

Clinical and Genetic Characteristics of Hypophosphatasia in Chinese Adults
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Biochemical Analysis
2.3. Radiography and Bone Densitometry
2.4. Genetic Analysis of ALPL
2.5. Statistics
3. Results
3.1. Clinical Features of Subjects
3.2. Biochemical Parameters and BMD Measurement
3.3. Mutation Analysis of ALPL
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bianchi, M.L.; Bishop, N.J.; Guañabens, N.; Hofmann, C.; Jakob, F.; Roux, C.; Zillikens, M.C. Hypophosphatasia in adolescents and adults: Overview of diagnosis and treatment. Osteoporos. Int. 2020, 31, 1445–1460. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia-aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef]
- Whyte, M.P. Atypical femoral fractures, bisphosphonates, and adult hypophosphatasia. J. Bone Miner. Res. 2009, 24, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Genest, F.; Seefried, L. Subtrochanteric and diaphyseal femoral fractures in hypophosphatasia-not atypical at all. Osteoporos. Int. 2018, 29, 1815–1825. [Google Scholar] [CrossRef]
- Christidis, G.; Martin, T.; Walter, F.; Lammert, F.; Krawczyk, M. Hypophosphatasia: An Underappreciated Cause of Atraumatic Stress Fractures. Am. J. Med. 2022, 135, e18–e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ke, Y.H.; Wang, C.; Yue, H.; Hu, W.W.; Gu, J.M.; Zhang, Z.L. Identification of the mutations in the tissue-nonspecific alkaline phosphatase gene in two Chinese families with hypophosphatasia. Arch. Med. Res. 2012, 43, 21–30. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- National Health Commission of the People’s Republic of China. Report on Nutrition and Chronic Disease Status of Chinese Residents (2020); People’s Medical Publishing House Co., Ltd.: Beijing, China, 2021. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Schmidt, T.; Mussawy, H.; Rolvien, T.; Hawellek, T.; Hubert, J.; Rüther, W.; Amling, M.; Barvencik, F. Clinical, radiographic and biochemical characteristics of adult hypophosphatasia. Osteoporos. Int. 2017, 28, 2653–2662. [Google Scholar] [CrossRef] [PubMed]
- Orimo, H.; Girschick, H.J.; Goseki-Sone, M.; Ito, M.; Oda, K.; Shimada, T. Mutational analysis and functional correlation with phenotype in German patients with childhood-type hypophosphatasia. J. Bone Miner. Res. 2001, 16, 2313–2319. [Google Scholar] [CrossRef]
- Taillandier, A.; Lia-Baldini, A.S.; Mouchard, M.; Robin, B.; Muller, F.; Simon-Bouy, B.; Serre, J.L.; Bera-Louville, A.; Bonduelle, M.; Eckhardt, J.; et al. Twelve novel mutations in the tissue-nonspecific alkaline phosphatase gene (ALPL) in patients with various forms of hypophosphatasia. Hum. Mutat. 2001, 18, 83–84. [Google Scholar] [CrossRef]
- Taillandier, A.; Domingues, C.; De Cazanove, C.; Porquet-Bordes, V.; Monnot, S.; Kiffer-Moreira, T.; Rothenbuhler, A.; Guggenbuhl, P.; Cormier, C.; Baujat, G.; et al. Molecular diagnosis of hypophosphatasia and differential diagnosis by targeted Next Generation Sequencing. Mol. Genet. Metab. 2015, 116, 215–220. [Google Scholar] [CrossRef]
- Dunnen, J.D. Available online: https://databases.lovd.nl/shared/variants/0000709880#00025251 (accessed on 27 March 2023).
- Braunstein, N.A. Multiple fractures, pain, and severe disability in a patient with adult-onset hypophosphatasia. Bone Rep. 2016, 4, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lia-Baldini, A.S.; Muller, F.; Taillandier, A.; Gibrat, J.F.; Mouchard, M.; Robin, B.; Simon-Bouy, B.; Serre, J.L.; Aylsworth, A.S.; Bieth, E.; et al. A molecular approach to dominance in hypophosphatasia. Hum. Genet. 2001, 109, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Dunnen, J.D. Available online: https://databases.lovd.nl/shared/variants/0000709882#00025251 (accessed on 27 March 2023).
- Orimo, H.; Goseki-Sone, M.; Hosoi, T.; Shimada, T. Functional assay of the mutant tissue-nonspecific alkaline phosphatase gene using U2OS osteoblast-like cells. Mol. Genet. Metab. 2008, 94, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Genetics, F. Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV002492124.1/ (accessed on 27 March 2023).
- Invitae. Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV002028553.3/ (accessed on 27 March 2023).
- Weiss, M.J.; Cole, D.E.; Ray, K.; Whyte, M.P.; Lafferty, M.A.; Mulivor, R.A.; Harris, H. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc. Natl. Acad. Sci. USA 1988, 85, 7666–7669. [Google Scholar] [CrossRef]
- Hofmann, C.; Girschick, H.J.; Mentrup, B.; Graser, S.; Seefried, L.; Liese, J.; Jakob, F. Clinical Aspects of Hypophosphatasia: An Update. Clin. Rev. Bone Mineral. Metab. 2013, 11, 60–70. [Google Scholar] [CrossRef]
- Fraser, D. Hypophosphatasia. Am. J. Med. 1957, 22, 730–746. [Google Scholar] [CrossRef]
- Mornet, E.; Yvard, A.; Taillandier, A.; Fauvert, D.; Simon-Bouy, B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann. Hum. Genet. 2011, 75, 439–445. [Google Scholar] [CrossRef]
- Mornet, E. Molecular Genetics of Hypophosphatasia and Phenotype-Genotype Correlations. Subcell. Biochem. 2015, 76, 25–43. [Google Scholar] [CrossRef]
- Mornet, E.; Taillandier, A.; Domingues, C.; Dufour, A.; Benaloun, E.; Lavaud, N.; Wallon, F.; Rousseau, N.; Charle, C.; Guberto, M.; et al. Hypophosphatasia: A genetic-based nosology and new insights in genotype-phenotype correlation. Eur. J. Hum. Genet. 2021, 29, 289–299. [Google Scholar] [CrossRef]
- Xu, L.; Pang, Q.; Jiang, Y.; Wang, O.; Li, M.; Xing, X.; Xia, W. Four novel mutations in the ALPL gene in Chinese patients with odonto, childhood, and adult hypophosphatasia. Biosci. Rep. 2018, 38, BSR20171377. [Google Scholar] [CrossRef]
- Zhang, Q.; Qin, Z.; Yi, S.; Wei, H.; Zhou, X.Z.; Shen, F. Case Report: Variations in the ALPL Gene in Chinese Patients with Hypophosphatasia. Front. Genet. 2021, 12, 732621. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, M.; Liang, X.; Wu, D.; Li, W.; Su, C.; Cao, B.; Chen, J.; Gong, C. Clinical and genetic characteristics of hypophosphatasia in Chinese children. Orphanet J. Rare Dis. 2021, 16, 159. [Google Scholar] [CrossRef] [PubMed]
- Dahir, K.M.; Seefried, L.; Kishnani, P.S.; Petryk, A.; Högler, W.; Linglart, A.; Martos-Moreno, G.; Ozono, K.; Fang, S.; Rockman-Greenberg, C. Clinical profiles of treated and untreated adults with hypophosphatasia in the Global HPP Registry. Orphanet J. Rare Dis. 2022, 17, 277. [Google Scholar] [CrossRef]
- Durrough, C.; Colazo, J.M.; Simmons, J.; Hu, J.R.; Hudson, M.; Black, M.; de Riesthal, M.; Dahir, K. Characterization of physical, functional, and cognitive performance in 15 adults with hypophosphatasia. Bone 2021, 142, 115695. [Google Scholar] [CrossRef]
- Martins, L.; de Almeida, A.B.; Dos Santos, E.J.L.; Foster, B.L.; Machado, R.A.; Kantovitz, K.R.; Coletta, R.D.; Nociti, F.H., Jr. A novel combination of biallelic ALPL mutations associated with adult hypophosphatasia: A phenotype-genotype association and computational analysis study. Bone 2019, 125, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Lefever, E.; Witters, P.; Gielen, E.; Vanclooster, A.; Meersseman, W.; Morava, E.; Cassiman, D.; Laurent, M.R. Hypophosphatasia in Adults: Clinical Spectrum and Its Association With Genetics and Metabolic Substrates. J. Clin. Densitom. 2020, 23, 340–348. [Google Scholar] [CrossRef]
- Fauvert, D.; Brun-Heath, I.; Lia-Baldini, A.S.; Bellazi, L.; Taillandier, A.; Serre, J.L.; de Mazancourt, P.; Mornet, E. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med. Genet. 2009, 10, 51. [Google Scholar] [CrossRef]
- Hepp, N.; Frederiksen, A.L.; Duno, M.; Jørgensen, N.R.; Jensen, J.B. Biochemical and clinical manifestations in adults with hypophosphatasia: A national cross-sectional study. Osteoporos. Int. 2022, 33, 2595–2605. [Google Scholar] [CrossRef]
- Quinn, H.B.; Busch, R.S.; Kane, M.P. The Occurrence and Burden of Hypophosphatasia in an Ambulatory Care Endocrinology Practice. Endocr. Pract. 2021, 27, 1189–1192. [Google Scholar] [CrossRef]
- Genest, F.; Claußen, L.; Rak, D.; Seefried, L. Bone mineral density and fracture risk in adult patients with hypophosphatasia. Osteoporos. Int. 2021, 32, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Jandl, N.M.; Schmidt, T.; Rolvien, T.; Stürznickel, J.; Chrysostomou, K.; von Vopelius, E.; Volk, A.E.; Schinke, T.; Kubisch, C.; Amling, M.; et al. Genotype-Phenotype Associations in 72 Adults with Suspected ALPL-Associated Hypophosphatasia. Calcif. Tissue Int. 2021, 108, 288–301. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, J.; Behanova, M.; Haschka, J.; Roetzer, K.; Uyanik, G.; Hadzimuratovic, B.; Witsch-Baumgartner, M.; Schett, G.; Zwerina, J.; Kocijan, R. Identifying adult hypophosphatasia in the rheumatology unit. Orphanet J. Rare Dis. 2022, 17, 435. [Google Scholar] [CrossRef] [PubMed]
- Karakostas, P.; Dolscheid-Pommerich, R.; Hass, M.D.; Weber, N.; Brossart, P.; Schäfer, V.S. Prevalence of hypophosphatasia in adult patients in rheumatology. Z. Rheumatol. 2022, 81, 513–519. [Google Scholar] [CrossRef]
- Ng, E.; Ashkar, C.; Seeman, E.; Schneider, H.G.; Nguyen, H.; Ebeling, P.R.; Sztal-Mazer, S. A low serum alkaline phosphatase may signal hypophosphatasia in osteoporosis clinic patients. Osteoporos. Int. 2023, 34, 327–337. [Google Scholar] [CrossRef]
- Alonso, N.; Larraz-Prieto, B.; Berg, K.; Lambert, Z.; Redmond, P.; Harris, S.E.; Deary, I.J.; Pugh, C.; Prendergast, J.; Ralston, S.H. Loss-of-Function Mutations in the ALPL Gene Presenting with Adult Onset Osteoporosis and Low Serum Concentrations of Total Alkaline Phosphatase. J. Bone Miner. Res. 2020, 35, 657–661. [Google Scholar] [CrossRef]
- Righetti, M.; Wach, J.; Desmarchelier, R.; Coury, F. Teriparatide treatment in an adult patient with hypophosphatasia exposed to bisphosphonate and revealed by bilateral atypical fractures. Jt. Bone Spine 2018, 85, 365–367. [Google Scholar] [CrossRef]
- Seefried, L.; Dahir, K.; Petryk, A.; Högler, W.; Linglart, A.; Martos-Moreno, G.; Ozono, K.; Fang, S.; Rockman-Greenberg, C.; Kishnani, P.S. Burden of Illness in Adults With Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J. Bone Miner. Res. 2020, 35, 2171–2178. [Google Scholar] [CrossRef]
- Zhou, W.; van Rooij, J.G.J.; Ebeling, P.R.; Verkerk, A.; Zillikens, M.C. The Genetics of Atypical Femur Fractures-a Systematic Review. Curr. Osteoporos. Rep. 2021, 19, 123–130. [Google Scholar] [CrossRef]
- Magdaleno, A.L.; Singh, S.; Venkataraman, S.; Perilli, G.A.; Lee, Y.Y. Adult-onset hypophosphatasia: Before and after tretment with asfotase alfa. AACE Clin. Case Rep. 2019, 5, e344–e348. [Google Scholar] [CrossRef]
- Stürznickel, J.; Schmidt, F.N.; von Vopelius, E.; Delsmann, M.M.; Schmidt, C.; Jandl, N.M.; Oheim, R.; Barvencik, F. Bone healing and reactivation of remodeling under asfotase alfa therapy in adult patients with pediatric-onset hypophosphatasia. Bone 2021, 143, 115794. [Google Scholar] [CrossRef]
- Klidaras, P.; Severt, J.; Aggers, D.; Payne, J.; Miller, P.D.; Ing, S.W. Fracture Healing in Two Adult Patients With Hypophosphatasia After Asfotase Alfa Therapy. JBMR Plus 2018, 2, 304–307. [Google Scholar] [CrossRef]
- Millán, J.L.; Whyte, M.P. Alkaline Phosphatase and Hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Mumm, S.; Deal, C. Adult hypophosphatasia treated with teriparatide. J. Clin. Endocrinol. Metab. 2007, 92, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Schalin-Jäntti, C.; Mornet, E.; Lamminen, A.; Välimäki, M.J. Parathyroid hormone treatment improves pain and fracture healing in adult hypophosphatasia. J. Clin. Endocrinol. Metab. 2010, 95, 5174–5179. [Google Scholar] [CrossRef]
- Cundy, T.; Michigami, T.; Tachikawa, K.; Dray, M.; Collins, J.F.; Paschalis, E.P.; Gamsjaeger, S.; Roschger, A.; Fratzl-Zelman, N.; Roschger, P.; et al. Reversible Deterioration in Hypophosphatasia Caused by Renal Failure With Bisphosphonate Treatment. J. Bone Miner. Res. 2015, 30, 1726–1737. [Google Scholar] [CrossRef]
- Laroche, M. Failure of teriparatide in treatment of bone complications of adult hypophosphatasia. Calcif. Tissue Int. 2012, 90, 250. [Google Scholar] [CrossRef]
- Gagnon, C.; Sims, N.A.; Mumm, S.; McAuley, S.A.; Jung, C.; Poulton, I.J.; Ng, K.W.; Ebeling, P.R. Lack of sustained response to teriparatide in a patient with adult hypophosphatasia. J. Clin. Endocrinol. Metab. 2010, 95, 1007–1012. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | Dental Issues | Fracture | Musculoskeletal Symptoms | Other | Misdiagnosis |
|---|---|---|---|---|---|---|---|
| FAM1-3 | male | 38 | early loss of deciduous teeth, abnormal teeth eruption | RF, mVF, pseudofracture | leg pain | growth retardation, bowing legs | - |
| FAM1-1 | male | 64 | - | - | - | fatigue | - |
| FAM1-2 | female | 64 | - | - | - | - | - |
| FAM1-4 | male | 36 | - | - | - | - | - |
| FAM2-4 | female | 70 | early loss of permanent teeth, dental caries | mVF | backache | - | |
| FAM2-5 | female | 63 | early loss of permanent teeth, periodontitis | - | arthralgia | - | OA * |
| PT-1 | female | 34 | - | - | muscle spasm | fatigue, short stature | |
| PT-2 | female | 32 | - | - | - | fatigue | OP |
| PT-3 | female | 62 | dental caries, early loss of permanent teeth | AF | arthralgia | fatigue | RA * |
| PT-4 | female | 43 | - | - | - | - | OP Δ |
| PT-5 | female | 58 | early loss of permanent teeth | - | backache | - | OP |
| PT-6 | female | 32 | - | - | - | fatigue | - |
| PT-7 | female | 62 | - | - | arthralgia | - | OP |
| PT-8 | female | 64 | - | MF | backache | - | OP Δ |
| PT-9 | female | 47 | - | - | fatigue | OP Δ | |
| PT-10 | female | 69 | early loss of permanent teeth | mVF | backache | short stature | OP Δ |
| PT-11 | female | 69 | - | VF | backache | - | OP Δ |
| PT-12 | female | 74 | early loss of permanent teeth | - | backache | CPPD | OP Δ, RA * |
| PT-13 | female | 54 | early loss of permanent teeth | FF, pseudofracture | leg pain | CPPD, short stature | - |
| Patients | ALP (U/L) | Calcium * (mmol/L) | Phosphate + (mmol/L) | PTH (pg/mL) | 25OHD (ng/mL) | β-CTX (ng/mL) | OC (ng/mL) | ||
|---|---|---|---|---|---|---|---|---|---|
| Recent | Average | Lowest | |||||||
| FAM1-3 | 34 | 30.7 | 28 | 2.34 | 1.43 | 30.37 | 27.05 | 285.5 | 13.33 |
| FAM1-1 | 40 | NA | NA | 2.3 | 1.31 | 38.37 | 22.35 | 328.5 | NA |
| FAM1-2 | 53 | NA | NA | 2.23 | 1.51 | 39.34 | 15.94 | 286.4 | NA |
| FAM1-4 | 32 | NA | NA | 2.33 | 1.25 | 34.2 | 12.97 | 84.96 | NA |
| FAM2-3 | 28 | 29.3 | 26 | 2.32 | 1.21 | 48.32 | 26.79 | 267 | 15.66 |
| FAM2-4 | 38 | 33.5 | 29 | 2.27 | 1.33 | 65.43 | 25.93 | 200.8 | 14.44 |
| PT-1 | 23 | 28 | 22 | 2.3 | 0.94 | 49.2 | 11.9 | 237.7 | 11.1 |
| PT-2 | 23 | 28 | 23 | 2.27 | 1.35 | 48.1 | 18.88 | 300.85 | 13.67 |
| PT-3 | 27.8 | 27.6 | 25 | 2.4 | 1.08 | 20.5 | 25 | 657 | 15.7 |
| PT-4 | 27 | 29 | 25 | 2.28 | 1.64 | 28.7 | 31.1 | 478.1 | 18.6 |
| PT-5 | 32 | 30 | 28 | 2.22 | 1.61 | 29.3 | 19.9 | NA | NA |
| PT-6 | 26 | 31.3 | 26 | 2.3 | 1.13 | 18.56 | 25.7 | 345 | 12.8 |
| PT-7 | 29 | 28 | 27 | NA | NA | NA | NA | NA | NA |
| PT-8 | 14 | 17 | 14 | 2.28 | 1.21 | 37.56 | 28.17 | 80.6 | 9.27 |
| PT-9 | 27 | 31 | 27 | 2.49 | 1.12 | 43.41 | 26.52 | 274.9 | 9.19 |
| PT-10 | 28 | 27.3 | 22 | 2.32 | 1.29 | 28.2 | 30.6 | 532 | 26.1 |
| PT-11 | 19.2 | 20.4 | 19.2 | 2.3 | 1.12 | 55.9 | 18 | 100 | 8.6 |
| PT-12 | 28 | 27.7 | 23 | 2.33 | 1.15 | 57.02 | 28.58 | 267.6 | 11.05 |
| PT-13 | 23 | 24.3 | 22 | 2.36 | 1.33 | 18.64 | 27.8 | 836.4 | 15.51 |
| With Fracture | Without Fracture | p | |
|---|---|---|---|
| Age, years | 66.8 ± 3.56 | 50.75 ± 15.06 | 0.004 |
| Female/all sex | 5/5 | 11/12 | - |
| ALP, U/L | 23.4 ± 6.47 | 31.50 ± 8.59 | 0.059 |
| Calcium, mmol/L | 2.32 ± 0.046 | 2.30 ± 0.069 | 0.416 |
| Phosphorus, mmol/L | 1.18 ± 0.046 | 1.30 ± 0.209 | 0.129 |
| PTH, pg/mL | 38.09 ± 14.40 | 40.77 ± 12.92 | 0.712 |
| 25OHD, ng/mL | 25.71 ± 4.77 | 22.33 ± 6.37 | 0.257 |
| β-CTX, ng/mL | 327.32 ± 238.25 | 280.48 ± 101.42 | 0.713 |
| OC, ng/mL | 15.07 ± 7.03 | 13.11 ± 2.90 | 0.578 |
| Patients | Age | LS BMD | Z Score | FN BMD | Z Score | TH BMD | Z Score |
|---|---|---|---|---|---|---|---|
| FAM1-3 | 38 | 0.744 | −1.8 | UM | - | UM | - |
| FM2-4 | 70 | 0.901 | NA | 0.452 | NA | 0.67 | NA |
| FM2-5 | 63 | 0.895 | NA | 0.544 | NA | 0.77 | NA |
| PT-1 | 34 | 0.804 | −1.7 | 0.611 | −1.8 | 0.719 | −1.8 |
| PT-2 | 32 | 1.111 | 0 | 0.711 | −1.8 | 0.785 | −1.5 |
| PT-3 | 62 | 0.83 | 0.4 | 0.529 | −1.3 | 0.704 | −0.2 |
| PT-4 | 43 | 0.892 | −1.0 | 0.751 | −0.5 | 0.869 | −0.4 |
| PT-9 | 47 | 0.792 | −2.5 | 0.764 | −0.9 | 0.66 | −2.1 |
| PT-13 | 54 | 0.921 | −1.1 | UM | - | UM | - |
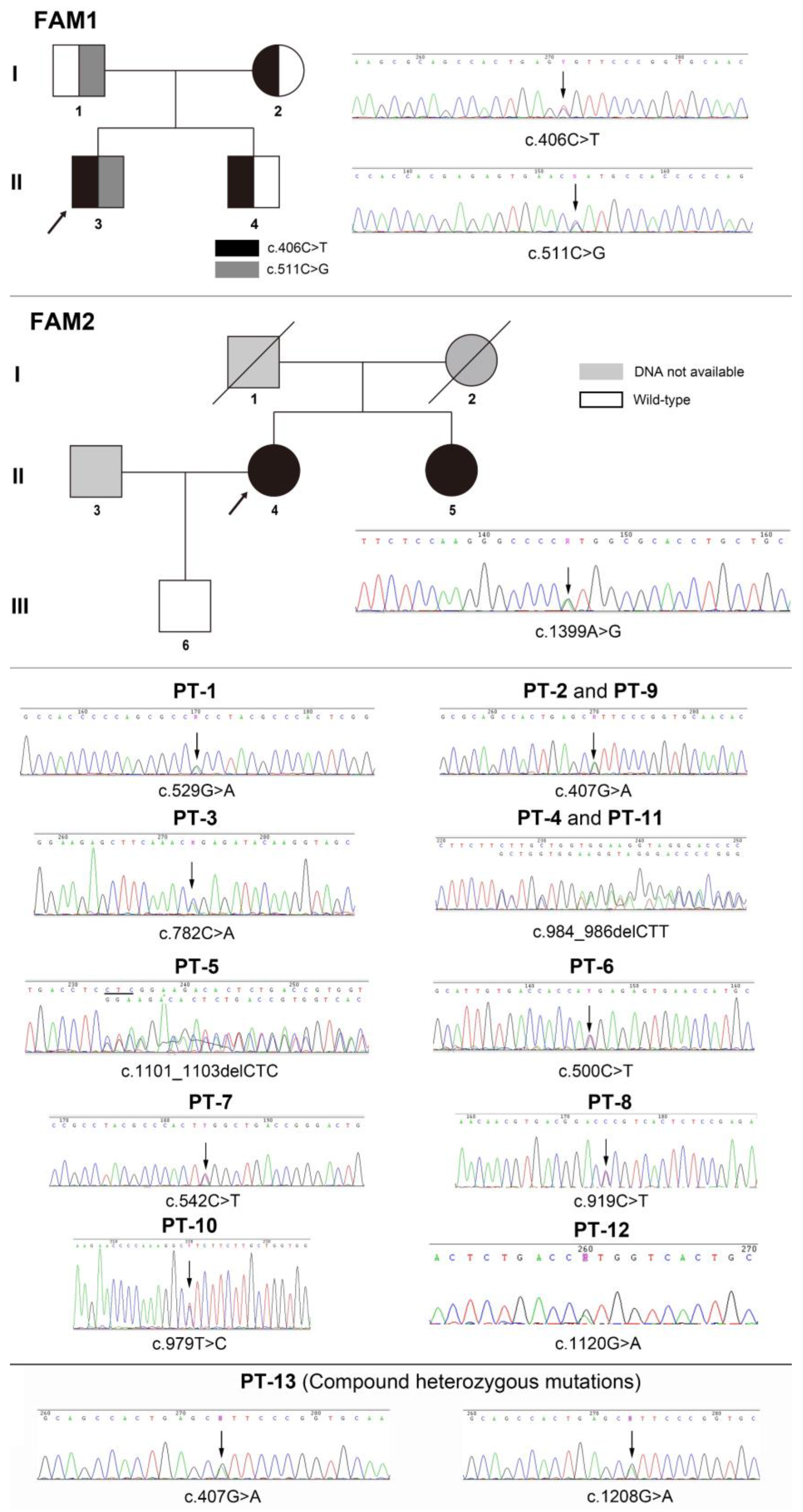
| Patients | Status | DNA Variations | Amino Acid Change | Reported | ACMG [9] Classification |
|---|---|---|---|---|---|
| FM1-3 PB | cHet/MS | c.406C>T | p.Arg136Cys | Yes | Pathogenic [10] |
| c.511C>G | p.His171Ala | No | LP a | ||
| FM1-1 PB’s farther | Het/MS | c.511C>G | p.His171Ala | No | LP a |
| FM1-2 PB’s mother | Het/MS | c.406C>T | p.Arg136Cys | Yes | Pathogenic [10] |
| FM1-4 PB’s brother | Het/MS | c.406C>T | p.Arg136Cys | Yes | Pathogenic [10] |
| FM2-3 PB | Het/MS | c.1399A>G | p.Met467Val | No | LP b |
| FM2-4 PB’s s sister | Het/MS | c.1399A>G | p.Met467Val | No | LP b |
| PT-1 | Het/MS | c.529G>A | p.Ala177Thr | Yes | Pathogenic [11] |
| PT-2 | Het/MS | c.407G>A | p.Arg136His | Yes | Pathogenic [12] |
| PT-3 | Het/MS | c.782C>A | p.Pro261Gln | No | US c |
| PT-4 | Het/DEL | c.984_986delCTT | p.Phe328del | Yes | Pathogenic [13] |
| PT-5 | Het/DEL | c.1101_1103delCTC | p.Ser368del | Yes | Pathogenic [14] |
| PT-6 | Het/MS | c.500C>T | p.Thr167Met | Yes | Pathogenic [15] |
| PT-7 | Het/MS | c.542C>T | p.Ser181Leu | Yes | Pathogenic [16] |
| PT-8 | Het/MS | c.919C>T | p.Pro307Ser | Yes | Pathogenic [17] |
| PT-9 | Het/MS | c.407G>A | p.Arg136His | Yes | Pathogenic [12] |
| PT-10 | Het/MS | c.979T>C | p.Phe327Leu | Yes | LP [18] |
| PT-11 | Het/DEL | c.984_986delCTT | p.Phe328del | Yes | Pathogenic [13] |
| PT-12 | Het/MS | c.1120G>A | p.Val327Met | Yes | LP [19] |
| PT-13 | cHet/MS | c.407G>A | p.Arg136His | Yes | Pathogenic [12] |
| c.1208G>A | p.Ser403Ala | Yes | US [20] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Ren, N.; Wang, Z.; Wang, Y.; Hu, Y.; Hu, W.; Gu, J.; Hong, W.; Zhang, Z.; Wang, C. Clinical and Genetic Characteristics of Hypophosphatasia in Chinese Adults. Genes 2023, 14, 922. https://doi.org/10.3390/genes14040922
Li X, Ren N, Wang Z, Wang Y, Hu Y, Hu W, Gu J, Hong W, Zhang Z, Wang C. Clinical and Genetic Characteristics of Hypophosphatasia in Chinese Adults. Genes. 2023; 14(4):922. https://doi.org/10.3390/genes14040922
Chicago/Turabian StyleLi, Xiang, Na Ren, Ziyuan Wang, Ya Wang, Yunqiu Hu, Weiwei Hu, Jiemei Gu, Wei Hong, Zhenlin Zhang, and Chun Wang. 2023. "Clinical and Genetic Characteristics of Hypophosphatasia in Chinese Adults" Genes 14, no. 4: 922. https://doi.org/10.3390/genes14040922
APA StyleLi, X., Ren, N., Wang, Z., Wang, Y., Hu, Y., Hu, W., Gu, J., Hong, W., Zhang, Z., & Wang, C. (2023). Clinical and Genetic Characteristics of Hypophosphatasia in Chinese Adults. Genes, 14(4), 922. https://doi.org/10.3390/genes14040922