Association between Stress and the HPA Axis in the Atopic Dermatitis

Abstract
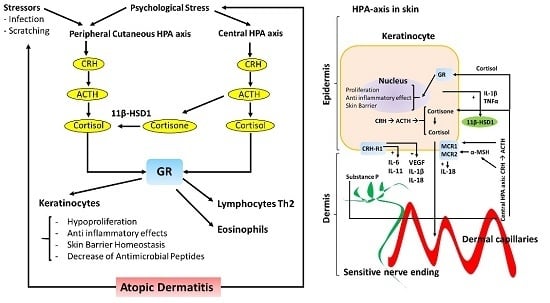
:
1. Introduction
2. Hypothalamic–Pituitary–Adrenal (HPA) Axis
3. The Peripheral HPA Axis
4. HPA Axis and Skin Inflammation
5. HPA Axis and Skin Barrier
6. The Effect of Stress in AD on the HPA Axis
7. Conclusions
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| AD | Atopic dermatitis |
| AMP | Antimicrobial peptide |
| α-MSH | Alpha melanocyte-stimulating hormone |
| CRH | Corticotrophin-releasing hormone |
| CRH-R | Corticotrophin-releasing hormone receptor |
| EKO | Epidermal knockout |
| GC | Glucocorticoid |
| GR | Glucocorticoid receptor |
| HPA | Hypothalamic–pituitary–adrenal |
| IL | Interleukin |
| IgE | Immunoglobulin E |
| MCR | Melanocortin receptor |
| POMC | Pro-opiomelanocortin |
| PS | Psychological stress |
| SC | Stratum corneum |
| SP | Substance P |
| TEWL | Transepidermal water loss |
| Th2 | T helper cell type 2 |
| TSLP | Thymic stromal lymphopoietin |
| TNF-α | Tumor necrosis factor alpha |
| 11β-HSD | 11 beta-hydroxysteroid dehydrogenase |
References
- Bieber, T. Atopic dermatitis. N. Engl. J. Med. 2008, 358, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Wakefield, J.S. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Clin. Rev. Allergy Immunol. 2011, 41, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Irvine, A.D.M.; Eichenfield, L.F.M.; Friedlander, S.F.M.; Simpson, E.L.M.M. Review of critical issues in the pathogenesis of atopic dermatitis. Semin. Cutan. Med. Surg. 2016, 35, S89–S91. [Google Scholar] [CrossRef] [PubMed]
- Ohtsu, H.; Seike, M. Histamine and histamine receptors in allergic dermatitis. Handb. Exp. Pharmacol. 2017, 241, 333–345. [Google Scholar] [PubMed]
- Senra, M.S.; Wollenberg, A. Psychodermatological aspects of atopic dermatitis. Br. J. Dermatol. 2014, 170, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Altemus, M.; Rao, B.; Dhabhar, F.S.; Ding, W.; Granstein, R.D. Stress-induced changes in skin barrier function in healthy women. J. Investig. Dermatol. 2001, 117, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Lack, G. Update on risk factors for food allergy. J. Allergy. Clin. Immunol. 2012, 129, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Naidu, Y. Perturbation of epidermal barrier function correlates with initiation of cytokine cascade in human skin. J. Am. Acad. Dermatol. 1994, 30, 535–546. [Google Scholar] [CrossRef]
- Hon, K.L.; Leung, A.K.; Barankin, B. Barrier repair therapy in atopic dermatitis: An overview. Am. J. Clin. Dermatol. 2013, 14, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.M.M.; Irvine, A.D.M.; Eichenfield, L.F.M.; Friedlander, S.F.M. Update on epidemiology, diagnosis, and disease course of atopic dermatitis. Semin. Cutan. Med. Surg. 2016, 35, S84–S88. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, S.; Kawai, M.; Kasajima, Y.; Hamamoto, T. Increased plasma tumour necrosis factor-α concentration in atopic dermatitis. Arch. Dis. Child. 1992, 67, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Engebretsen, K.A.; Thyssen, J.P. Skin barrier function and allergens. Curr. Probl. Dermatol. 2016, 49, 90–102. [Google Scholar] [PubMed]
- Demehri, S.; Liu, Z.; Lee, J.; Lin, M.H.; Crosby, S.D.; Roberts, C.J.; Grigsby, P.W.; Miner, J.H.; Farr, A.G.; Kopan, R. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008, 6, e123. [Google Scholar]
- Dumortier, A.; Durham, A.D.; Di Piazza, M.; Vauclair, S.; Koch, U.; Ferrand, G.; Ferrero, I.; Demehri, S.; Song, L.L.; Farr, A.G.; et al. Atopic dermatitis-like disease and associated lethal myeloproliferative disorder arise from loss of Notch signaling in the murine skin. PLoS ONE 2010, 5, e9258. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Dubus, P.; Hovnanian, A. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Oyoshi, M.K.; Garibyan, L.; Kumar, L.; Ziegler, S.F.; Geha, R.S. TSLP acts on infiltrating effector T cells to drive allergic skin inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 11875–11880. [Google Scholar] [CrossRef] [PubMed]
- Segawa, R.; Yamashita, S.; Mizuno, N.; Shiraki, M.; Hatayama, T.; Satou, N.; Hiratsuka, M.; Hide, M.; Hirasawa, N. Identification of a cell line producing high levels of TSLP: Advantages for screening of anti-allergic drugs. J. Immunol. Methods. 2014, 402, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.; Omori, M.; Gyarmati, D.; Zhou, B.; Aye, T.; Brewer, A.; Comeau, M.R.; Campbell, D.J.; Ziegler, S.F. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J. Exp. Med. 2005, 202, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Tatsuno, K.; Fujiyama, T.; Yamaguchi, H.; Waki, M.; Tokura, Y. TSLP directly interacts with skin-homing TH2 cells highly expressing its receptor to enhance IL-4 production in atopic dermatitis. J. Investig. Dermatol. 2015, 135, 3017–3024. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, U.; Hvid, M.; Johansen, C.; Buchner, M.; Folster-Holst, R.; Deleuran, M.; Vestergaard, C. TSLP, IL-31, IL-33 and sST2 are new biomarkers in endophenotypic profiling of adult and childhood atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.I.; Heuze, M.L.; Martinez-Cingolani, C.; Volpe, E.; Donnadieu, M.H.; Piel, M.; Homey, B.; Lennon-Dumenil, A.M.; Soumelis, V. The human cytokine TSLP triggers a cell-autonomous dendritic cell migration in confined environments. Blood 2011, 118, 3862–3869. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Schaper, K.; Rossbach, K.; Kother, B.; Stark, H.; Kietzmann, M.; Werfel, T.; Gutzmer, R. Stimulation of the histamine 4 receptor upregulates thymic stromal lymphopoietin (TSLP) in human and murine keratinocytes. Pharmacol. Res. 2016, 113, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Werfel, T.; Allam, J.P.; Biedermann, T.; Eyerich, K.; Gilles, S.; Guttman-Yassky, E.; Hoetzenecker, W.; Knol, E.; Simon, H.U.; Wollenberg, A.; et al. Cellular and molecular immunologic mechanisms in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Ebrecht, M.; Hellhammer, D.H. Blunted HPA axis responsiveness to stress in atopic patients is associated with the acuity and severeness of allergic inflammation. Brain Behav. Immun. 2010, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Hellhammer, D.H. Endocrine and immune responses to stress in chronic inflammatory skin disorders. Ann. N. Y. Acad. Sci. 2003, 992, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, S. Neuroimmune communication in skin: Far from peripheral. J. Investig. Dermatol. 2008, 128, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Priftis, K.N.; Papadimitriou, A.; Nicolaidou, P.; Chrousos, G.P. The hypothalamic–pituitary–adrenal axis in asthmatic children. Trends Endocrinol. Metab. 2008, 19, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P. The hypothalamic–pituitary–adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 1995, 332, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Chrousos, G.P. Stress system—Organization, physiology and immunoregulation. Neuroimmunomodulation 2006, 13, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Ketchesin, K.D.; Stinnett, G.S.; Seasholtz, A.F. Corticotropin-releasing hormone-binding protein and stress: From invertebrates to humans. Stress 2017, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Zbytek, B.; Tobin, D.J.; Theoharides, T.C.; Rivier, J. Key role of CRF in the skin stress response system. Endocr. Rev. 2013, 34, 827–884. [Google Scholar] [CrossRef] [PubMed]
- Millington, G.W. Proopiomelanocortin (POMC): The cutaneous roles of its melanocortin products and receptors. Clin. Exp. Dermatol. 2006, 31, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Airaghi, L.; Colombo, G.; Lipton, J.M. α-Melanocyte-stimulating hormone in normal human physiology and disease states. Trends Endocrinol. Metab. 2000, 11, 304–308. [Google Scholar] [CrossRef]
- Slominski, A.T.; Zmijewski, M.A.; Skobowiat, C.; Zbytek, B.; Slominski, R.M.; Steketee, J.D. Sensing the environment: Regulation of local and global homeostasis by the skin’s neuroendocrine system. Adv. Anat. Embryol. Cell Biol. 2012, 212, 1–115. [Google Scholar]
- Kim, J.E.; Cho, B.K.; Cho, D.H.; Park, H.J. Expression of hypothalamic–pituitary–adrenal axis in common skin diseases: Evidence of its association with stress-related disease activity. Acta. Derm. Venereol. 2013, 93, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, G. HPA axis responsiveness to stress: Implications for healthy aging. Exp. Gerontol. 2011, 46, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Zen, M.; Canova, M.; Campana, C.; Bettio, S.; Nalotto, L.; Rampudda, M.; Ramonda, R.; Iaccarino, L.; Doria, A. The kaleidoscope of glucorticoid effects on immune system. Autoimmun. Rev. 2011, 10, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Ebrecht, M.; Kern, S.; Hellhammer, D.H. Endocrine stress responses in TH1-mediated chronic inflammatory skin disease (psoriasis vulgaris)—Do they parallel stress-induced endocrine changes in TH2-mediated inflammatory dermatoses (atopic dermatitis)? Psychoneuroendocrinology 2006, 31, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Kalogeromitros, D. The critical role of mast cells in allergy and inflammation. Ann. N. Y. Acad. Sci. 2006, 1088, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Ito, T.; Kromminga, A.; Bettermann, A.; Takigawa, M.; Kees, F.; Straub, R.H.; Paus, R. Human hair follicles display a functional equivalent of the hypothalamic–pituitary–adrenal axis and synthesize cortisol. FASEB J. 2005, 19, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Tuckey, R.C.; Paus, R. Differential expression of HPA axis homolog in the skin. Mol. Cell Endocrinol. 2007, 265–266, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Peters, E.M.; Liezmann, C.; Klapp, B.F.; Kruse, J. The neuroimmune connection interferes with tissue regeneration and chronic inflammatory disease in the skin. Ann. N. Y. Acad. Sci. 2012, 1262, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Rohleder, N. Acute and chronic stress induced changes in sensitivity of peripheral inflammatory pathways to the signals of multiple stress systems—2011 Curt Richter Award Winner. Psychoneuroendocrinology 2012, 37, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Harvima, I.T.; Nilsson, G.; Naukkarinen, A. Role of mast cells and sensory nerves in skin inflammation. G. Ital. Dermatol. Venereol. 2010, 145, 195–204. [Google Scholar] [PubMed]
- Dhabhar, F.S.; Malarkey, W.B.; Neri, E.; McEwen, B.S. Stress-induced redistribution of immune cells—From barracks to boulevards to battlefields: A tale of three hormones—Curt Richter Award winner. Psychoneuroendocrinology 2012, 37, 1345–1368. [Google Scholar] [CrossRef] [PubMed]
- Arck, P.C.; Slominski, A.; Theoharides, T.C.; Peters, E.M.; Paus, R. Neuroimmunology of stress: Skin takes center stage. J. Investig. Dermatol. 2006, 126, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Pisarchik, A.; Slominski, A. Molecular and functional characterization of novel CRFR1 isoforms from the skin. Eur. J. Biochem. 2004, 271, 2821–2830. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Luger, T.; Paus, R.; Solomon, S. Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol. Rev. 2000, 80, 979–1020. [Google Scholar] [PubMed]
- Gantz, I.; Fong, T.M. The melanocortin system. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E468–E474. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Zmijewski, M.; Slominski, R.M.; Kauser, S.; Wortsman, J.; Tobin, D.J. Corticotropin releasing hormone and the skin. Front. Biosci. 2006, 11, 2230–2248. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Pisarchik, A.; Tobin, D.J.; Mazurkiewicz, J.E.; Wortsman, J. Differential expression of a cutaneous corticotropin-releasing hormone system. Endocrinology 2004, 145, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Nagata, H.; Umemura, S.; Kawana, S.; Osamura, R.Y. In situ expression of corticotropin-releasing hormone (CRH) and proopiomelanocortin (POMC) genes in human skin. FASEB J. 2001, 15, 2297–2299. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Chrousos, G.P. Stress Hormones, Th1/Th2 patterns, Pro/Anti-inflammatory Cytokines and Susceptibility to Disease. Trends. Endocrinol. Metab. 1999, 10, 359–368. [Google Scholar] [CrossRef]
- Cemil, B.C.; Canpolat, F.; Yilmazer, D.; Eskioglu, F.; Alper, M. The association of PASI scores with CRH-R1 expression in patients with psoriasis. Arch. Dermatol. Res. 2012, 304, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Donelan, J.M.; Papadopoulou, N.; Cao, J.; Kempuraj, D.; Conti, P. Mast cells as targets of corticotropin-releasing factor and related peptides. Trends Pharmacol. Sci. 2004, 25, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J.; Pisarchik, A.; Zbytek, B.; Linton, E.A.; Mazurkiewicz, J.E.; Wei, E.T. Cutaneous expression of corticotropin-releasing hormone (CRH), urocortin, and CRH receptors. FASEB J. 2001, 15, 1678–1693. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Zbytek, B.; Pisarchik, A.; Slominski, R.M.; Zmijewski, M.A.; Wortsman, J. CRH functions as a growth factor/cytokine in the skin. J. Cell Physiol. 2006, 206, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Pikula, M.; Slominski, R.M.; Mysliwski, A.; Wei, E.; Wortsman, J.; Slominski, A.T. Corticotropin-releasing hormone triggers differentiation in HaCaT keratinocytes. Br. J. Dermatol. 2005, 152, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Slominski, A.T. Corticotropin-releasing hormone induces keratinocyte differentiation in the adult human epidermis. J. Cell Physiol. 2005, 203, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Zbytek, B.; Mysliwski, A.; Slominski, A.; Wortsman, J.; Wei, E.T.; Mysliwska, J. Corticotropin-releasing hormone affects cytokine production in human HaCaT keratinocytes. Life Sci. 2002, 70, 1013–1021. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Singh, L.K.; Boucher, W.; Pang, X.; Letourneau, R.; Webster, E.; Chrousos, G. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology 1998, 139, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.L.; Yu, X.J.; Chen, L.M.; Jiang, H.; Li, C.Y. Corticotropin-releasing hormone attenuates vascular endothelial growth factor release from human HaCaT keratinocytes. Regul. Pept. 2010, 160, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.T.; Gao, G.C. Corticotropin-releasing factor: An inhibitor of vascular leakage in rat skeletal muscle and brain cortex after injury. Regul. Pept. 1991, 33, 93–104. [Google Scholar] [CrossRef]
- Zbytek, B.; Pfeffer, L.M.; Slominski, A.T. CRH inhibits NF-κB signaling in human melanocytes. Peptides 2006, 27, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Itoi, S.; Terao, M.; Murota, H.; Katayama, I. 11β-Hydroxysteroid dehydrogenase 1 contributes to the pro-inflammatory response of keratinocytes. Biochem. Biophys. Res. Commun. 2013, 440, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11β-Hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Seckl, J.R. 11β-Hydroxysteroid dehydrogenases: Changing glucocorticoid action. Curr. Opin. Pharmacol. 2004, 4, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Murota, H.; Kimura, A.; Kato, A.; Ishikawa, A.; Igawa, K.; Miyoshi, E.; Katayama, I. 11β-Hydroxysteroid dehydrogenase-1 is a novel regulator of skin homeostasis and a candidate target for promoting tissue repair. PLoS ONE 2011, 6, e25039. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Walker, E.A.; Hardy, R.S.; Mayes, A.E.; Stewart, P.M. Localization, age- and site-dependent expression, and regulation of 11β-hydroxysteroid dehydrogenase type 1 in skin. J. Investig. Dermatol. 2011, 131, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, N.; Prime, S.S. Keratinocytes synthesize and activate cortisol. J. Cell Biochem. 2011, 112, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Hupe, M.; Uchida, Y.; Mauro, T.; Elias, P.M.; Holleran, W.M. Increased glucocorticoid activation during mouse skin wound healing. J. Endocrinol. 2014, 221, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Youm, J.K.; Park, K.; Uchida, Y.; Chan, A.; Mauro, T.M.; Holleran, W.M.; Elias, P.M. Local blockade of glucocorticoid activation reverses stress- and glucocorticoid-induced delays in cutaneous wound healing. Wound Repair Regen. 2013, 21, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, E.M. Neuroendocrine regulation of autoimmune/inflammatory disease. J. Endocrinol. 2001, 169, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection versus immunopathology. Allergy Asthma Clin. Immunol. 2008, 4, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S.; McEwen, B.S. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: A potential role for leukocyte trafficking. Brain Behav. Immun. 1997, 11, 286–306. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, H.J.; Lee, J.Y.; Cho, B.K.; Gallo, R.L.; Cho, D.H. Adrenocorticotropin hormone stimulates interleukin-18 expression in human HaCaT keratinocytes. J. Investig. Dermatol. 2007, 127, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, H.J.; Lee, J.H.; Lee, J.Y.; Cho, B.K.; Kang, J.S.; Kang, H.; Yang, Y.; Cho, D.H. Corticotropin-releasing hormone (CRH) downregulates interleukin-18 expression in human HaCaT keratinocytes by activation of p38 mitogen-activated protein kinase (MAPK) pathway. J. Investig. Dermatol. 2005, 124, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Lonne-Rahm, S.B.; Rickberg, H.; El-Nour, H.; Marin, P.; Azmitia, E.C.; Nordlind, K. Neuroimmune mechanisms in patients with atopic dermatitis during chronic stress. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Schut, C.; Weik, U.; Tews, N.; Gieler, U.; Deinzer, R.; Kupfer, J. Psychophysiological effects of stress management in patients with atopic dermatitis: A randomized controlled trial. Acta Derm. Venereol. 2013, 93, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Latorre, V.; Sanchis, A.; Perez, P. Epidermal inactivation of the glucocorticoid receptor triggers skin barrier defects and cutaneous inflammation. J. Investig. Dermatol. 2013, 133, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Man, M.Q.; Lin, T.K.; Santiago, J.L.; Celli, A.; Zhong, L.; Huang, Z.M.; Roelandt, T.; Hupe, M.; Sundberg, J.P.; Silva, K.A.; et al. Basis for enhanced barrier function of pigmented skin. J. Investig. Dermatol. 2014, 134, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Man, W.Y.; Lv, C.Z.; Song, S.P.; Shi, Y.J.; Elias, P.M.; Man, M.Q. Epidermal permeability barrier recovery is delayed in vitiligo-involved sites. Skin Pharmacol. Physiol. 2010, 23, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bigliardi, P.L.; Sumanovski, L.T.; Buchner, S.; Rufli, T.; Bigliardi-Qi, M. Different expression of mu-opiate receptor in chronic and acute wounds and the effect of β-endorphin on transforming growth factor β type II receptor and cytokeratin 16 expression. J. Investig. Dermatol. 2003, 120, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Eichenfield, L.F.; Tom, W.L.; Chamlin, S.L.; Feldman, S.R.; Hanifin, J.M.; Simpson, E.L.; Berger, T.G.; Bergman, J.N.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: Section 1. Diagnosis and assessment of atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.Y.; Jung, M.; Kim, D.H.; Lee, H.J.; Choi, E.H. Topical glucocorticoid or pimecrolimus treatment suppresses thymic stromal lymphopoietin-related allergic inflammatory mechanism in an oxazolone-induced atopic dermatitis murine model. Arch. Dermatol. Res. 2015, 307, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Le, T.A.; Takai, T.; Vu, A.T.; Kinoshita, H.; Ikeda, S.; Ogawa, H.; Okumura, K. Glucocorticoids inhibit double-stranded RNA-induced thymic stromal lymphopoietin release from keratinocytes in an atopic cytokine milieu more effectively than tacrolimus. Int. Arch. Allergy Immunol. 2010, 153, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, O.; Lee, B.; Vouthounis, C.; Vukelic, S.; Pastar, I.; Blumenberg, M.; Brem, H.; Tomic-Canic, M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: Inhibition of apoptosis, interferon-γ pathway, and wound healing along with promotion of terminal differentiation. J. Biol. Chem. 2007, 282, 4021–4034. [Google Scholar] [CrossRef] [PubMed]
- Mooney, E.; Rademaker, M.; Dailey, R.; Daniel, B.S.; Drummond, C.; Fischer, G.; Foster, R.; Grills, C.; Halbert, A.; Hill, S.; et al. Adverse effects of topical corticosteroids in paediatric eczema: Australasian consensus statement. Australas. J. Dermatol. 2015, 56, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Terao, M.; Itoi, S.; Murota, H.; Katayama, I. Expression profiles of cortisol-inactivating enzyme, 11β-hydroxysteroid dehydrogenase-2, in human epidermal tumors and its role in keratinocyte proliferation. Exp. Dermatol. 2013, 22, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Denda, S.; Kumamoto, J.; Denda, M. Low environmental humidity induces synthesis and release of cortisol in an epidermal organotypic culture system. Exp. Dermatol. 2013, 22, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.; Feingold, K.R.; Komuves, L.G.; Elias, P.M.; Muglia, L.J.; Majzoub, J.A.; Williams, M.L. Glucocorticoid deficiency delays stratum corneum maturation in the fetal mouse. J. Investig. Dermatol. 1998, 111, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, H.; Takigawa, M. Anxiety in allergy and atopic dermatitis. Curr. Opin. Allergy Clin. Immunol. 2006, 6, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Kojima, R.; Matsuda, A.; Nomura, I.; Matsubara, O.; Nonoyama, S.; Ohya, Y.; Saito, H.; Matsumoto, K. Salivary cortisol response to stress in young children with atopic dermatitis. Pediatr. Dermatol. 2013, 30, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Haeck, I.M.; Timmer-de Mik, L.; Lentjes, E.G.; Buskens, E.; Hijnen, D.J.; Guikers, C.; Bruijnzeel-Koomen, C.A.; de Bruin-Weller, M.S. Low basal serum cortisol in patients with severe atopic dermatitis: Potent topical corticosteroids wrongfully accused. Br. J. Dermatol. 2007, 156, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Harbuz, M.S.; Stephanou, A.; Knight, R.A.; Chover-Gonzalez, A.J.; Lightman, S.L. Action of interleukin-2 and interleukin-4 on CRF mRNA in the hypothalamus and POMC mRNA in the anterior pituitary. Brain Behav. Immun. 1992, 6, 214–222. [Google Scholar] [CrossRef]
- Watanabe, T.; Fujioka, T.; Hashimoto, M.; Nakamura, S. Stress and brain angiotensin II receptors. Crit. Rev. Neurobiol. 1998, 12, 305–317. [Google Scholar] [CrossRef] [PubMed]
- DiMicco, J.A.; Sarkar, S.; Zaretskaia, M.V.; Zaretsky, D.V. Stress-induced cardiac stimulation and fever: Common hypothalamic origins and brainstem mechanisms. Auton. Neurosci. 2006, 126–127, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic–pituitary–adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Stephens, M.A.; Wand, G. Stress and the HPA axis: Role of glucocorticoids in alcohol dependence. Alcohol Res. 2012, 34, 468–483. [Google Scholar] [PubMed]
- Fukada, M.; Kaidoh, T.; Ito, A.; Yano, T.; Hayashibara, C.; Watanabe, T. “Green odor” inhalation reduces the skin-barrier disruption induced by chronic restraint stress in rats: Physiological and histological examinations. Chem. Senses 2007, 32, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Aioi, A.; Okuda, M.; Matsui, M.; Tonogaito, H.; Hamada, K. Effect of high population density environment on skin barrier function in mice. J. Dermatol. Sci. 2001, 25, 189–197. [Google Scholar] [CrossRef]
- Choi, E.H.; Brown, B.E.; Crumrine, D.; Chang, S.; Man, M.Q.; Elias, P.M.; Feingold, K.R. Mechanisms by which psychologic stress alters cutaneous permeability barrier homeostasis and stratum corneum integrity. J. Investig. Dermatol. 2005, 124, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Demerjian, M.; Crumrine, D.; Brown, B.E.; Mauro, T.; Elias, P.M.; Feingold, K.R. Glucocorticoid blockade reverses psychological stress-induced abnormalities in epidermal structure and function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R1657–R1662. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Man, M.Q.; Santiago, J.L.; Scharschmidt, T.C.; Hupe, M.; Martin-Ezquerra, G.; Youm, J.K.; Zhai, Y.; Trullas, C.; Feingold, K.R.; et al. Paradoxical benefits of psychological stress in inflammatory dermatoses models are glucocorticoid mediated. J. Investig. Dermatol. 2014, 134, 2890–2897. [Google Scholar] [CrossRef] [PubMed]
- Buske-Kirschbaum, A.; Gierens, A.; Hollig, H.; Hellhammer, D.H. Stress-induced immunomodulation is altered in patients with atopic dermatitis. J. Neuroimmunol. 2002, 129, 161–167. [Google Scholar] [CrossRef]
- Buske-Kirschbaum, A.; Geiben, A.; Hollig, H.; Morschhauser, E.; Hellhammer, D. Altered responsiveness of the hypothalamus-pituitary-adrenal axis and the sympathetic adrenomedullary system to stress in patients with atopic dermatitis. J. Clin. Endocrinol. Metab. 2002, 87, 4245–4251. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Lee, S.H.; Oh, D.S.; Kim, Y.U. Melatonin inhibits neuronal dysfunction-associated with neuroinflammation by atopic psychological stress in NC/Nga atopic-like mouse models. J. Pineal. Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann. N. Y. Acad. Sci. 1998, 851, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Tsigos, C.; Chrousos, G.P. Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Kellner, M.; Yassouridis, A.; Manz, B.; Steiger, A.; Holsboer, F.; Wiedemann, K. Corticotropin-releasing hormone inhibits melatonin secretion in healthy volunteers—A potential link to low-melatonin syndrome in depression? Neuroendocrinology 1997, 65, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Zmijewski, M.A.; Semak, I.; Kim, T.K.; Janjetovic, Z.; Slominski, R.M.; Zmijewski, J.W. Melatonin, mitochondria, and the skin. Cell Mol. Life Sci. 2017, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.; Jarrett, P.; Broadbent, E. The effects of relaxation before or after skin damage on skin barrier recovery: A preliminary study. Psychosom. Med. 2015, 77, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Aberg, K.M.; Radek, K.A.; Choi, E.H.; Kim, D.K.; Demerjian, M.; Hupe, M.; Kerbleski, J.; Gallo, R.L.; Ganz, T.; Mauro, T.; et al. Psychological stress downregulates epidermal antimicrobial peptide expression and increases severity of cutaneous infections in mice. J. Clin. Investig. 2007, 117, 3339–3349. [Google Scholar] [CrossRef] [PubMed]



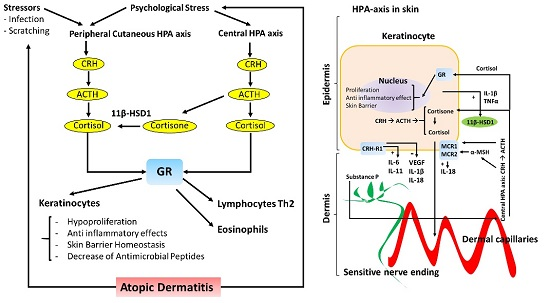{kind=link}
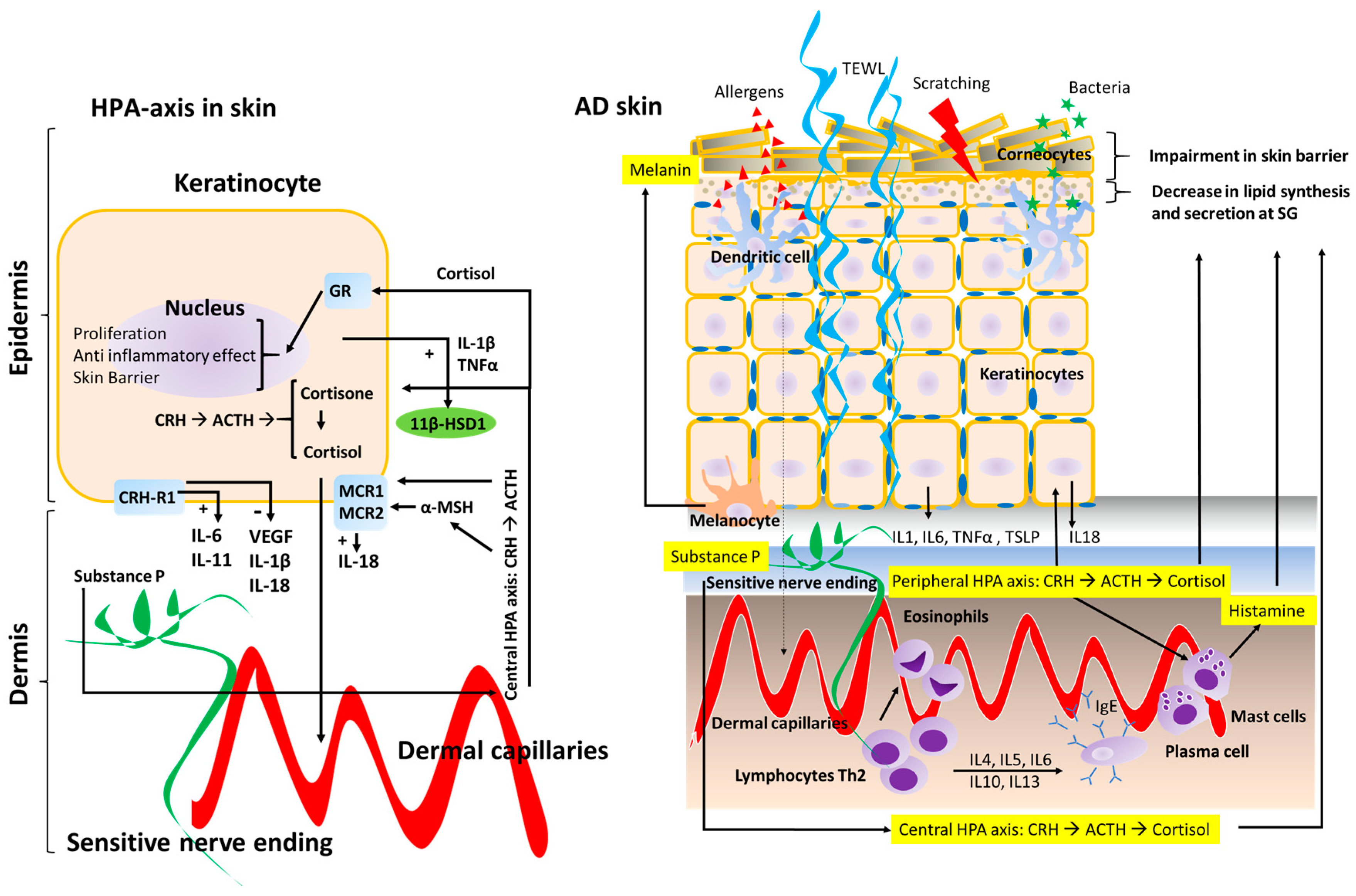{kind=link}
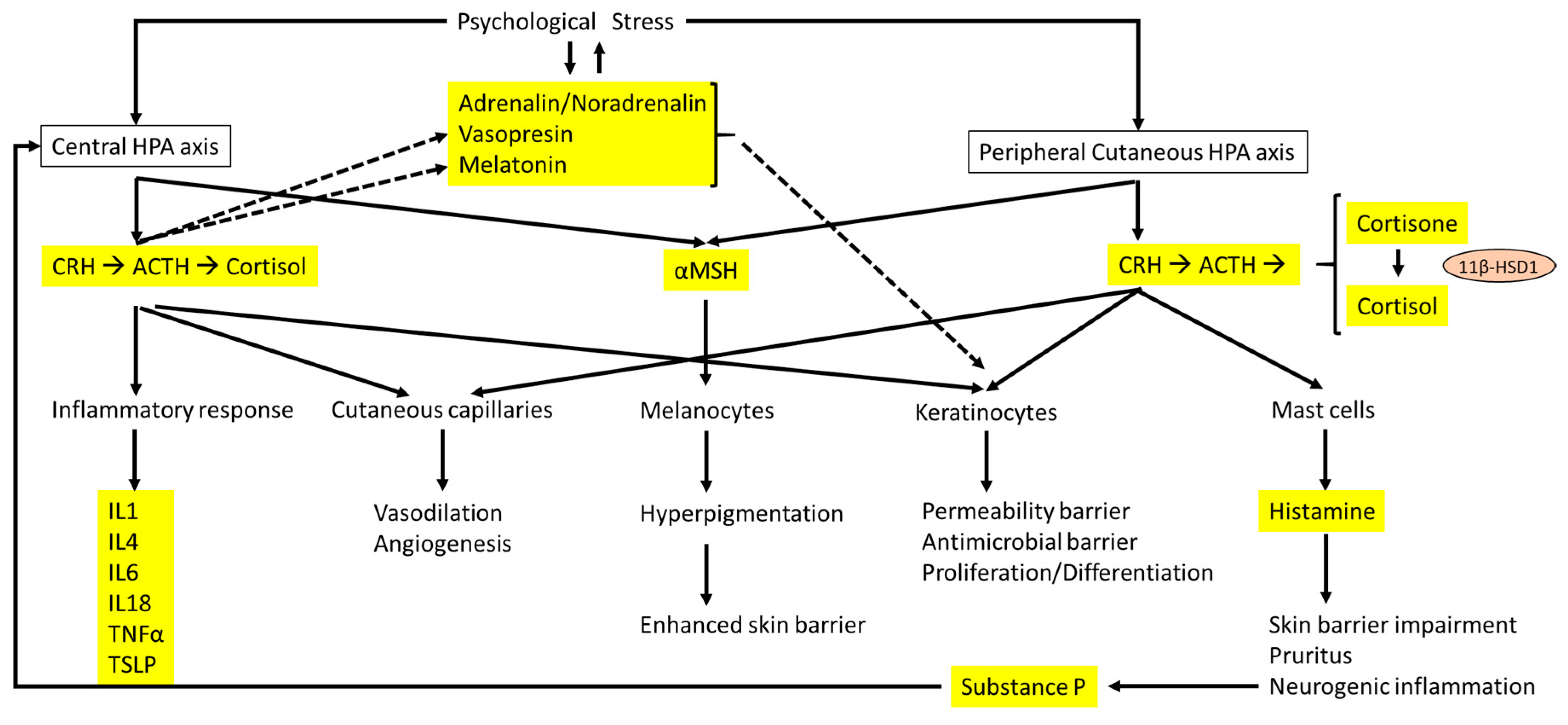{kind=link}
| Elements | Psychological Effects | Impacts on Skin Barrier |
|---|---|---|
| ACTH | Melanin production | Enhancement |
| α-MSH | Melanin production | Enhancement |
| Strong anti-inflammatory effects | Protection | |
| β-endorphin | Enhances the epidermal turnover rate | Protection |
| Cortisol | Acute PS: anti-inflammatory effects | Protection |
| Chronic PS: Downregulation of AMPs | Compromising | |
| 11β-HSD1 | Cortisol production | Delay in wound healing |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, T.-K.; Zhong, L.; Santiago, J.L. Association between Stress and the HPA Axis in the Atopic Dermatitis. Int. J. Mol. Sci. 2017, 18, 2131. https://doi.org/10.3390/ijms18102131
Lin T-K, Zhong L, Santiago JL. Association between Stress and the HPA Axis in the Atopic Dermatitis. International Journal of Molecular Sciences. 2017; 18(10):2131. https://doi.org/10.3390/ijms18102131
Chicago/Turabian StyleLin, Tzu-Kai, Lily Zhong, and Juan Luis Santiago. 2017. "Association between Stress and the HPA Axis in the Atopic Dermatitis" International Journal of Molecular Sciences 18, no. 10: 2131. https://doi.org/10.3390/ijms18102131
APA StyleLin, T.-K., Zhong, L., & Santiago, J. L. (2017). Association between Stress and the HPA Axis in the Atopic Dermatitis. International Journal of Molecular Sciences, 18(10), 2131. https://doi.org/10.3390/ijms18102131