
Anti-Drug Antibodies in the Biological Therapy of Autoimmune Rheumatic Diseases
 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Autoimmune Rheumatic Diseases and Biological Therapy
2.1. ARDs Classification
2.2. Biologic Therapy in ARDs
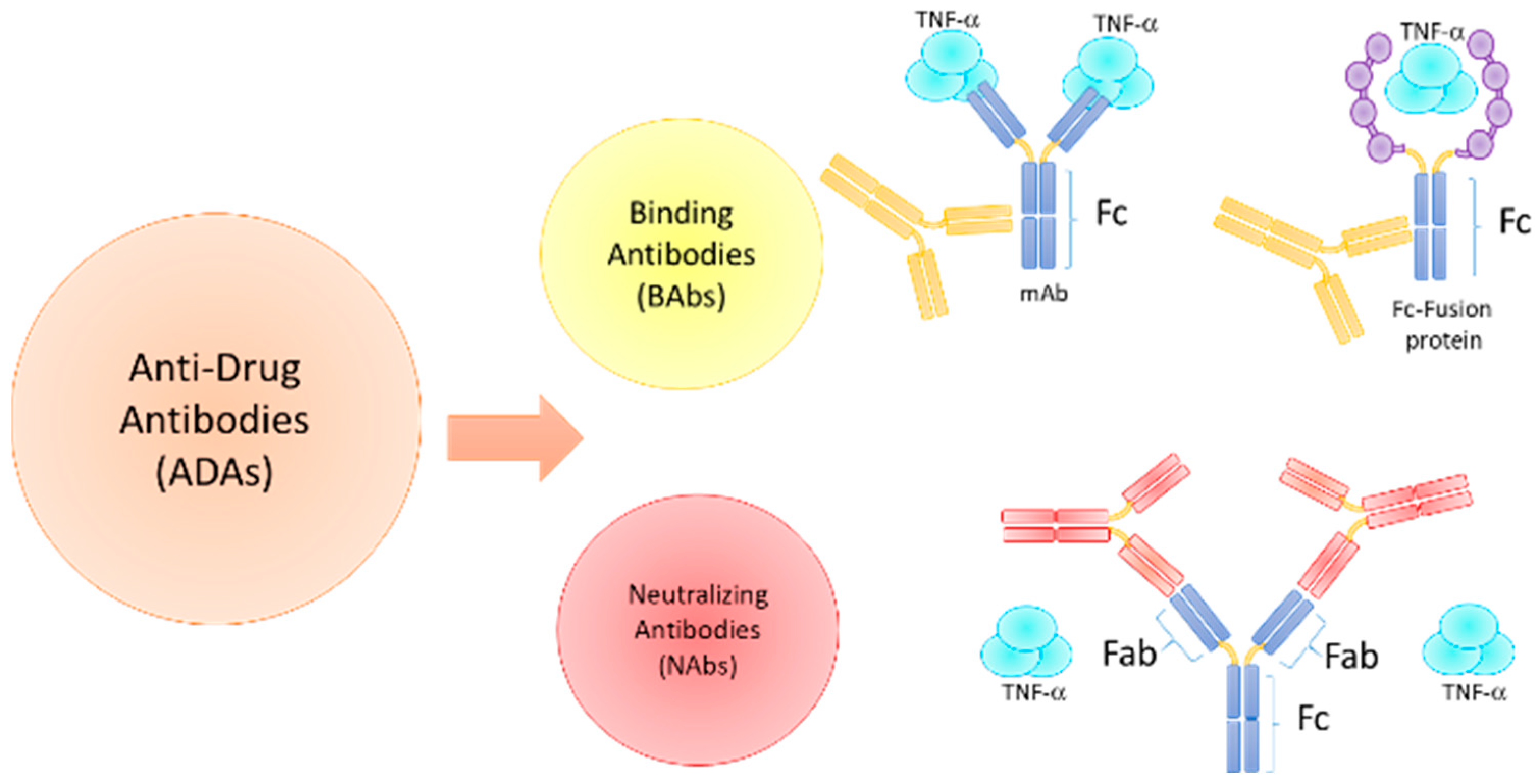
3. Immunogenicity of Biologic Agents in ARDs
3.1. Immunogenicity Factors and ADAs Development
3.2. TNF Inhibitors
3.2.1. Infliximab (IFX)
3.2.2. Adalimumab (ADL)
3.2.3. Golimumab (GLM)
3.2.4. Etanercept (ETA)
3.2.5. Certolizumab Pegol (CZP)
3.3. B Cell-Targeting Biologics
3.3.1. Rituximab (RTX)
3.3.2. Belimumab (BLM)
3.4. T Cell Activation Inhibitor/Co-Stimulation Modulator
Abatacept (ABT)
3.5. IL-6R Inhibitors
3.5.1. Tocilizumab (TCZ)
3.5.2. Sarilumab (SLM)
3.6. IL-17A Inhibitors
3.6.1. Secukinumab (SCK)
3.6.2. Ixekizumab (IXK)
3.6.3. Brodalumab (BDL)
3.7. IL-12/23 and IL-23p19 Inhibitors
3.7.1. Ustekinumab (UTK)
3.7.2. Guselkumab (GKM), Risankizumab (RZM) and Tildrakizumab (TZM)
3.8. IL-1 Inhibitors
3.8.1. Anakinra (ANA)
3.8.2. Canakinumab (CKM)
4. ADAs Detection and Clinical Implications
4.1. Assays for ADAs Detection
4.2. Clinical Implications and Therapeutic Monitoring
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moutsopoulos, H.M. Autoimmune rheumatic diseases: One or many diseases? J. Transl. Autoimmun. 2021, 4, 100129. [Google Scholar] [CrossRef]
- Bays, A.M.; Gardner, G. Pharmacologic Therapies for Rheumatologic and Autoimmune Conditions. Med. Clin. North. Am. 2016, 100, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Strand, V.; Goncalves, J.; Isaacs, J.D. Immunogenicity of biologic agents in rheumatology. Nat. Rev. Rheumatol. 2021, 17, 81–97. [Google Scholar] [CrossRef]
- Schaeverbeke, T.; Truchetet, M.E.; Kostine, M.; Barnetche, T.; Bannwarth, B.; Richez, C. Immunogenicity of biologic agents in rheumatoid arthritis patients: Lessons for clinical practice. Rheumatology 2016, 55, 210–220. [Google Scholar] [CrossRef]
- Strand, V.; Balsa, A.; Al-Saleh, J.; Barile-Fabris, L.; Horiuchi, T.; Takeuchi, T.; Lula, S.; Hawes, C.; Kola, B.; Marshall, L. Immunogenicity of Biologics in Chronic Inflammatory Diseases: A Systematic Review. BioDrugs 2017, 31, 299–316. [Google Scholar] [CrossRef]
- Maini, R.N.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Davis, D.; Macfarlane, J.D.; Antoni, C.; Leeb, B.; Elliott, M.J.; Woody, J.N.; et al. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor alpha monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis Rheum. 1998, 41, 1552–1563. [Google Scholar] [CrossRef]
- Krieckaert, C.L.; Nurmohamed, M.T.; Wolbink, G.J. Methotrexate reduces immunogenicity in adalimumab treated rheumatoid arthritis patients in a dose dependent manner. Ann. Rheum. Dis. 2012, 71, 1914–1915. [Google Scholar] [CrossRef]
- Garces, S.; Demengeot, J.; Benito-Garcia, E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: A systematic review of the literature with a meta-analysis. Ann. Rheum. Dis. 2013, 72, 1947–1955. [Google Scholar] [CrossRef]
- US Department of Health and Human Services. Immunogenicity Testing of Therapeutic Protein Products—Developing and Validating Assays for Anti-Drug Antibody Detection. FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-testing-therapeutic-protein-products-developing-and-validating-assays-anti-drug (accessed on 22 February 2022).
- Guideline on Immunogenicity Assessment of Therapeutic Protein. EMEA/CHMP/BMWP/42832/2005. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf (accessed on 22 February 2022).
- Strand, V.; Goncalves, J.; Hickling, T.P.; Jones, H.E.; Marshall, L.; Isaacs, J.D. Immunogenicity of Biosimilars for Rheumatic Diseases, Plaque Psoriasis, and Inflammatory Bowel Disease: A Review from Clinical Trials and Regulatory Documents. BioDrugs 2020, 34, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Barturen, G.; Beretta, L.; Cervera, R.; Van Vollenhoven, R.; Alarcon-Riquelme, M.E. Moving towards a molecular taxonomy of autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Ringold, S.; Khanna, D.; Neogi, T.; Johnson, S.R.; Miller, A.; Brunner, H.I.; Ogawa, R.; Felson, D.; Ogdie, A.; et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res. 2015, 67, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Zhang, J.; Zhang, Z.J.; Zhang, X. Systemic Autoimmune Diseases 2014. J. Immunol. Res. 2015, 2015, 183591. [Google Scholar] [CrossRef]
- Giacomelli, R.; Afeltra, A.; Alunno, A.; Bartoloni-Bocci, E.; Berardicurti, O.; Bombardieri, M.; Bortoluzzi, A.; Caporali, R.; Caso, F.; Cervera, R.; et al. Guidelines for biomarkers in autoimmune rheumatic diseases—Evidence based analysis. Autoimmun. Rev. 2019, 18, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.E.; Weber, S.; Jakob, R.; Chute, C.G. ICD-11: An international classification of diseases for the twenty-first century. BMC Med. Inform. Decis. Mak. 2021, 21, 206. [Google Scholar] [CrossRef]
- Selmi, C.; Generali, E.; Massarotti, M.; Bianchi, G.; Scire, C.A. New treatments for inflammatory rheumatic disease. Immunol. Res. 2014, 60, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Buer, J.K. A history of the term “DMARD”. Inflammopharmacology 2015, 23, 163–171. [Google Scholar] [CrossRef]
- Patane, M.; Ciriaco, M.; Chimirri, S.; Ursini, F.; Naty, S.; Grembiale, R.D.; Gallelli, L.; De Sarro, G.; Russo, E. Interactions among Low Dose of Methotrexate and Drugs Used in the Treatment of Rheumatoid Arthritis. Adv. Pharmacol. Sci. 2013, 2013, 313858. [Google Scholar] [CrossRef] [PubMed]
- Araujo, A.G.S.; Borba, H.H.L.; Tonin, F.S.; Lenzi, L.; Venson, R.; Pontarolo, R.; Wiens, A. Safety of Biologics Approved for the Treatment of Rheumatoid Arthritis and Other Autoimmune Diseases: A Disproportionality Analysis from the FDA Adverse Event Reporting System (FAERS). BioDrugs 2018, 32, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, D. Infectious risks associated with biologics. Adv. Exp. Med. Biol. 2013, 764, 151–158. [Google Scholar]
- Valenzuela, F.; Flores, R. Immunogenicity to biological drugs in psoriasis and psoriatic arthritis. Clinics 2021, 76, e3015. [Google Scholar] [CrossRef]
- Mayrhofer, P.; Kunert, R. Nomenclature of humanized mAbs: Early concepts, current challenges and future perspectives. Hum. Antibodies 2019, 27, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Tao, M.; Wang, Q.; Lu, X.; Yuan, H. Fusion proteins of biologic agents in the treatment of rheumatoid arthritis (RA): A network meta-analysis. Medicine 2021, 100, e26350. [Google Scholar] [CrossRef]
- Jani, M.; Barton, A.; Warren, R.B.; Griffiths, C.E.; Chinoy, H. The role of DMARDs in reducing the immunogenicity of TNF inhibitors in chronic inflammatory diseases. Rheumatology 2014, 53, 213–222. [Google Scholar] [CrossRef]
- Jarvi, N.L.; Balu-Iyer, S.V. Immunogenicity Challenges Associated with Subcutaneous Delivery of Therapeutic Proteins. BioDrugs 2021, 35, 125–146. [Google Scholar] [CrossRef] [PubMed]
- Fathallah, A.M.; Bankert, R.B.; Balu-Iyer, S.V. Immunogenicity of subcutaneously administered therapeutic proteins--a mechanistic perspective. AAPS J. 2013, 15, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Vaisman-Mentesh, A.; Gutierrez-Gonzalez, M.; DeKosky, B.J.; Wine, Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment with Monoclonal Antibodies. Front. Immunol. 2020, 11, 1951. [Google Scholar] [CrossRef]
- Carrascosa, J.M. Immunogenicity in biologic therapy: Implications for dermatology. Actas Dermosifiliogr. 2013, 104, 471–479. [Google Scholar] [CrossRef]
- Atiqi, S.; Hooijberg, F.; Loeff, F.C.; Rispens, T.; Wolbink, G.J. Immunogenicity of TNF-Inhibitors. Front. Immunol. 2020, 11, 312. [Google Scholar] [CrossRef]
- Croft, M.; Siegel, R.M. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat. Rev. Rheumatol. 2017, 13, 217–233. [Google Scholar] [CrossRef]
- Lichtenstein, L.; Ron, Y.; Kivity, S.; Ben-Horin, S.; Israeli, E.; Fraser, G.M.; Dotan, I.; Chowers, Y.; Confino-Cohen, R.; Weiss, B. Infliximab-Related Infusion Reactions: Systematic Review. J. Crohns Colitis 2015, 9, 806–815. [Google Scholar] [CrossRef]
- Jani, M.; Dixon, W.G.; Chinoy, H. Drug safety and immunogenicity of tumour necrosis factor inhibitors: The story so far. Rheumatology 2018, 57, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Krintel, S.B.; Grunert, V.P.; Hetland, M.L.; Johansen, J.S.; Rothfuss, M.; Palermo, G.; Essioux, L.; Klause, U. The frequency of anti-infliximab antibodies in patients with rheumatoid arthritis treated in routine care and the associations with adverse drug reactions and treatment failure. Rheumatology 2013, 52, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Arstikyte, I.; Kapleryte, G.; Butrimiene, I.; Venalis, A. Influence of Immunogenicity on the Efficacy of Long-Term Treatment with TNF alpha Blockers in Rheumatoid Arthritis and Spondyloarthritis Patients. Biomed. Res. Int. 2015, 2015, 604872. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, S.; Greenwald, D. Golimumab. MAbs 2009, 1, 422–431. [Google Scholar] [CrossRef]
- Singh, J.A.; Noorbaloochi, S.; Singh, G. Golimumab for rheumatoid arthritis: A systematic review. J. Rheumatol. 2010, 37, 1096–1104. [Google Scholar] [CrossRef]
- Thomas, S.S.; Borazan, N.; Barroso, N.; Duan, L.; Taroumian, S.; Kretzmann, B.; Bardales, R.; Elashoff, D.; Vangala, S.; Furst, D.E. Comparative Immunogenicity of TNF Inhibitors: Impact on Clinical Efficacy and Tolerability in the Management of Autoimmune Diseases. A Systematic Review and Meta-Analysis. BioDrugs 2015, 29, 241–258. [Google Scholar] [CrossRef]
- Bader-Meunier, B.; Krzysiek, R.; Lemelle, I.; Pajot, C.; Carbasse, A.; Poignant, S.; Melki, I.; Quartier, P.; Choupeaux, L.; Henry, E.; et al. Etanercept concentration and immunogenicity do not influence the response to Etanercept in patients with juvenile idiopathic arthritis. Semin. Arthritis Rheum. 2019, 48, 1014–1018. [Google Scholar] [CrossRef]
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.C.; Al-Maini, M.H.; Pavelka, K.; Mahgoub, E.; Kotak, S.; Korth-Bradley, J.; et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE 2017, 12, e0175207. [Google Scholar]
- Hsu, L.; Snodgrass, B.T.; Armstrong, A.W. Antidrug antibodies in psoriasis: A systematic review. Br. J. Dermatol. 2014, 170, 261–273. [Google Scholar] [CrossRef]
- Tyring, S.; Gordon, K.B.; Poulin, Y.; Langley, R.G.; Gottlieb, A.B.; Dunn, M.; Jahreis, A. Long-term safety and efficacy of 50 mg of etanercept twice weekly in patients with psoriasis. Arch. Dermatol. 2007, 143, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Scott, L.J. Certolizumab Pegol: A Review in Moderate to Severe Plaque Psoriasis. BioDrugs 2020, 34, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Certolizumab Pegol: A Review in Inflammatory Autoimmune Diseases. BioDrugs 2016, 30, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Forger, F.; Abraham, B.; Flynn, A.D.; Molto, A.; Flipo, R.M.; van Tubergen, A.; Shaughnessy, L.; Simpson, J.; Teil, M.; et al. Lack of placental transfer of certolizumab pegol during pregnancy: Results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann. Rheum. Dis. 2018, 77, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Wolf, D.C.; Kosutic, G.; Parker, G.; Schreiber, S.; Lee, S.D.; Abraham, B.; Afzali, A.; Arsenescu, R.I.; Gutierrez, A.; et al. Effects of Transient and Persistent Anti-drug Antibodies to Certolizumab Pegol: Longitudinal Data from a 7-Year Study in Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 1047–1056. [Google Scholar] [CrossRef]
- van Schouwenburg, P.A.; Rispens, T.; Wolbink, G.J. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 164–172. [Google Scholar] [CrossRef]
- Dunn, N.; Juto, A.; Ryner, M.; Manouchehrinia, A.; Piccoli, L.; Fink, K.; Piehl, F.; Fogdell-Hahn, A. Rituximab in multiple sclerosis: Frequency and clinical relevance of anti-drug antibodies. Mult. Scler. 2018, 24, 1224–1233. [Google Scholar] [CrossRef]
- Oomen, I.; Nassar-Sheikh Rashid, A.; Bouts, A.H.M.; Gouw, S.C.; Kuijpers, T.W.; Rispens, T.; de Vries, A.; Wolbink, G.; Berg, J.M.V.D.; Schonenberg-Meinema, D. Anti-rituximab antibodies affect pharmacokinetics and pharmacodynamics of rituximab in children with immune-mediated diseases. Clin. Exp. Rheumatol. 2022, 40, 183–190. [Google Scholar] [CrossRef]
- Blair, H.A.; Duggan, S.T. Belimumab: A Review in Systemic Lupus Erythematosus. Drugs 2018, 78, 355–366. [Google Scholar] [CrossRef]
- Haggerty, H.G.; Abbott, M.A.; Reilly, T.P.; DeVona, D.A.; Gleason, C.R.; Tay, L.; Dodge, R.; Aranda, R. Evaluation of immunogenicity of the T cell costimulation modulator abatacept in patients treated for rheumatoid arthritis. J. Rheumatol. 2007, 34, 2365–2373. [Google Scholar]
- Nash, P.; Nayiager, S.; Genovese, M.C.; Kivitz, A.J.; Oelke, K.; Ludivico, C.; Palmer, W.; Rodriguez, C.; Delaet, I.; Elegbe, A.; et al. Immunogenicity, safety, and efficacy of abatacept administered subcutaneously with or without background methotrexate in patients with rheumatoid arthritis: Results from a phase III, international, multicenter, parallel-arm, open-label study. Arthritis Care Res. 2013, 65, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Covarrubias, A.; Leon, G.; Mysler, E.; Keiserman, M.; Valente, R.; Nash, P.; Simon-Campos, J.A.; Porawska, W.; Box, J.; et al. Subcutaneous abatacept versus intravenous abatacept: A phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum. 2011, 63, 2854–2864. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Choy, E.; Kivitz, A.; Ogata, A.; Bao, M.; Nomura, A.; Lacey, S.; Pei, J.; Reiss, W.; Pethoe-Schramm, A.; et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1078–1085. [Google Scholar] [CrossRef]
- Burmester, G.R.; Rubbert-Roth, A.; Cantagrel, A.; Hall, S.; Leszczynski, P.; Feldman, D.; Rangaraj, M.J.; Roane, G.; Ludivico, C.; Lu, P.; et al. A randomised, double-blind, parallel-group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease-modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann. Rheum. Dis. 2014, 73, 69–74. [Google Scholar]
- Tanaka, Y.; Takahashi, T.; Sumi, M.; Hagino, O.; Van Hoogstraten, H.; Xu, C.; Kato, N.; Kameda, H. Immunogenicity of sarilumab and impact on safety and efficacy in Japanese patients with rheumatoid arthritis: Analysis of two Phase 3 randomised clinical trials. Mod. Rheumatol. 2022, 32, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.F.; Parrino, J.; Mangan, E.K.; Paccaly, A.; Lin, Y.; Xu, C.; Fan, C.; Graham, N.M.H.; van Hoogstraten, H.; Torri, A. Immunogenicity of Sarilumab Monotherapy in Patients with Rheumatoid Arthritis Who Were Inadequate Responders or Intolerant to Disease-Modifying Antirheumatic Drugs. Rheumatol. Ther. 2019, 6, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.W.; Lee, E.B.; Wu, J.J. Systematic review of anti-drug antibodies of IL-17 inhibitors for psoriasis. J. Dermatol. Treat. 2019, 30, 110–116. [Google Scholar] [CrossRef]
- Deodhar, A.; Gladman, D.D.; McInnes, I.B.; Spindeldreher, S.; Martin, R.; Pricop, L.; Porter, B.; Safi, J.; Shete, A.; Bruin, G. Secukinumab Immunogenicity over 52 Weeks in Patients with Psoriatic Arthritis and Ankylosing Spondylitis. J. Rheumatol. 2020, 47, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Spindeldreher, S.; Maillere, B.; Correia, E.; Tenon, M.; Karle, A.; Jarvis, P.; Kolbinger, F. Secukinumab Demonstrates Significantly Lower Immunogenicity Potential Compared to Ixekizumab. Dermatol. Ther. 2018, 8, 57–68. [Google Scholar] [CrossRef]
- Gordon, K.B.; Colombel, J.F.; Hardin, D.S. Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. N. Engl. J. Med. 2016, 375, 2102. [Google Scholar] [CrossRef]
- Bagel, J.; Lebwohl, M.; Israel, R.J.; Jacobson, A. Immunogenicity and skin clearance recapture in clinical studies of brodalumab. J. Am. Acad. Dermatol. 2020, 82, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.; Moon, J.Y.; Javadi, S.S.; Munawar, L.; Maul, J.T.; Wu, J.J. Anti-drug antibodies of IL-23 inhibitors for psoriasis: A systematic review. J. Eur. Acad. Dermatol. Venereol. 2022, 36, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Canete, J.D. Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opin. Drug Saf. 2018, 17, 727–732. [Google Scholar] [CrossRef]
- Fleischmann, R.M.; Schechtman, J.; Bennett, R.; Handel, M.L.; Burmester, G.R.; Tesser, J.; Modafferi, D.; Poulakos, J.; Sun, G. Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: A large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 2003, 48, 927–934. [Google Scholar] [CrossRef]
- Sun, H.; Van, L.M.; Floch, D.; Jiang, X.; Klein, U.R.; Abrams, K.; Sunkara, G. Pharmacokinetics and Pharmacodynamics of Canakinumab in Patients with Systemic Juvenile Idiopathic Arthritis. J. Clin. Pharmacol. 2016, 56, 1516–1527. [Google Scholar] [CrossRef] [PubMed]
- Tavakolpour, S.; Alesaeidi, S.; Darvishi, M.; GhasemiAdl, M.; Darabi-Monadi, S.; Akhlaghdoust, M.; Behjati, S.E.; Jafarieh, A. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin. Rheumatol. 2019, 38, 2977–2994. [Google Scholar] [CrossRef]
- Leandro, M.; Isenberg, D.A. Rituximab—The first twenty years. Lupus 2021, 30, 371–377. [Google Scholar] [CrossRef]
- Faustini, F.; Dunn, N.; Kharlamova, N.; Ryner, M.; Bruchfeld, A.; Malmstrom, V.; Fogdell-Hahn, A.; Gunnarsson, I. First exposure to rituximab is associated to high rate of anti-drug antibodies in systemic lupus erythematosus but not in ANCA-associated vasculitis. Arthritis Res. Ther. 2021, 23, 211. [Google Scholar] [CrossRef]
- Levy, R.A.; Gonzalez-Rivera, T.; Khamashta, M.; Fox, N.L.; Jones-Leone, A.; Rubin, B.; Burriss, S.W.; Gairy, K.; van Maurik, A.; Roth, D.A. 10 Years of belimumab experience: What have we learnt? Lupus 2021, 30, 1705–1721. [Google Scholar] [CrossRef]
- Pombo-Suarez, M.; Gomez-Reino, J.J. Abatacept for the treatment of rheumatoid arthritis. Expert Rev. Clin. Immunol. 2019, 15, 319–326. [Google Scholar] [CrossRef]
- Blair, H.A.; Deeks, E.D. Abatacept: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef]
- Rubbert-Roth, A.; Furst, D.E.; Nebesky, J.M.; Jin, A.; Berber, E. A Review of Recent Advances Using Tocilizumab in the Treatment of Rheumatic Diseases. Rheumatol. Ther. 2018, 5, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, M.; Laskou, F.; Stapleton, P.P.; Hadavi, S.; Dasgupta, B. Tocilizumab (Actemra). Hum. Vaccines Immunother. 2017, 13, 1972–1988. [Google Scholar] [CrossRef] [PubMed]
- Karle, A.; Spindeldreher, S.; Kolbinger, F. Secukinumab, a novel anti-IL-17A antibody, shows low immunogenicity potential in human in vitro assays comparable to other marketed biotherapeutics with low clinical immunogenicity. MAbs 2016, 8, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Blauvelt, A.; Armstrong, A.; Langley, R.G.; de Vera, A.; Kolbinger, F.; Spindeldreher, S.; Ren, M.; Bruin, G. Secukinumab, a fully human anti-interleukin-17A monoclonal antibody, exhibits low immunogenicity in psoriasis patients treated up to 5 years. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Reich, K.; Blauvelt, A.; Armstrong, A.; Langley, R.G.; Fox, T.; Huang, J.; Papavassilis, C.; Liang, E.; Lloyd, P.; Bruin, G. Secukinumab, a fully human anti-interleukin-17A monoclonal antibody, exhibits minimal immunogenicity in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2017, 176, 752–758. [Google Scholar] [CrossRef]
- Syed, Y.Y. Ixekizumab: A Review in Moderate to Severe Plaque Psoriasis. Am. J. Clin. Dermatol. 2017, 18, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef]
- Mease, P.J.; McInnes, I.B.; Kirkham, B.; Kavanaugh, A.; Rahman, P.; van der Heijde, D.; Landewé, R.; Nash, P.; Pricop, L.; Yuan, J.; et al. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N. Engl. J. Med. 2015, 373, 1329–1339. [Google Scholar] [CrossRef]
- Rafael-Vidal, C.; Perez, N.; Altabas, I.; Garcia, S.; Pego-Reigosa, J.M. Blocking IL-17: A Promising Strategy in the Treatment of Systemic Rheumatic Diseases. Int. J. Mol. Sci. 2020, 21, 7100. [Google Scholar] [CrossRef]
- Fiorino, G.; Allocca, M.; Correale, C.; Roda, G.; Furfaro, F.; Loy, L.; Zilli, A.; Peyrin-Biroulet, L.; Danese, S. Positioning ustekinumab in moderate-to-severe ulcerative colitis: New kid on the block. Expert Opin. Biol. Ther. 2020, 20, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Gensler, L.S.; Yang, Z.; Gasink, C.; Chakravarty, S.D.; Farahi, K.; Ramachandran, P.; Ott, E.; Strober, B.E. Ustekinumab Safety in Psoriasis, Psoriatic Arthritis, and Crohn’s Disease: An Integrated Analysis of Phase II/III Clinical Development Programs. Drug Saf. 2019, 42, 751–768. [Google Scholar] [CrossRef]
- Hanauer, S.B.; Sandborn, W.J.; Feagan, B.G.; Gasink, C.; Jacobstein, D.; Zou, B.; Johanns, J.; Adedokun, O.J.; Sands, B.E.; Rutgeerts, P.; et al. IM-UNITI: Three-year Efficacy, Safety, and Immunogenicity of Ustekinumab Treatment of Crohn’s Disease. J. Crohns Colitis 2020, 14, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Rutgeerts, P.; Gasink, C.; Jacobstein, D.; Zou, B.; Johanns, J.; Sands, B.E.; Hanauer, S.B.; Targan, S.; Ghosh, S.; et al. Long-term efficacy and safety of ustekinumab for Crohn’s disease through the second year of therapy. Aliment. Pharmacol. Ther. 2018, 48, 65–77. [Google Scholar] [CrossRef]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Li, G.G.; Liu, Q.; Niu, X.; Li, R.; Ma, H. Short-Term Efficacy and Safety of IL-17, IL-12/23, and IL-23 Inhibitors Brodalumab, Secukinumab, Ixekizumab, Ustekinumab, Guselkumab, Tildrakizumab, and Risankizumab for the Treatment of Moderate to Severe Plaque Psoriasis: A Systematic Review and Network Meta-Analysis of Randomized Controlled Trials. J. Immunol. Res. 2019, 2019, 2546161. [Google Scholar]
- Stefania, S.; Colia, R.; Cinzia, R.; Corrado, A.; Cantatore, F.P. Off-label use of anti-IL-1 drugs in rheumatic diseases. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211006584. [Google Scholar] [CrossRef]
- Mertens, M.; Singh, J.A. Anakinra for rheumatoid arthritis. Cochrane Database Syst. Rev. 2009, CD005121. [Google Scholar] [CrossRef]
- Kotter, I.; Schedel, J.; Kummerle-Deschner, J.B. Periodic fever syndrome/autoinflammatory syndrome. Z. Rheumatol. 2009, 68, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.; Horneff, G.; Brik, R.; McCann, L.; Kasapcopur, O.; Rutkowska-Sak, L.; et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2396–2406. [Google Scholar] [CrossRef]
- Dhimolea, E. Canakinumab. MAbs 2010, 2, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Betrains, A.; Staels, F.; Vanderschueren, S. Efficacy and safety of canakinumab treatment in schnitzler syndrome: A systematic literature review. Semin. Arthritis Rheum. 2020, 50, 636–642. [Google Scholar] [CrossRef]
- Wulffraat, N.M. A safety evaluation of canakinumab for the treatment of systemic onset juvenile idiopathic arthritis. Expert Opin. Drug Saf. 2015, 14, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Cota-Arce, J.M.; Cota, J.; De Leon-Nava, M.A.; Hernandez-Caceres, A.; Moncayo-Salazar, L.I.; Valle-Alvarado, F.; Cordero-Moreno, V.L.; Bonfil-Solis, K.L.; Bichara-Figueroa, J.E.; Hernández-Hernández, J.; et al. Efficacy and safety of canakinumab in the treatment of adult-onset Still’s disease: A systematic review. Semin. Arthritis Rheum. 2021, 51, 1282–1290. [Google Scholar] [CrossRef]
- Benucci, M.; Grossi, V.; Manfredi, M.; Damiani, A.; Infantino, M.; Moscato, P.; Cinquanta, L.; Gremese, E.; Tolusso, B.; Petricca, L.; et al. Laboratory Monitoring of Biological Therapies in Rheumatology: The Role of Immunogenicity. Ann. Lab. Med. 2020, 40, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wala, I.; Han, H.; Nagatani, J.; Barger, T.; Civoli, F.; Kaliyaperumal, A.; Zhuang, Y.; Gupta, S. Comparison of cell-based and non-cell-based assay platforms for the detection of clinically relevant anti-drug neutralizing antibodies for immunogenicity assessment of therapeutic proteins. J. Immunol. Methods 2015, 419, 1–8. [Google Scholar] [CrossRef]
- Jolicoeur, P.; Tacey, R.L. Development and validation of cell-based assays for the detection of neutralizing antibodies to drug products: A practical approach. Bioanalysis 2012, 4, 2959–2970. [Google Scholar] [CrossRef]
- Maneiro, J.R.; Salgado, E.; Gomez-Reino, J.J. Immunogenicity of monoclonal antibodies against tumor necrosis factor used in chronic immune-mediated Inflammatory conditions: Systematic review and meta-analysis. JAMA Intern. Med. 2013, 173, 1416–1428. [Google Scholar] [CrossRef]
- Kostine, M.; Finckh, A.; Bingham, C.O.; Visser, K.; Leipe, J.; Schulze-Koops, H.; Choy, E.H.; Benesova, K.; Radstake, T.R.D.J.; Cope, A.P.; et al. EULAR points to consider for the diagnosis and management of rheumatic immune-related adverse events due to cancer immunotherapy with checkpoint inhibitors. Ann. Rheum. Dis. 2021, 80, 36–48. [Google Scholar] [CrossRef]
- CEBM. Oxford Centre for Evidence-Based Medicine—Levels of Evidence (March 2009). 2009. Available online: https://www.cebm.net/2009/06/oxford-centre-evidencebasedmedicine-levels-evidence-march-2009/ (accessed on 18 April 2023).
- van Bezooijen, J.S.; Koch, B.C.; van Doorn, M.B.; Prens, E.P.; van Gelder, T.; Schreurs, M.W. Comparison of Three Assays to Quantify Infliximab, Adalimumab, and Etanercept Serum Concentrations. Ther. Drug Monit. 2016, 38, 432–438. [Google Scholar] [CrossRef]
- Nasser, Y.; Labetoulle, R.; Harzallah, I.; Berger, A.E.; Roblin, X.; Paul, S. Comparison of Point-of-Care and Classical Immunoassays for the Monitoring Infliximab and Antibodies Against Infliximab in IBD. Dig. Dis. Sci. 2018, 63, 2714–2721. [Google Scholar] [CrossRef]
- Laserna-Mendieta, E.J.; Salvador-Martin, S.; Marin-Jimenez, I.; Menchen, L.A.; Lopez-Cauce, B.; Lopez-Fernandez, L.A.; Lucendo, A.J. Comparison of a new rapid method for determination of serum anti-adalimumab and anti-infliximab antibodies with two established ELISA kits. J. Pharm. Biomed. Anal. 2021, 198, 114003. [Google Scholar] [CrossRef] [PubMed]
- Krieckaert, C.L.; van Tubergen, A.; Gehin, J.E.; Hernandez-Breijo, B.; Le Meledo, G.; Balsa, A.; Böhm, P.; Cucnik, S.; Elkayam, O.; Goll, G.L.; et al. EULAR points to consider for therapeutic drug monitoring of biopharmaceuticals in inflammatory rheumatic and musculoskeletal diseases. Ann. Rheum. Dis. 2023, 82, 65–73. [Google Scholar] [CrossRef]
- Jani, M.; Chinoy, H.; Warren, R.B.; Griffiths, C.E.; Plant, D.; Fu, B.; Morgan, A.W.; Wilson, A.G.; Isaacs, J.D.; Hyrich, K.; et al. Clinical utility of random anti-tumor necrosis factor drug-level testing and measurement of antidrug antibodies on the long-term treatment response in rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Siljehult, F.; Arlestig, L.; Eriksson, C.; Rantapaa-Dahlqvist, S. Concentrations of infliximab and anti-drug antibodies in relation to clinical response in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2018, 47, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Teresa, J.; Chamaida, P.R.; Ana, M.F.; Victoria, N.C.; Theo, R.; Annick, V.; Karien, B.; Eva-María, O.; Cristina, D.; Alejandro, V.; et al. Predictive Value of Serum Infliximab Levels at Induction Phase in Rheumatoid Arthritis Patients. Open Rheumatol. J. 2017, 11, 75–87. [Google Scholar]
- Jamnitski, A.; Bartelds, G.M.; Nurmohamed, M.T.; van Schouwenburg, P.A.; van Schaardenburg, D.; Stapel, S.O.; Dijkmans, B.A.C.; Aarden, L.; Wolbink, G.J. The presence or absence of antibodies to infliximab or adalimumab determines the outcome of switching to etanercept. Ann. Rheum. Dis. 2011, 70, 284–288. [Google Scholar] [CrossRef]
- Bartelds, G.M.; Wijbrandts, C.A.; Nurmohamed, M.T.; Stapel, S.; Lems, W.F.; Aarden, L.; Dijkmans, B.A.C.; Tak, P.P.; Wolbink, G.J. Anti-infliximab and anti-adalimumab antibodies in relation to response to adalimumab in infliximab switchers and anti-tumour necrosis factor naive patients: A cohort study. Ann. Rheum. Dis. 2010, 69, 817–821. [Google Scholar] [CrossRef]


{kind=link}
| Target | Biological Agent | Format | Indication |
|---|---|---|---|
| TNF-α | Infliximab | Chimeric mAb | CD, UC, PsA, PsO, AS, RA |
| TNF-α | Adalimumab | Fully human mAb | RA, PsA, PsO, AS, CD, JIA, UV |
| TNF-α | Golimumab | Fully human mAb | AS, RA, PsA |
| TNF-α | Etanercept | Fusion protein | RA, pJIA, PsA, AS, PsO |
| TNF-α | Certolizumab pegol | Humanized mAb | CD, RA, PsA, AS, nr-axSpA |
| CD20 | Rituximab | Chimeric mAb | RA, SLE |
| BLyS | Belimumab | Fully human mAb | SLE |
| CTLA-4 | Abatacept | Fusion protein | RA, PsA |
| IL-6R | Tocilizumab | Humanized mAb | RA, sJIA, pJIA |
| IL-6R | Sarilumab | Fully human mAb | RA |
| IL-17A | Secukinumab | Fully human mAb | AS, RA, PsA, PsO, uveitis, AxSpA, nr-AxSpA |
| IL-17A | Ixekizumab | Humanized mAb | PsO, PsA, and AS |
| IL-17RA | Brodalumab | Fully human mAb | PsA |
| IL-12R/IL-23R | Ustekinumab | Human mAb | PsO, CD, UC |
| IL-23 p19 | Risankizumab | Humanized mAb | PsO |
| IL-23 p19 | Tildrakizumab | Humanized mAb | PsO |
| IL-23 p19 | Guselkumab | Fully human mAb | PsO |
| IL-1RA | Anakinra | Fusion protein | RA and autoinflammatory fever syndrome |
| IL1-β | Canakinumab | Fully human mAb | AR, sJIA |
| Biological Agent | Abbreviation | ADAs% Min–Max | Nabs% Min–Max | Ref | |
|---|---|---|---|---|---|
| TNFi | Infliximab | IFX | 0–83.0 | Not reported | [5,42] |
| Adalimumab | ADL | 0–54.0 | Not reported | [5,42] | |
| Golimumab | GLM | 0–19.0 | Not reported | [5] | |
| Etanercept | ETA | 0–18.3 | Not reported | [5,42] | |
| Certolizumab pegol | CZP | 3–37.0 | Not reported | [5] | |
| B cell-targeting biologics | Rituximab | RTX | 23.1–50.0 | Not reported | [49,50] |
| Belimumab | BLM | 0–4.8 | Not reported | [51] | |
| T cell activation inhibitor/co-stimulation modulator | Abatacept | ABT | 0.9–4.1 | 0–0.4 | [52,53,54] |
| IL-6R inhibitors | Tocilizumab | TCZ | 0.7–2.0 | 0.8–1.3 | [55,56] |
| Sarilumab | SLM | 1.4–12.3 | 0–10.8 | [57,58] | |
| IL-17A inhibitors | Secukinumab | SCK | 0–1.0 | Not reported | [5,59,60,61] |
| Ixekizumab | IXK | 1.7–9.0 | Not reported | [62] | |
| Brodalumab | BDL | 1.4–2.7 | 0 | [59,63] | |
| IL-12/23 and IL-23p19 inhibitors | Ustekinumab | UTK | 1.0–11.0 | Not reported | [5,42] |
| Guselkumab | GKM | 4.1–14.7 | 0.1–0.6 | [64] | |
| Risankizumab | RZM | 14.1–31.0 | 2.1–16.0 | [64] | |
| Tildrakizumab | TZM | 6.51–18.0 | 2.5–3.2 | [64] | |
| IL-1 inhibitors | Anakinra | ANA | <1 | Not reported | [65,66] |
| Canakinumab | CKM | 3.1 | 0 | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pizano-Martinez, O.; Mendieta-Condado, E.; Vázquez-Del Mercado, M.; Martínez-García, E.A.; Chavarria-Avila, E.; Ortuño-Sahagún, D.; Márquez-Aguirre, A.L. Anti-Drug Antibodies in the Biological Therapy of Autoimmune Rheumatic Diseases. J. Clin. Med. 2023, 12, 3271. https://doi.org/10.3390/jcm12093271
Pizano-Martinez O, Mendieta-Condado E, Vázquez-Del Mercado M, Martínez-García EA, Chavarria-Avila E, Ortuño-Sahagún D, Márquez-Aguirre AL. Anti-Drug Antibodies in the Biological Therapy of Autoimmune Rheumatic Diseases. Journal of Clinical Medicine. 2023; 12(9):3271. https://doi.org/10.3390/jcm12093271
Chicago/Turabian StylePizano-Martinez, Oscar, Edgar Mendieta-Condado, Mónica Vázquez-Del Mercado, Erika Aurora Martínez-García, Efrain Chavarria-Avila, Daniel Ortuño-Sahagún, and Ana Laura Márquez-Aguirre. 2023. "Anti-Drug Antibodies in the Biological Therapy of Autoimmune Rheumatic Diseases" Journal of Clinical Medicine 12, no. 9: 3271. https://doi.org/10.3390/jcm12093271
APA StylePizano-Martinez, O., Mendieta-Condado, E., Vázquez-Del Mercado, M., Martínez-García, E. A., Chavarria-Avila, E., Ortuño-Sahagún, D., & Márquez-Aguirre, A. L. (2023). Anti-Drug Antibodies in the Biological Therapy of Autoimmune Rheumatic Diseases. Journal of Clinical Medicine, 12(9), 3271. https://doi.org/10.3390/jcm12093271