortenson, P.N.; Murray, C.W. Assessing the lipophilicity of fragments and early hits. J. Comput. Mol. Des. 2011, 25, 663–667. [Google Scholar] [CrossRef]
- Kumar, A.; Voet, A.; Zhang, K. Fragment Based Drug Design: From Experimental to Computational Approaches. Curr. Med. Chem. 2012, 19, 5128–5147. [Google Scholar] [CrossRef]
- El Hassab, M.A.; Shoun, A.A.; Al-Rashood, S.T.; Al-Warhi, T.; Eldehna, W.M. Identification of a New Potential SARS-COV-2 RNA-Dependent RNA Polymerase Inhibitor via Combining Fragment-Based Drug Design, Docking, Molecular Dynamics, and MM-PBSA Calculations. Front. Chem. 2020, 8, 584894. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef]
- Evensen, E.; Joseph-McCarthy, D.; Karplus, M. MCSS Version 2.1; Harvard University: Cambridge, MA, USA, 1997. [Google Scholar]
- Rimmert, B.; Sabet, S.; Ackad, E.; Yousef, M.S. A 3D structural model and dynamics of hepatitis C virus NS3/4A protease (genotype 4a, strain ED43) suggest conformational instability of the catalytic triad: Implications in catalysis and drug resistivity. J. Biomol. Struct. Dyn. 2013, 32, 950–958. [Google Scholar] [CrossRef] [PubMed]
- El-Hasab, M.A.E.-M.; El-Bastawissy, E.E.; El-Moselhy, T.F. Identification of potential inhibitors for HCV NS3 genotype 4a by combining protein–ligand interaction fingerprint, 3D pharmacophore, docking, and dynamic simulation. J. Biomol. Struct. Dyn. 2018, 36, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- El-Hassab, M.A.E.-M.; El-Bastawissy, E.E.; El-Moselhy, T.F. Identification of potential inhibitors for HCV NS5b of genotype 4a by combining dynamic simulation, protein–ligand interaction fingerprint, 3D pharmacophore, docking and 3D QSAR. J. Biomol. Struct. Dyn. 2019, 38, 4521–4535. [Google Scholar] [CrossRef]
- Nagarajan, H.; Narayanaswamy, S.; Vetrivel, U. Mutational landscape screening of methylene tetrahydrofolate reductase to predict homocystinuria associated variants: An integrative computational approach. Mutat. Res. Mol. Mech. Mutagen. 2020, 819–820, 111687. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.C. Open Source Drug Discovery. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. BIOVIA Workbook, Release 2016, BIOVIA Pipeline Pilot, Release 2016; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Böhm, H.-J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Caflisch, A.; Miranker, A.; Karplus, M. Multiple copy simultaneous search and construction of ligands in binding sites: Application to inhibitors of HIV-1 aspartic proteinase. J. Med. Chem. 1993, 36, 2142–2167. [Google Scholar] [CrossRef]
- Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Scholz, C.; Knorr, S.; Hamacher, K.; Schmidt, B. DOCKTITE A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment. J. Chem. Inf. Model. 2015, 23, 398–406. [Google Scholar] [CrossRef]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.-W.; Pandit, S.A.; Scott, H.L.; Jakobsson, E. An Improved United Atom Force Field for Simulation of Mixed Lipid Bilayers. J. Phys. Chem. B 2009, 113, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.H.; Cabeza de Vaca, I.; Liosi, M.E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Havranek, B.; Islam, S.M. Anin silicoapproach for identification of novel inhibitors as potential therapeutics targeting COVID-19 main protease. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogen Bond Name | Average Distance (A0) ± SD |
|---|---|
| Hydrogen bond with Tyrosine54 | 3.17 ± 0.06 |
| Hydrogen bond with Glutamic166 | 3.05 ± 0.12 |
| Hydrogen bond with Glutamic166 | 3.05 ± 0.08 |
| Hydrogen bond with Glutamic166 | 3.21 ± 0.1 |
| Hydrogen bond with Glutamic166 | 3.31 ± 0.12 |
| Hydrogen bond with Phenylalanine 140 | 2.98 ± 0.07 |
| Hydrogen bond with Serine 144 | 3.17 ± 0.11 |
| Hydrogen bond with Serine 144 | 3.18 ± 0.09 |
| Hydrogen bond with Glycine 143 | 3.25 ± 0.15 |
| Hydrogen bond with Glycine 143 | 3.29 ± 0.13 |
| Hydrogen bond with Threonine 26 | 2.43 ± 0.04 |
| Hydrogen bond with Threonine 26 | 2.58 ± 0.17 |
| Hydrogen bond with Histidine 41 | 3.02 ± 0.14 |
| Hydrogen bond with Histidine 164 | 3.00 ± 0.16 |
| Hydrogen bond with Cysteine 145 | 2.71 ± 0.07 |
| Complex | ΔE binding (kj/mol) | ΔE Electrostatic (kj/mol) | ΔE Van der Waals’ (kj/mol) | ΔE polar solvation (kj/mol) | SASA (kJ/mol) |
|---|---|---|---|---|---|
| RMH148–Mpro | −420 ± 21 | −149 ± 18 | −365 ± 26 | 131 ± 18 | −37 ± 2 |
| O6K–Mpro | −388 ± 20 | −138 ± 19 | −333 ± 23 | 117 ± 16 | −34 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassab, M.A.E.; Fares, M.; Amin, M.K.A.-H.; Al-Rashood, S.T.; Alharbi, A.; Eskandrani, R.O.; Alkahtani, H.M.; Eldehna, W.M. Toward the Identification of Potential α-Ketoamide Covalent Inhibitors for SARS-CoV-2 Main Protease: Fragment-Based Drug Design and MM-PBSA Calculations. Processes 2021, 9, 1004. https://doi.org/10.3390/pr9061004
Hassab MAE, Fares M, Amin MKA-H, Al-Rashood ST, Alharbi A, Eskandrani RO, Alkahtani HM, Eldehna WM. Toward the Identification of Potential α-Ketoamide Covalent Inhibitors for SARS-CoV-2 Main Protease: Fragment-Based Drug Design and MM-PBSA Calculations. Processes. 2021; 9(6):1004. https://doi.org/10.3390/pr9061004
Chicago/Turabian StyleHassab, Mahmoud A. El, Mohamed Fares, Mohammed K. Abdel-Hamid Amin, Sara T. Al-Rashood, Amal Alharbi, Razan O. Eskandrani, Hamad M. Alkahtani, and Wagdy M. Eldehna. 2021. "Toward the Identification of Potential α-Ketoamide Covalent Inhibitors for SARS-CoV-2 Main Protease: Fragment-Based Drug Design and MM-PBSA Calculations" Processes 9, no. 6: 1004. https://doi.org/10.3390/pr9061004
APA StyleHassab, M. A. E., Fares, M., Amin, M. K. A.-H., Al-Rashood, S. T., Alharbi, A., Eskandrani, R. O., Alkahtani, H. M., & Eldehna, W. M. (2021). Toward the Identification of Potential α-Ketoamide Covalent Inhibitors for SARS-CoV-2 Main Protease: Fragment-Based Drug Design and MM-PBSA Calculations. Processes, 9(6), 1004. https://doi.org/10.3390/pr9061004