
Depolymerization and Hydrogenation of Organosolv Eucalyptus Lignin by Using Nickel Raney Catalyst

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
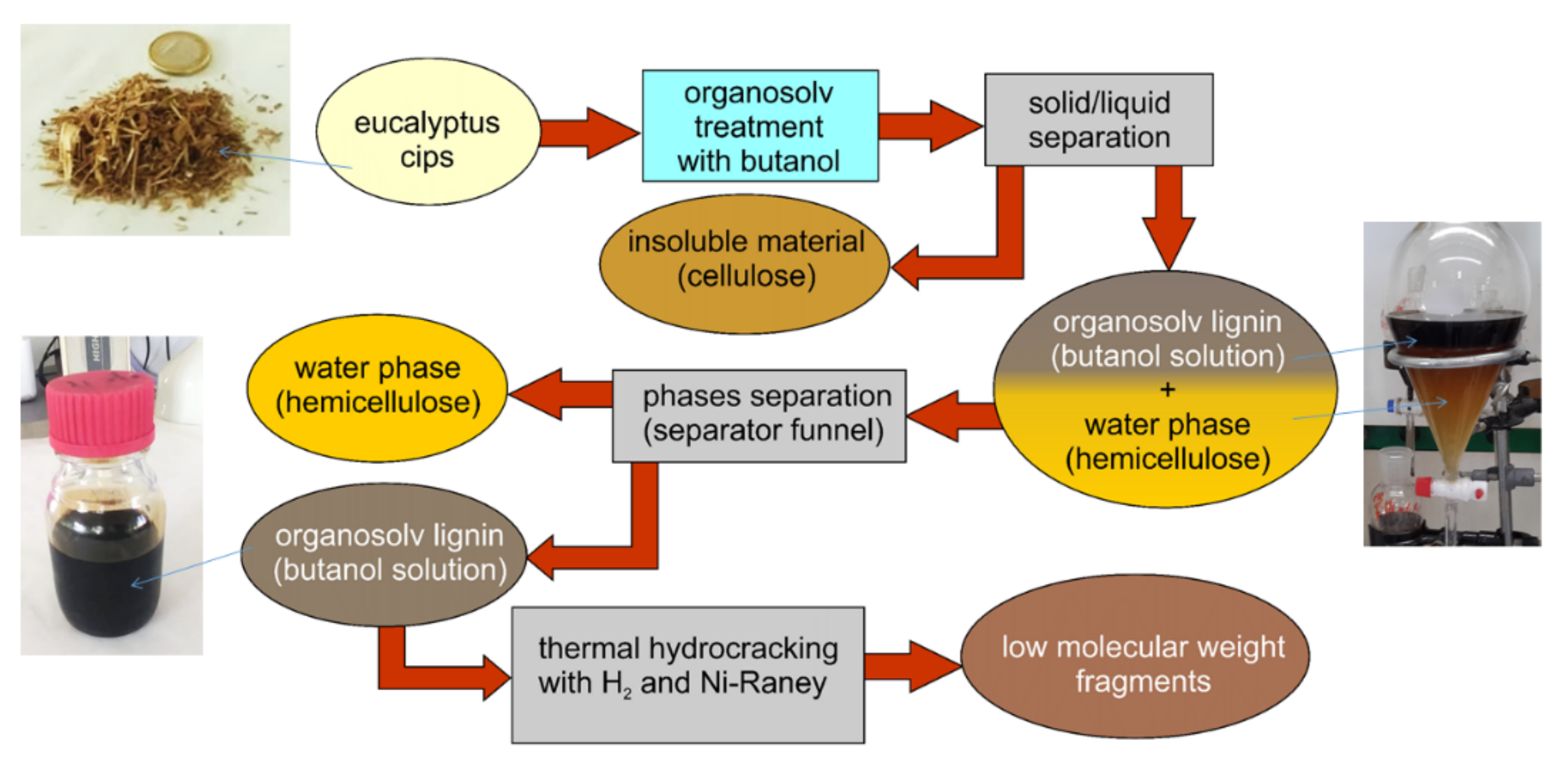
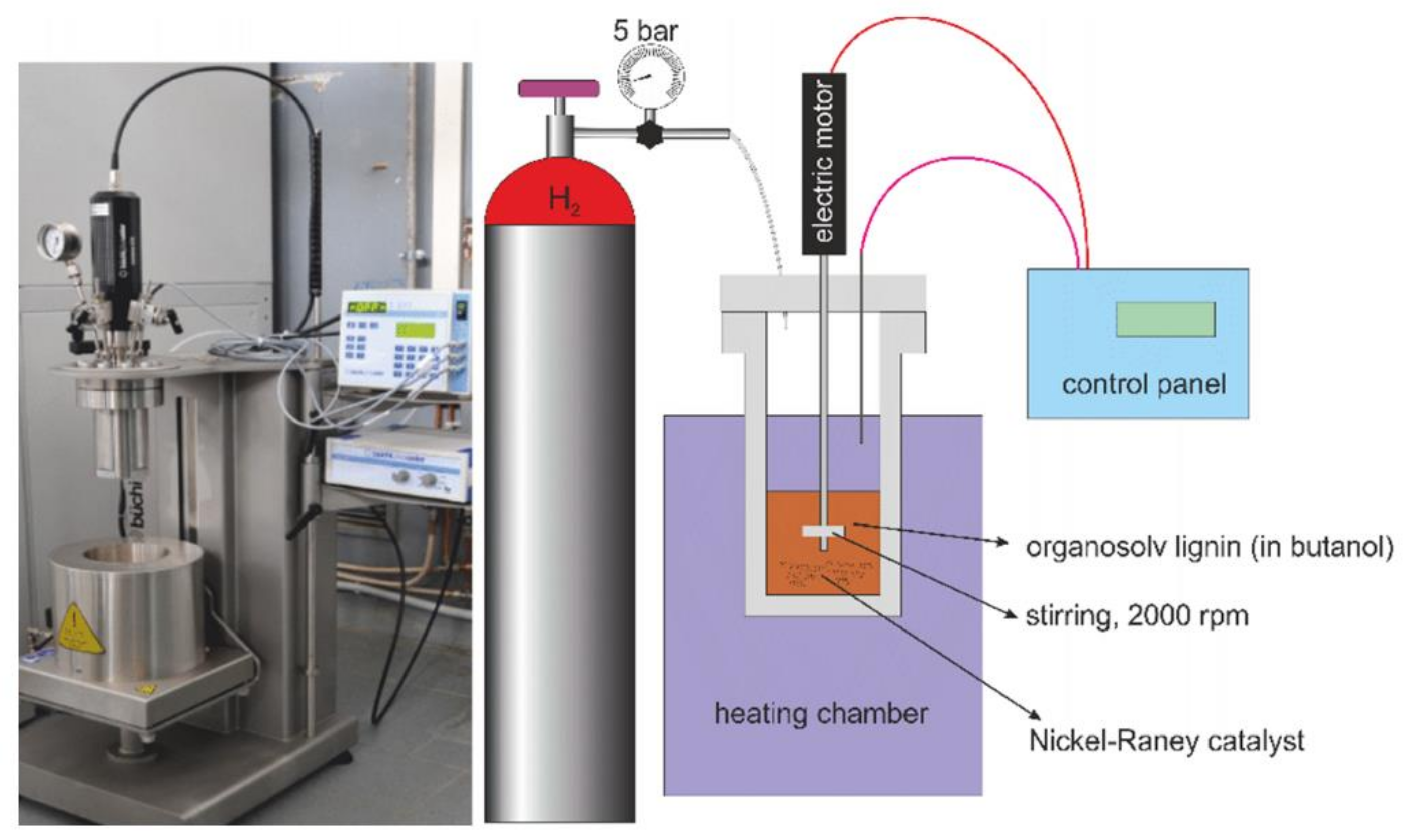
2.1. Lignin Extraction and Reaction System
2.2. Analytical Methods
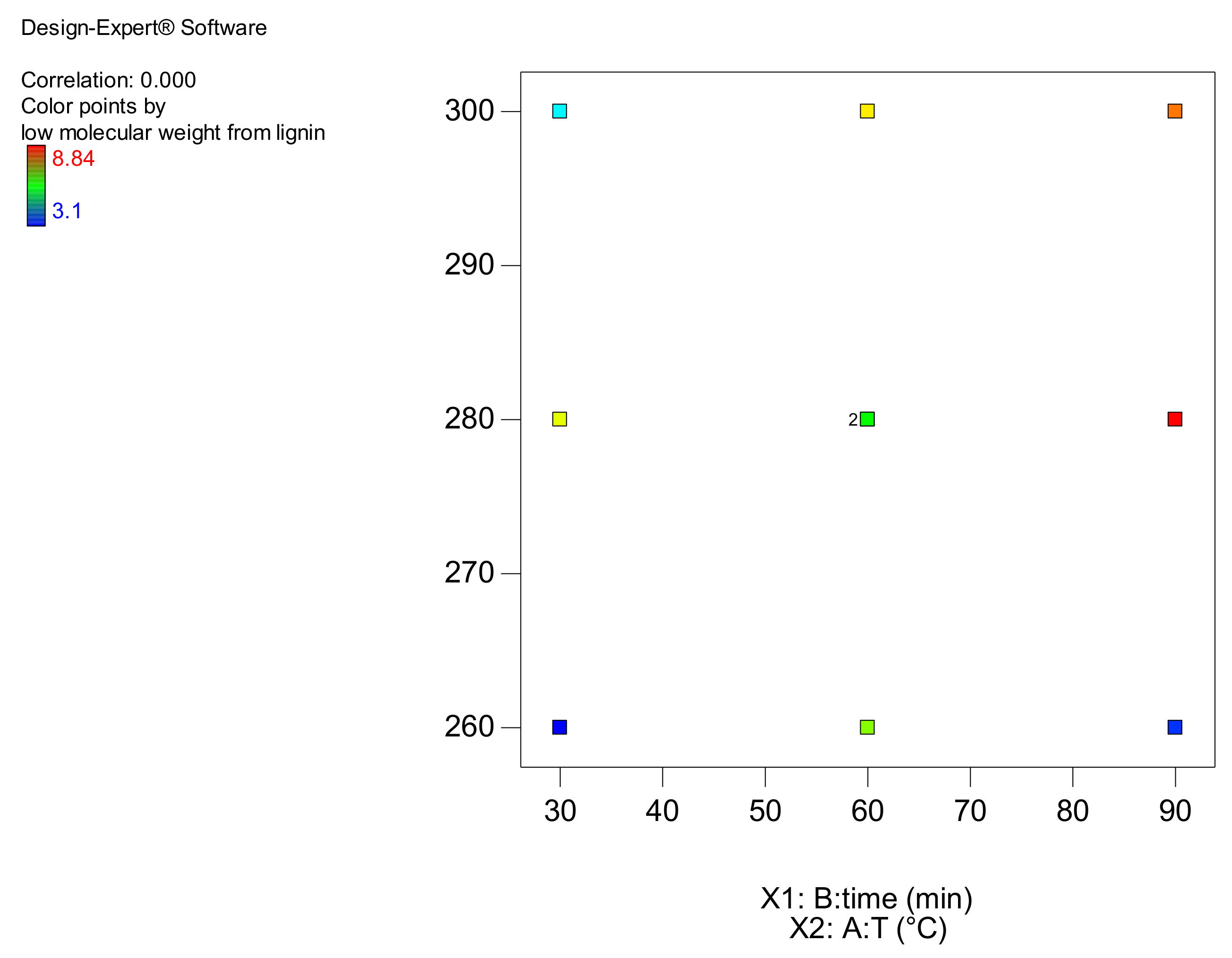
2.3. Experimental Design
- Temperature, °C, in the range 260–300;
- Time, minutes, in the range 30–90.
- Study Type: Response Surface;
- Design Type: Central Composite;
- Subtype: Randomized;
- Design Model: Quadratic.
3. Results and Discussion
3.1. Response 1: Diphenylmethane 4-ethyl
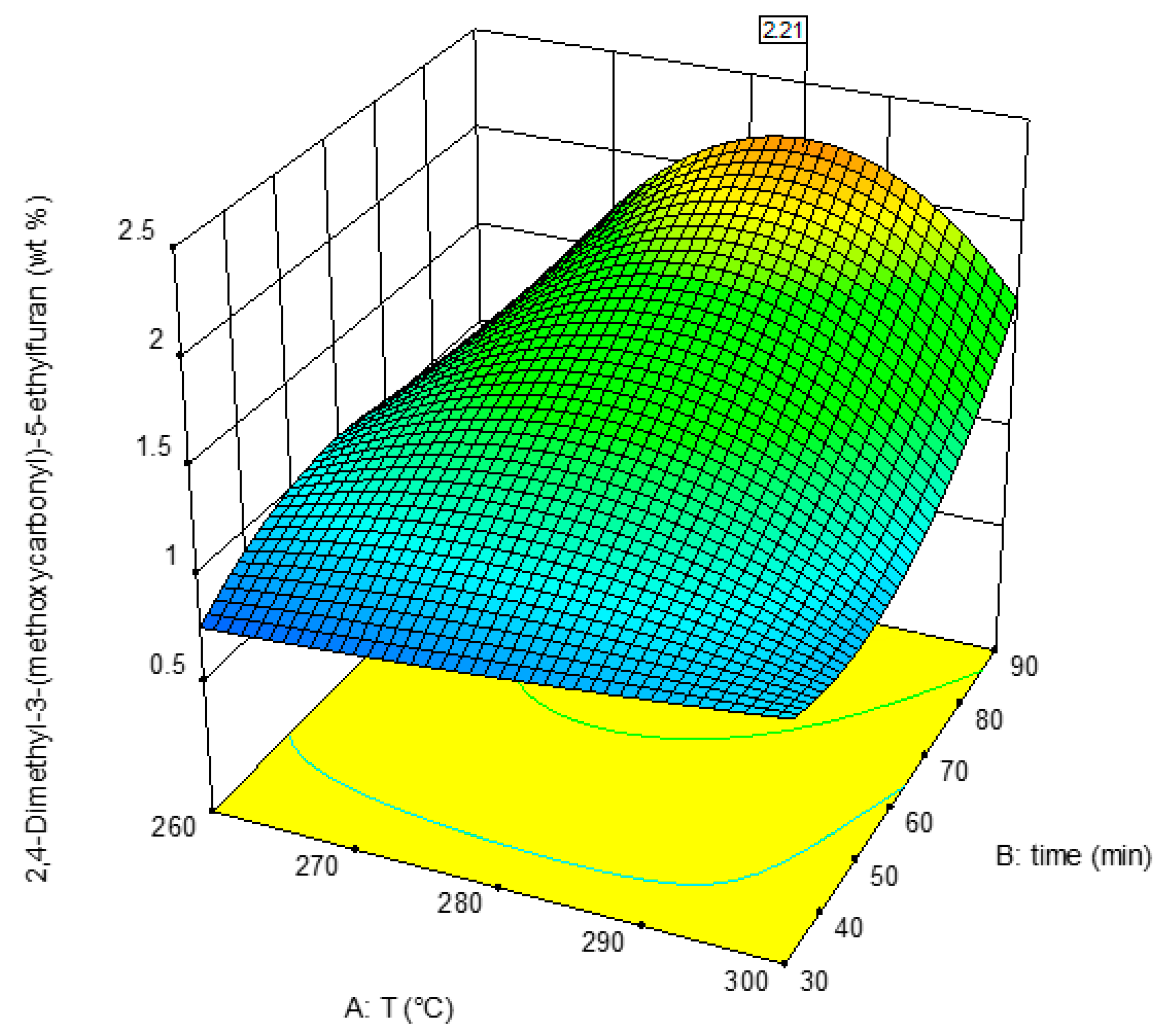
3.2. Response 2: 2,4-Dimethyl-3-(methoxycarbonyl)-5-ethylfuran
3.3. Response 3: 1,2,3-Trimethoxy Benzene
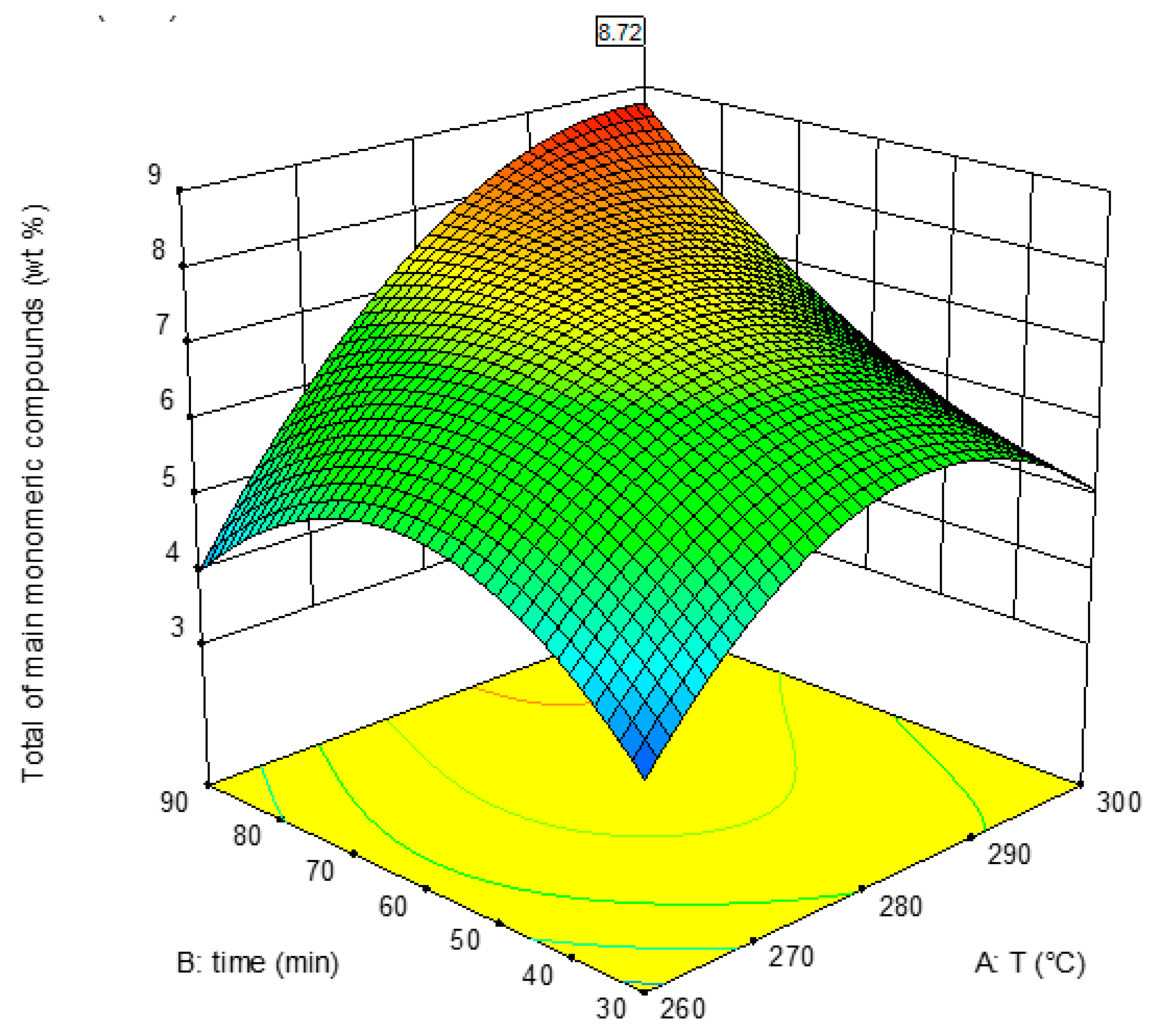
3.4. Response 4: Total of Main Monomeric Compounds
3.5. Response 5: Molecules Derived from Solvent
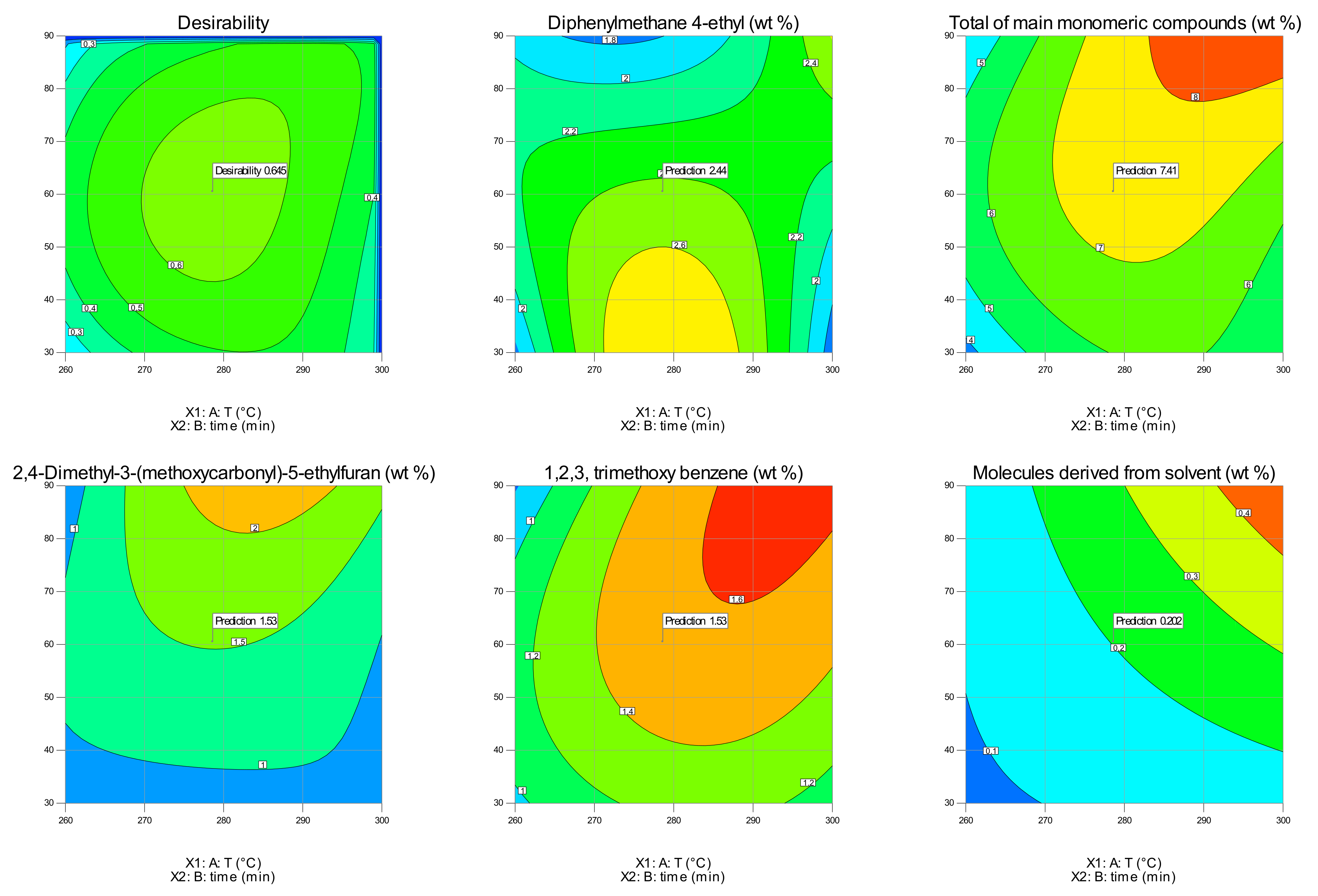
3.6. Optimization
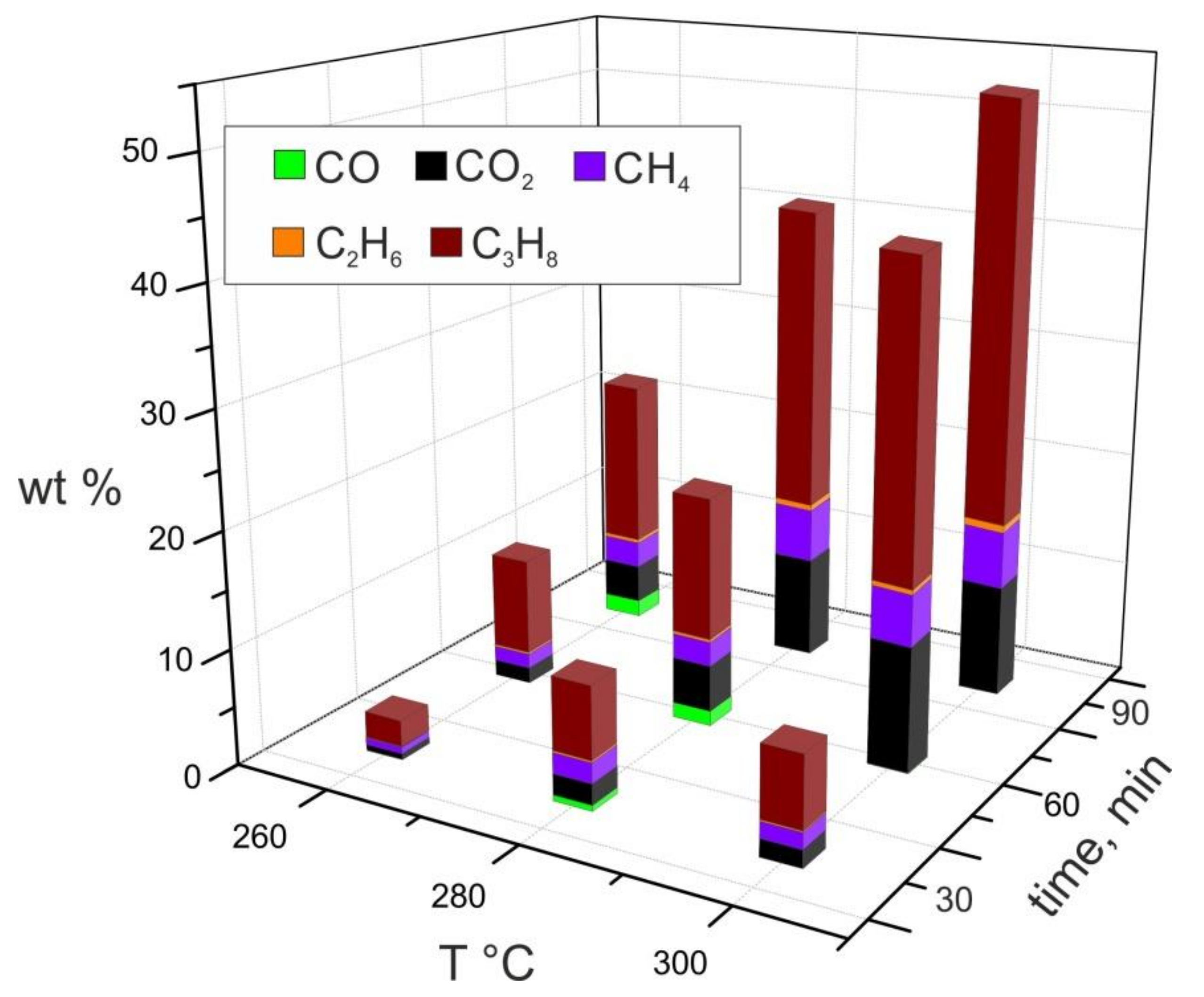
3.7. Gas Analyses
3.8. HPLC-GPC Analyses
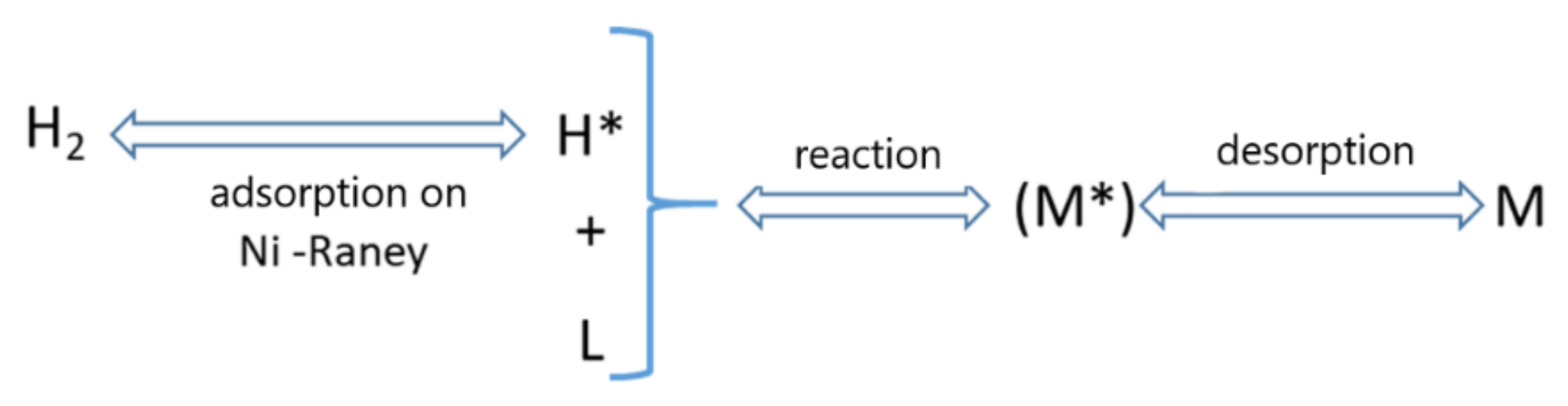
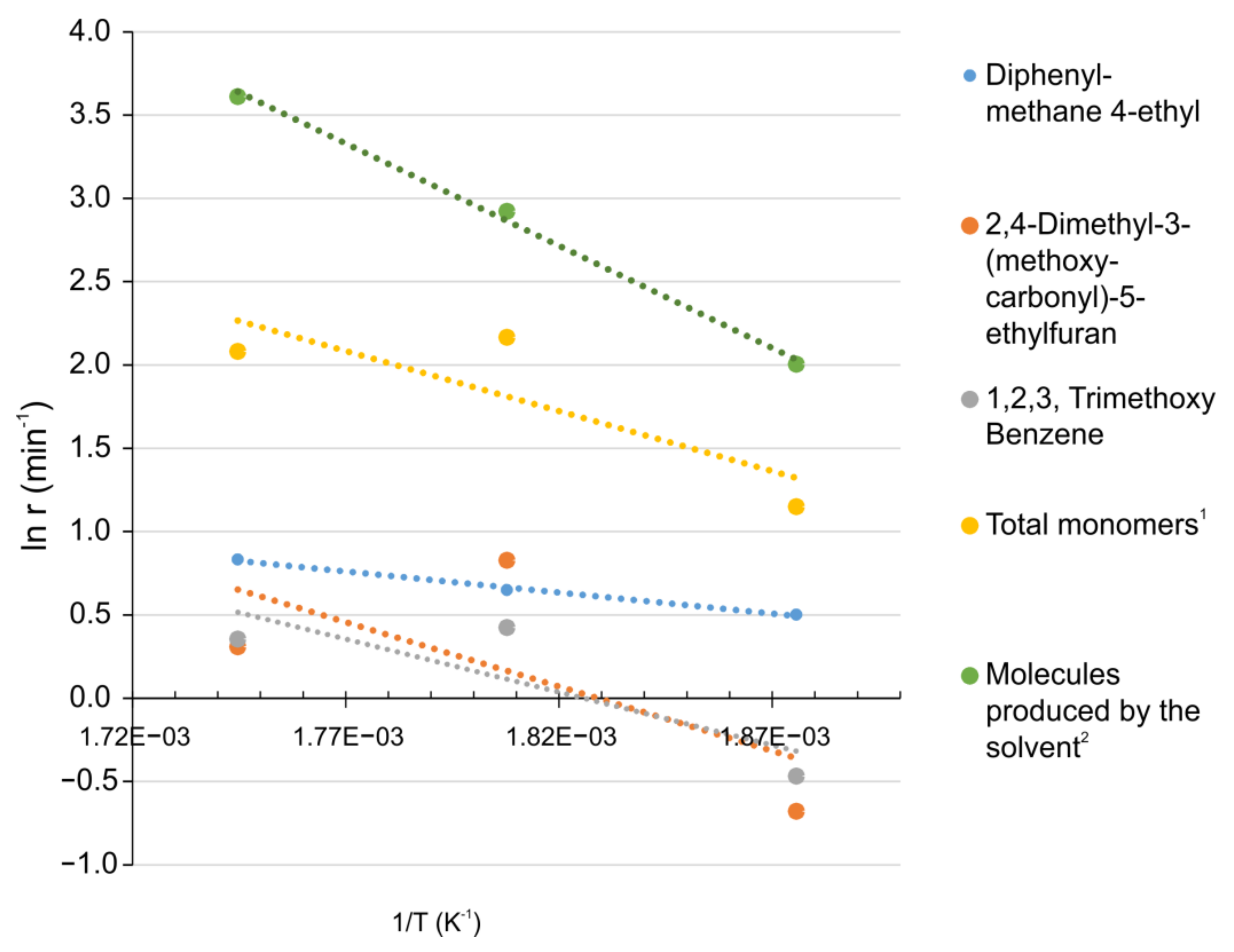
3.9. Kinetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, A.A.; Rapoport, A.; Kunze, G.; Kumar, S.; Singh, D.; Singh, B. Multifarious pretreatment strategies for the lignocellulosic substrates for the generation of renewable and sustainable biofuels: A review. Renew. Energy 2020, 160, 1228–1252. [Google Scholar]
- Qaseem, M.F.; Shaheen, H.; Wu, A.-M. Cell wall hemicellulose for sustainable industrial utilization. Renew. Sustain. Energy Rev. 2021, 144, 110996. [Google Scholar] [CrossRef]
- Banwell, M.G.; Pollard, B.; Liu, X.; Connal, L.A. Exploiting Nature’s Most Abundant Polymers: Developing New Pathways for the Conversion of Cellulose, Hemicellulose, Lignin and Chitin into Platform Molecules (and Beyond). Chem. Asian J. 2021, 16, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Robak, K.; Balcerek, M. Current state-of-the-art in ethanol production from lignocellulosic feedstocks. Microbiol. Res. 2020, 240, 126534. [Google Scholar] [CrossRef] [PubMed]
- Meneses, D.B.; de Oca-Vásquez, G.M.; Vega-Baudrit, J.R.; Rojas-Álvarez, M.; Corrales-Castillo, J.; Murillo-Araya, L.C. Pretreatment Methods of Lignocellulosic Wastes into Value-Added Products: Recent Advances and Possibilities. Biomass Convers. Biorefinery 2020, 1, 1–18. [Google Scholar]
- Banoub, J.; Delmas, G., Jr.; Joly, N.; Mackenzie, G.; Cachet, N.; Benjelloun-Mlayah, B.; Delmas, M. A critique on the structural analysis of lignins and application of novel tandem mass spectrometric strategies to determine lignin sequencing. J. Mass Spectrom. 2015, 50, 5–48. [Google Scholar] [CrossRef]
- Achyuthan, K.E.; Achyuthan, A.M.; Adams, P.D.; Harper, S.M.J.C.; Simmons, B.A.; Singh, A.K. Supramolecular Self-Assembled Chaos: Polyphenolic Lignin’s Barrier to Cost-Effective Lignocellulosic Biofuels. Molecules 2010, 15, 8641–8688. [Google Scholar] [CrossRef] [Green Version]
- Abejón, R.; Pérez-Acebo, H.; Clavijo, L. Alternatives for Chemical and Biochemical Lignin Valorization: Hot Topics from a Bibliometric Analysis of the Research Published During the 2000–2016 Period. Process 2018, 6, 98. [Google Scholar] [CrossRef] [Green Version]
- Poveda-Giraldo, J.A.; Solarte-Toro, J.C.; Alzate, C.A.C. The potential use of lignin as a platform product in biorefineries: A review. Renew. Sustain. Energy Rev. 2021, 138, 110688. [Google Scholar] [CrossRef]
- Dikshit, P.K.; Jun, H.-B.; Kim, B.S. Biological conversion of lignin and its derivatives to fuels and chemicals. Korean J. Chem. Eng. 2020, 37, 387–401. [Google Scholar] [CrossRef]
- Singh, S.K. Biological treatment of plant biomass and factors affecting bioactivity. J. Clean. Prod. 2021, 279, 123546. [Google Scholar] [CrossRef]
- Dashtban, M.; Schraft, H.; Syed, T.A.; Qin, W. Fungal biodegradation and enzymatic modification of lignin. Int. J. Biochem. Mol. Biol. 2010, 1, 36–50. [Google Scholar] [PubMed]
- Bugg, T.D.; Ahmad, M.; Hardiman, E.M.; Singh, R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr. Opin. Biotechnol. 2011, 22, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Galebach, P.; Mcclelland, D.J.; Eagan, N.M.; Wittrig, A.M.; Buchanan, J.S.; Dumesic, J.A.; Gasser, C.A.; Hommes, G.; Schäffer, A.; Corvini, P.F.X. Multi-catalysis reactions: New prospects and challenges of biotechnology to valorize lignin. Appl. Microbiol. Biot 2012, 95, 1115–1134. [Google Scholar]
- Brown, M.E.; Chang, M.C. Exploring bacterial lignin degradation. Curr. Opin. Chem. Biol. 2014, 19, 1–7. [Google Scholar] [CrossRef]
- Roth, S.; Spiess, A.C. Laccases for biorefinery applications: A critical review on challenges and perspectives. Bioprocess. Biosyst. Eng. 2015, 38, 2285–2313. [Google Scholar] [CrossRef]
- Husarcíková, J.; Voß, H.; Domínguez de María, P.; Schallmey, A. Microbial β-etherases and glutathione lyases for lignin valorisation in biorefineries: Current state and future perspectives. Appl. Microbiol. Biotechnol. 2018, 102, 5391–5401. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, K.; Liu, P.; Han, H.; Zhao, S.; Kakade, A.; Khan, A.; Du, D.; Li, X. Lignin Depolymerization and Utilization by Bacteria. Bioresour. Technol. 2018, 269, 557–566. [Google Scholar] [CrossRef]
- Chio, C.; Sain, M.; Qin, W. Lignin utilization: A review of lignin depolymerization from various aspects. Renew. Sustain. Energ. Rev. 2019, 107, 232–249. [Google Scholar] [CrossRef]
- Weng, C.; Peng, X.; Han, Y. Depolymerization and conversion of lignin to value-added bioproducts by microbial and enzymatic catalysis. Biotechnol. Biofuels 2021, 14, 84. [Google Scholar] [CrossRef]
- Pandey, M.P.K.; Kim, C.S. Lignin depolymerization and conversion: A review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energ. Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Davis, K.; Rover, M.; Brown, R.; Bai, X.; Wen, Z.; Jarboe, L. Recovery and Utilization of Lignin Monomers as Part of the Biorefinery Approach. Energies 2016, 9, 808. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.J.; Zhang, Y.; Fu, Y. Advances in upgrading lignin pyrolysis vapors by ex situ catalytic fast pyrolysis. Energy Technol. 2017, 5, 30–51. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.H.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea, M.O.; Agirre, I.; Iriondo, A.; Lopez-Urionabarrenechea, A.; Requies, J.M.; Agirrezabal-Telleria, I.; Bizkarra, K.; Barrio, V.L.; Cambra, J.F. Heterogeneous Catalyzed Thermochemical Conversion of Lignin Model Compounds: An Overview. Top. Curr. Chem. 2019, 377, 36. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Wu, C.W.; Saha, B. Emerging strategies for breaking the 3D amorphous network of lignin. Catal. Sci. Technol. 2014, 4, 3785–3799. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef]
- Ma, R.; Xu, Y.; Zhang, X. Catalytic oxidation of biorefinery lignin to value-added chemicals to support sustainable biofuel production. ChemSusChem 2015, 8, 24–51. [Google Scholar] [CrossRef]
- Cheng, C.; Wang, J.; Shen, D.; Xue, J.; Guan, S.; Gu, S.; Luo, K.H. Catalytic oxidation of lignin in solvent systems for production of renewable chemicals: A review. Polymers 2017, 9, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright side of lignin depolymerization: Toward new platform chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- QMargellou, A.; Triantafyllidis, K.S. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts 2019, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Dong, L.; Guo, Y.; Liu, X.; Wang, Y. Chemicals from lignin: A review of the catalytic conversion involving hydrogen. ChemSusChem 2020, 13, 4181–4198. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Duolikun, T.; Babu, P.S.; Rokhum, L.; Johan, M.R. Recent developments in selective catalytic conversion of lignin into aromatics and their derivatives. Biomass Convers. Bior. 2020, 10, 873–883. [Google Scholar] [CrossRef]
- Shivhare, A.; Jampaiah, D.; Bhargava, S.K.; Lee, A.F.; Srivastava, R.; Wilson, K. Hydrogenolysis of Lignin-Derived Aromatic Ethers over Heterogeneous Catalysts. ACS Sustain. Chem. Eng. 2021, 9, 3379–3407. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Li, X.; Chen, L.; Li, G.; Li, X. Lignin Valorization: A Novel in Situ Catalytic Hydrogenolysis Method in Alkaline Aqueous Solution. Energy Fuels 2018, 32, 7643–7651. [Google Scholar] [CrossRef]
- Shu, R.; Xu, Y.; Chen, P.; Ma, L.; Zhang, Q.; Zhou, L.; Wang, C. Mild Hydrogenation of Lignin Depolymerization Products Over Ni/SiO2 Catalyst. Energy Fuels 2017, 31, 7208–7213. [Google Scholar] [CrossRef]
- Kristianto, I.; Limarta, S.O.; Lee, H.; Ha, J.M.; Suh, D.J.; Jae, J. Effective depolymerization of concentrated acid hydrolysis lignin using a carbon-supported ruthenium catalyst in ethanol/formic acid media. Bioresour. Technol. 2017, 234, 424–431. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Wang, H.; Ma, Q.; Li, S.; Chang, H.; Jameel, H. Liquefaction of kraft lignin by hydrocracking with simultaneous use of a novel dual acid-base catalyst and a hydrogenation catalyst. Bioresour. Technol. 2017, 243, 100–106. [Google Scholar] [CrossRef]
- Shu, R.; Long, J.; Xu, Y.; Ma, L.; Zhang, Q.; Wang, T.; Wang, C.; Yuan, Z.; Wu, Q. Investigation on the structural effect of lignin during the hydrogenolysis process. Bioresour. Technol. 2016, 200, 14–22. [Google Scholar] [CrossRef]
- Joffres, B.; Nguyen, M.T.; Laurenti, D.; Souchon, V.; Charon, N.; Daudin, A.; Quignard, A.; Geantet, C. Lignin hydroconversion on MoS2-based supported catalyst: Comprehensive analysis of products and reaction scheme. Appl. Catal. B Environ. 2016, 184, 153–162. [Google Scholar] [CrossRef]
- McVeigh, A.; Bouxin, F.P.; Jarvis, M.C.; Jackson, S.D. Catalytic depolymerisation of isolated lignin to fine chemicals: Part 2—process optimisation. Catal. Sci. Technol. 2016, 6, 4142–4150. [Google Scholar] [CrossRef] [Green Version]
- Struven, J.O.; Meier, D. Hydrocracking of organosolv lignin in subcritical water to useful phenols employing various Raney nickel catalysts. ACS Sustain. Chem. Eng. 2016, 4, 3712–3721. [Google Scholar] [CrossRef]
- Meier, D.; Berns, J.; Faix, O.; Balfanz, U.; Baldauf, W. Hydrocracking of organocell lignin for phenol production. Biomass Bioenergy 1994, 7, 99–105. [Google Scholar] [CrossRef]
- Zhang, J.; Teo, J.; Chen, X.; Asakura, H.; Tanaka, T.; Teramura, K.; Yan, N. A series of NiM (M = Ru, Rh, and Pd) bimetallic catalysts for effective lignin hydrogenolysis in water. ACS Catal. 2014, 4, 1574–1583. [Google Scholar] [CrossRef]
- Barta, K.; Warner, G.R.; Beach, E.S.; Anastas, P.T. Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides. Green. Chem. 2014, 16, 191–196. [Google Scholar] [CrossRef]
- Kloekhorst, A.; Heeres, H.J. Catalytic hydrotreatment of alcell lignin using supported Ru, Pd, and Cu catalysts. ACS Sustain. Chem. Eng. 2015, 3, 1905–1914. [Google Scholar] [CrossRef]
- Kasakov, S.; Shi, H.; Camaioni, D.M.; Zhao, C.; Baráth, E.; Jentys, A.; Lercher, J.A. Reductive deconstruction of organosolv lignin catalyzed by zeolite supported nickel nanoparticles. Green Chem. 2015, 17, 5079–5090. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Webb, J.D.; Sorek, H.; Wemmer, D.E.; Hartwig, J.F. Fragmentation of Lignin Samples with Commercial Pd/C under Ambient Pressure of Hydrogen. ACS Catal. 2016, 6, 7385–7392. [Google Scholar] [CrossRef]
- Hakonen, K.J.; González Escobedo, J.L.; Meriö-Talvio, H.; Hashmi, S.F.; Karinen, R.S.; Lehtonen, J. Ethanol organosolv lignin depolymerization with hydrogen over a Pd/C catalyst. ChemistrySelect 2018, 3, 1761–1771. [Google Scholar] [CrossRef]
- Tymchyshyn, M.; Yuan, Z.; Zhang, Y.; Xu, C.C. Catalytic hydrodeoxygenation of guaiacol for organosolv lignin depolymerization—Catalyst screening and experimental validation. Fuel 2019, 254, 115664. [Google Scholar] [CrossRef]
- Figueirêdo, M.B.; Venderbosch, R.H.; Deuss, P.J.; Heeres, H.J. A Two-Step Approach for the Conversion of Technical Lignins to Biofuels. Adv. Sustain. Syst. 2020, 4, 1900147. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, W.; Lv, W.; Xu, Y.; Liu, J.; Wang, H.; Wang, C.; Ma, L. The protection of C−O bond of pine lignin in different organic solvent systems. Chemistry 2020, 5, 3850–3858. [Google Scholar] [CrossRef]
- Skaates, J.M.; Kay, W.B. The phase relations of binary systems that form azeotropes N-alkyl alcohol-benzene systems: Methanol through n-butanol. Chem. Eng. Sci. 1964, 19, 431–444. [Google Scholar] [CrossRef]
- Levenspiel, O. Reazioni catalizzate da solidi. In Ingegneria Delle Reazioni Chimiche, 2nd ed.; Ambrosiana: Milan, Italy, 1978; p. 471, Chapter 14. [Google Scholar]
- Bjelić, A.; Grilc, M.; Likozar, B. Hydrogenation and hydrodeoxygenation of aromatic lignin monomers overMe/C catalysts: Mechanisms, reaction micro-kinetic modelling and quantitative structure-activity relationships. Chem. Eng. 2019, 359, 305–320. [Google Scholar] [CrossRef]
- Bowker, M.; Petts, R.W.; Waugh, K.C. Temperature-programmed desorption studies of alcohol decomposition on ZnO: 1-propanol, 1-butanol and 2-butanol. J. Catal. 1986, 99, 53–61. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, L.; Zhang, F.; Wang, Z.; Cheng, Z.; Yuan, W.; Qi, F. Experimental and kinetic modeling study of n-butanol pyrolysis and combustion. Energy Fuels 2012, 26, 5550–5568. [Google Scholar] [CrossRef]
- Bimbela, F.; Oliva, M.; Ruiz, J.; García, L.; Arauzo, J. Catalytic steam reforming of model compounds of biomass pyrolysis liquids in fixed bed: Acetol and n-butanol. J. Anal. Appl. Pyrolysis 2009, 85, 204–213. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor 1 | Factor 2 | Response 1 | Response 2 | Response 3 | Response 4 | Response 5 | |
|---|---|---|---|---|---|---|---|
| Run | T | time | Diphenyl-methane 4-ethyl | 2,4-Dimethyl- 3-(methoxy- carbonyl)- 5-ethylfuran | 1,2,3-Trimethoxy benzene | Total of main monomeric compounds 2 | Molecules derived from solvent 3 |
| °C | min | wt % | wt % | wt % | wt % | wt % | |
| 1 | 260 | 30 | 1.62 | 0.61 | 0.87 | 3.1 | 0.0432 |
| 2 | 260 | 60 | 2.43 | 1.37 | 1.29 | 6.74 | 0.124 |
| 3 | 280 | 90 | 2.08 | 2.48 | 1.67 | 8.84 | 0.281 |
| 4 | 260 | 90 | 1.8 | 0.56 | 0.691 | 3.39 | 0.125 |
| 5 | 300 | 90 | 2.49 | 1.49 | 1.56 | 8.18 | 0.478 |
| 6 | 300 | 60 | 2.36 | 1.27 | 1.61 | 7.5 | 0.307 |
| 7 | 280 | 30 | 3.05 | 1.15 | 1.42 | 7.27 | 0.175 |
| 8 | 280 | 60 | 2.4 | 1.11 | 1.3 | 6.51 | 0.25 |
| 9 | 300 | 30 | 1.56 | 0.853 | 1.03 | 4.51 | 0.116 |
| 10 | 280 | 60 | 1.99 | 1.34 | 1.44 | 6 | 0.182 |
| Constraints | ||||
|---|---|---|---|---|
| Lower | Upper | |||
| Name | Goal | Limit | Limit | Importance |
| A:T | minimize | 260 | 300 | 1 |
| B:time | minimize | 30 | 90 | 1 |
| Diphenylmethane 4-ethyl | maximize | 1.56 | 3.05 | 5 |
| 2,4-Dimethyl-3-(methoxycarbonyl)-5-ethylfuran | maximize | 0.56 | 2.48 | 5 |
| 1,2,3-Trimethoxy benzene | maximize | 0.691 | 1.67 | 5 |
| Total of main monomeric compounds | maximize | 3.1 | 8.84 | 5 |
| Molecules derived from solvent | minimize | 0.0432 | 0.478 | 3 |
| T | time | Resp1 | Resp2 | Resp3 | Resp4 | Resp5 | Desirability |
|---|---|---|---|---|---|---|---|
| 278.6 | 60.5 | 2.444 | 7.412 | 1.529 | 1.527 | 0.202 | 0.645 |
| Run | T, °C | t, min | Solution Recovery 1, wt % | Produced Gas 2, wt % | Mass Balance 3 |
|---|---|---|---|---|---|
| 1 | 260 | 30 | 90.5 | 3.4 | 0.939 |
| 2 | 260 | 60 | 83.6 | 10.6 | 0.942 |
| 3 | 260 | 90 | 84.0 | 9.2 | 0.932 |
| 4 | 280 | 30 | 84.2 | 11.3 | 0.95.5 |
| 5 | 280 | 60 | 60.6 | 24.1 | 0.847 |
| 6 | 280 | 60 | 64.2 | 16.5 | 0.807 |
| 7 | 280 | 90 | 47.5 | 43.2 | 0.907 |
| 8 | 300 | 30 | 51.6 | 22.7 | 0.743 |
| 9 | 300 | 60 | 44.4 | 41.6 | 0.856 |
| 10 | 300 | 90 | 36.4 | 52.6 | 0.890 |
| H2 (bar) | T, (°C) | Time, (min) | Catalyst | Solvent | Products | Yield 1 (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 5 | 279 | 60 | Raney Nickel | butanol | monomers | 7.4 | This work |
| 40 | 280 | 300 | Pd/C, Pd/C+CrCl3 | methanol | monomers | ~45 | 40 |
| 75 | 360 | 180 | Raney Nickel | water | monomers | 25 | 43 |
| 180 | 400 | 120 | NiMo | oils | phenols | 12.8 | 44 |
| 10 | 130 | 120 | Ni85Ru15 | water | monomers | 80 | 45 |
| 40 | 140 | 1200 | Cu-PMO | methanol | catechols | 63.7 | 46 |
| 100 | 400 | 240 | Ru/TiO2 | − | Lignin oil | 78 | 47 |
| 20 | 320 | 360 | Ni/zeolites | hexadecane | hydrocarbons | 70 | 48 |
| 50 | 400 | 49 | Pd/C | ethanol | monomers | 13 | 50 |
| 20 | 240 | 240 | Pd/C | ethanol | monomers | ~17 | 53 |
| Diphenyl-methane 4-ethyl | 2,4-Dimethyl-3-(methoxy-carbonyl)-5-ethylfuran | 1,2,3-Trimethoxy Benzene | Total Monomers 1 | Molecules Produced by the Solvent 2 | |
|---|---|---|---|---|---|
| Ea, kJ/mol | 21.0 | 64.3 | 53.0 | 60.0 | 102.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgana, M.; Viola, E.; Zimbardi, F.; Cerone, N.; Romanelli, A.; Valerio, V. Depolymerization and Hydrogenation of Organosolv Eucalyptus Lignin by Using Nickel Raney Catalyst. Processes 2021, 9, 1093. https://doi.org/10.3390/pr9071093
Morgana M, Viola E, Zimbardi F, Cerone N, Romanelli A, Valerio V. Depolymerization and Hydrogenation of Organosolv Eucalyptus Lignin by Using Nickel Raney Catalyst. Processes. 2021; 9(7):1093. https://doi.org/10.3390/pr9071093
Chicago/Turabian StyleMorgana, Massimo, Egidio Viola, Francesco Zimbardi, Nadia Cerone, Assunta Romanelli, and Vito Valerio. 2021. "Depolymerization and Hydrogenation of Organosolv Eucalyptus Lignin by Using Nickel Raney Catalyst" Processes 9, no. 7: 1093. https://doi.org/10.3390/pr9071093
APA StyleMorgana, M., Viola, E., Zimbardi, F., Cerone, N., Romanelli, A., & Valerio, V. (2021). Depolymerization and Hydrogenation of Organosolv Eucalyptus Lignin by Using Nickel Raney Catalyst. Processes, 9(7), 1093. https://doi.org/10.3390/pr9071093