1. Introduction
Corrosion of substances and materials that are exposed to open air and its pollutants, as opposed to submerged in fluid, is called atmospheric or air corrosion [
1]. The ever-increasing dissatisfaction with atmospheric corrosion is leading in expense and weight loss of material above any other type of material degradation processes. Nevertheless, atmospheric corrosion is also accountable for half the corrosion damage every year [
2]. However, some metals that form non-permeable layers give rise to the formation of oxides, which, in turn, achieve limited thickness through the dispersion of hardly noticeable oxide layers at room temperatures, unseen by the naked eye. In the case of metals such as chromium, titanium, and weathering steel, these limited thickness layers give an incredible shield over the metal surface.
Day-to-day enhancements in environmental contaminants have become a global concern. Different levels of pollutants entering the environment affect the corrosion rate. Few pollutants affecting the corrosion rate have been reported. Sulfur dioxide (SO
2) derived from the burning of petroleum products containing sulfur is estimated to be a leading factor in environmental corrosion [
3,
4]. The corrosion rates of non-coated metallic materials in the surrounding atmosphere are amplified by sulfur dioxide pollutants. Sulfur dioxide has higher dissolvability in water, giving rise to the formation of H
2SO
4 over the surface in the form of surface dampness thin layer [
5,
6]. Comparatively, levels of NOx discharged from exhaust and combustion (street activities, traffic, and production of energy) are greater than emissions of sulfur dioxide [
7,
8]. Increasing environmental saltiness improves the electrolyte arrangement of the surface by using hygroscopic salts (e.g., MgCl
2 and NaCl), which, in turn, enhance atmospheric corrosion rates. Several studies have stated that the core reason behind environmental corrosion is chloride ions [
9,
10,
11]. Nonetheless, hydrogen peroxide (H
2O
2) is another pollutant available in the atmosphere, and it is the predisposition of hydrogen peroxide to be responsive to all iron alloys and composites, resulting in the production of HOX from its decomposition. Covering and steeping the surface of a target metal with a solution containing hydrogen peroxide results in the production of a passive layer [
12,
13]. Several studies [
14,
15] have revealed that most environmental changes and transformations of the atmosphere related to chlorinated hydrocarbons also contribute to fresh air purification levels.
Weathering steel (WS) comprises 1–2.5% alloying elements (Cr, Cu, Si, and P) and is the most common type of structural steel, widely used for bridges, buildings, etc. [
16]. Moreover, it tends to form rust at a rate depending on the level of contact with oxygen in the presence of moisture and air. Researchers [
17,
18] demonstrated that the main products of environmental corrosion of steel are magnetite (Fe
3O
4), amorphous ferric oxy-hydroxide (FeOx(OH)
3-2x), and crystalline rust (α-FeOOH or ɣ-FeOOH) as well as β-FeOOH in marine environments. According to Kamimura et. al. [
18], corrosion begins with the anodic dissolution of iron into ferrous ions (Fe
2+). These ions react with the moisture (hydrolysis) on the surface of the steel to form FeOH
+. The FeOH
+, in turn, reacts with the oxygen (oxidation) in the atmosphere to form ɣ-FeOOH, which crystallizes and precipitates out of the system (the rate of crystallization and precipitation is increased if drying cycle occurs). Moisture mixed with pollutants such as sulfur dioxide (SO
2) has a relatively low pH. In contact with this mixture, the crystalline ɣ-FeOOH dissolves to form amorphous FeOx (OH)
3-2x, which precipitates again. Lastly, the FeO
x (OH)
3-2x undergoes a solid-state transformation (deprotonation hydroxyl ions from rainwater) to become α-FeOOH [
19,
20,
21].
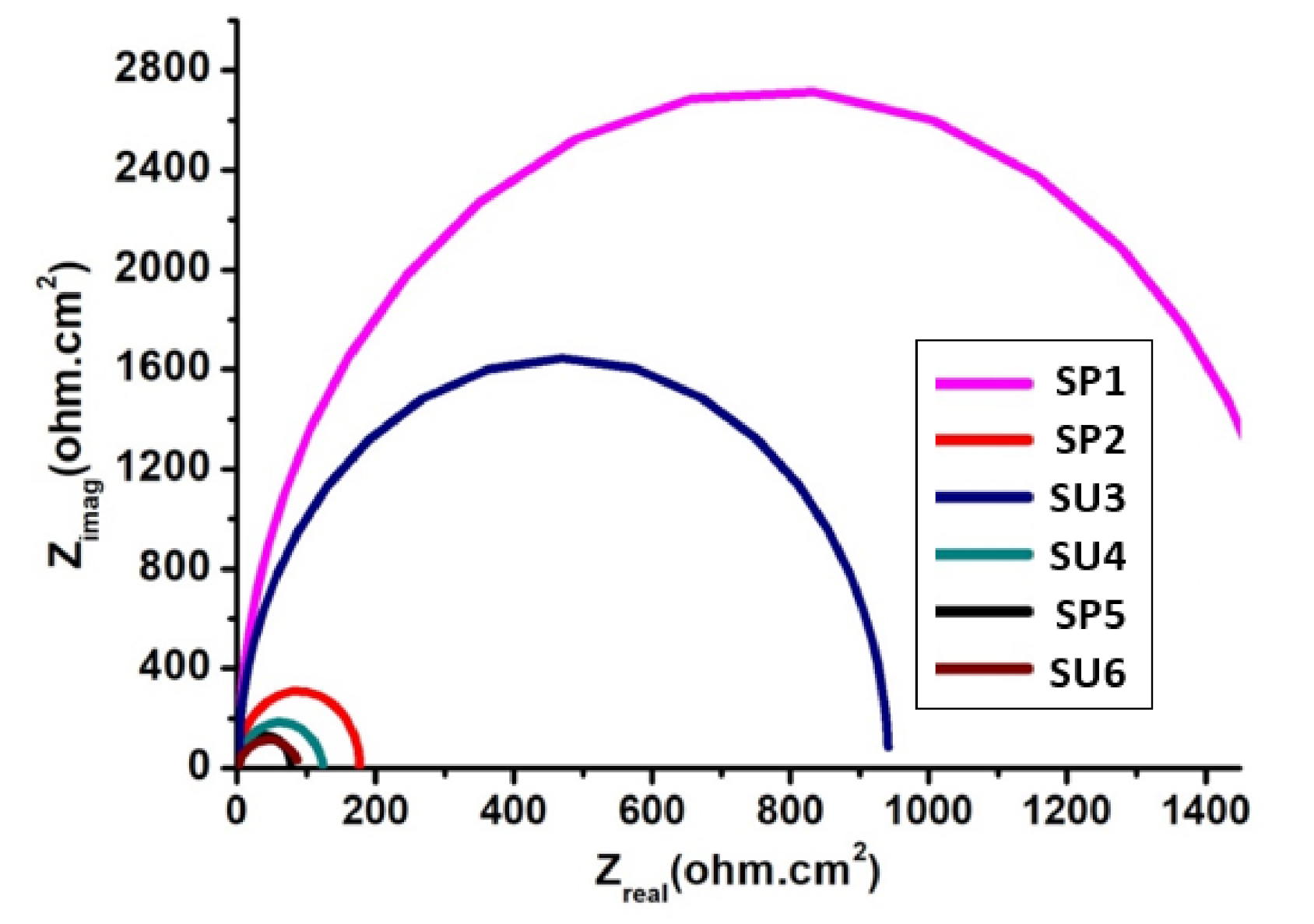
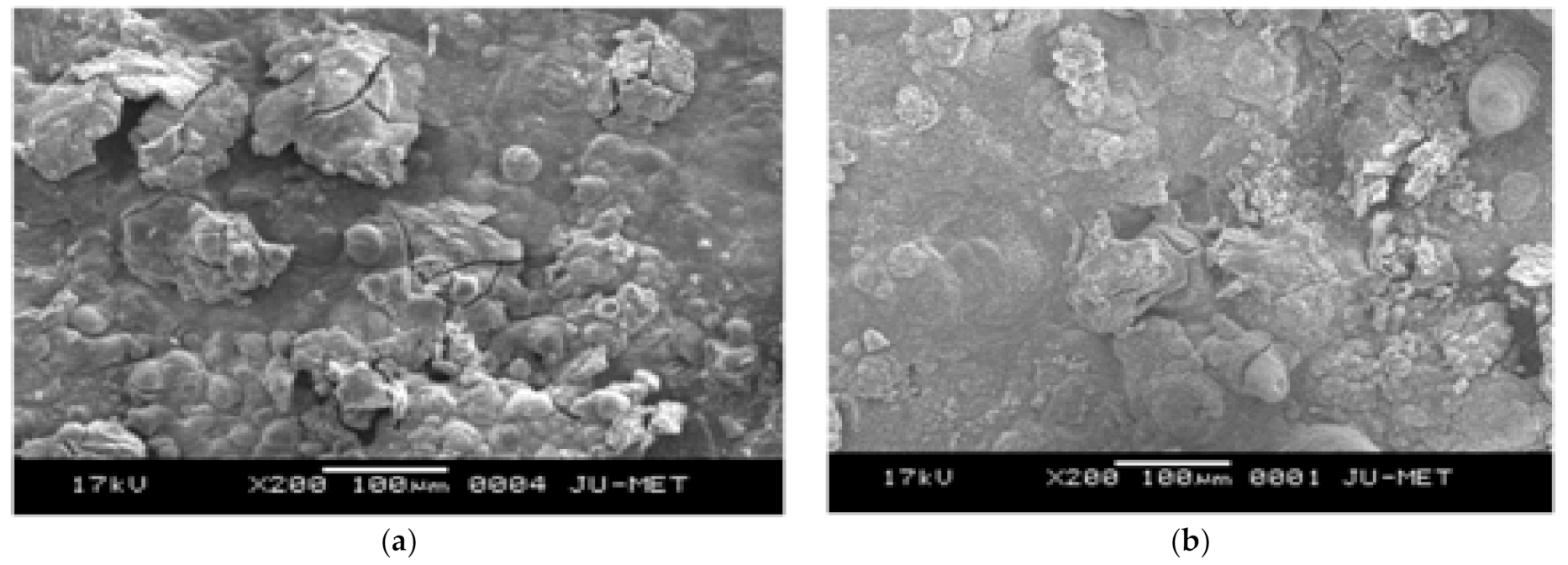
Using controlled passivation, the rust on weathering steel (WS), comparable to the rust formed on weathering steel due to a long period of exposure in Digha, was simulated. The simulation of the data was carried out by anodically polarizing the WS with a nobler metal, as the usage of a potentiostat would have been expensive and confined. Previous studies of Digha stated that 0.01 M KCl is found to be stable for simulating the data. Therefore, weathering steel was connected with nobler metals, such as Cu and graphite, and dipped in 0.01 M KCl. The corrosion rate of weathering steel that was anodically polarized with Cu or graphite showed a significant decrease in corrosion rate, confirming that the progressive rusting of weathering steel leads to a progressive decrease in the corrosion rate. The rust manufactured by this process was characterized by SEM and analyzed by EDX. The corrosion rate of WS obtained by this method was compared and rust characterized with data from 18-month field exposed WS in Digha. Thus, the study revealed that the protective layer of weathering that was produced after 18 months’ exposure could easily be countered in the laboratory by connecting WS with nobler metals and dipping it in an appropriate solution. Using this inexpensive method, the corrosion rate of WS was also decreased.
Generally, this work aims to improve the atmospheric corrosion effect of commercial weathering steel in the localized region of Digha, a sea resort of West Bengal, India, using anodic polarization protection technique. The use of this technology ensures that the best anti-corrosion behavior of commercial weathering steel is obtained in the specified region, which has not been investigated before.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}