


The Self-Disproportionation of Enantiomers (SDE) of α-Pinene via Evaporation off Silica Gel and Foam Fractionation—Validation of the Plausibility of SDE via Gas Chromatography (GC) for α-Pinene


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
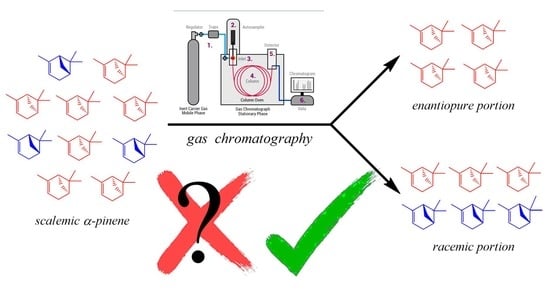
2.1. Attempted Replication of SDE via GC (SDEvGC)
2.2. NMR and IR Spectroscopic Analysis
2.3. Attempted SDE via Distillation (SDEvD)
2.4. SDE via Evaporation (SDEvE)
2.5. SDE via Adsorptive Bubble Separation (SDEvABS)
2.6. Context of the SDE
3. Conclusions
4. Experimental
4.1. Materials
4.2. GC
4.3. NMR
4.4. IR
4.5. Evaporation off Adsorbents
4.6. Foam Fractionation
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soloshonok, V.A. Remarkable amplification of the self-disproportionation of enantiomers on achiral-phase chromatography columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A.E. Self-disproportionation of enantiomers via achiral chromatography: A warning and an extra dimension in optical purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in asymmetric synthesis and problems with the accurate determination of the stereochemical outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Wzorek, A.; Kołbus, A.; Urbaniak, M.; Han, J.; Soloshonok, V.A.; Klika, K.D. Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry 2021, 13, 543. [Google Scholar] [CrossRef]
- Abás, S.; Arróniz, C.; Molins, E.; Escolano, C. Access to the enantiopure pyrrolobenzodiazepine (PBD) dilactam nucleus via self-disproportionation of enantiomers. Tetrahedron 2018, 74, 867–871. [Google Scholar] [CrossRef]
- Olbrycht, M.; Gumieniak, J.; Mruc, P.; Balawejder, M.; Piątkowski, W.; Antos, D. Separation of non-racemic mixtures of enantiomers by achiral chromatography. J. Chromatogr. A 2023, 1693, 463977. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical Purifications via Self-Disproportionation of Enantiomers by Achiral Chromatography: Case Study of a Series of α-CF3-containing Secondary Alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef]
- Carman, R.M.; Klika, K.D. The Optical Fractionation of a Partially Racemic Natural Product by Chromatography over an Achiral Substrate. Aust. J. Chem. 1991, 44, 895–896. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Soloshonok, V.A. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Berbasov, D.O. Self-Disproportionation of Enantiomers on Achiral Phase Chromatography. One More Example of Fluorine’s Magic Powers. Chim. Oggi-Chem. Today 2006, 24, 44–47. [Google Scholar]
- Soloshonok, V.A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J.S.; Singleton, D.A. Phenomenon of Optical Self-Purification of Chiral Non-Racemic Compounds. J. Am. Chem. Soc. 2007, 129, 12112–12113. [Google Scholar] [CrossRef] [PubMed]
- Ueki, H.; Yasumoto, M.; Soloshonok, V.A. Rational application of self-disproportionation of enantiomers via sublimation—A novel methodological dimension for enantiomeric purifications. Tetrahedron Asymmetry 2010, 21, 1396–1400. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Carman, R.M.; Klika, K.D. Partially Racemic Compounds as Brushtail Possum Urinary Metabolites. Aust. J. Chem. 1992, 45, 651–657. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Klika, K.D. Terminology related to the phenomenon ‘self-disproportionation of enantiomers’ (SDE). Helv. Chem. Acta 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Katagiri, T.; Yoda, C.; Furuhashi, K.; Ueki, K.; Kubota, T. Separation of an enantiomorph and its racemate by distillation: Strong chiral recognizing ability of trifluorolactates. Chem. Lett. 1996, 25, 115–116. [Google Scholar] [CrossRef]
- Koppenhoefer, B.; Trettin, U. Is it possible to affect the enantiomeric composition by a simple distillation process? Fresenius Z. Anal. Chem. 1989, 333, 750. [Google Scholar] [CrossRef]
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates, and Resolutions; Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]
- Zenkevich, I.G.; Pavlovskii, A.A. Effects of the Dynamic Modification of Stationary Phases by Sorbates in Gas Chromatography: The Possibility of Separating Enantiomers in Achiral Systems. Russ. J. Phys. Chem. A 2016, 90, 2110–2118. [Google Scholar] [CrossRef]
- Davankov, V.A. Can Enantiomers Be Separated in Achiral Chromatographic Systems. Russ. J. Phys. Chem. A 2016, 90, 2119–2121. [Google Scholar] [CrossRef]
- Baumann, A.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D.; Miller, A.K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106. [Google Scholar] [CrossRef]
- Szakács, Z.; Sánta, Z.; Lomoschitz, A.; Szántay, C., Jr. Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis. Trends Anal. Chem. 2018, 109, 180–197. [Google Scholar] [CrossRef]
- Nieminen, V.; Murzin, D.Y.; Klika, K.D. NMR and molecular modeling of the dimeric self-association of the enantiomers of 1,1′-bi-2-naphthol and 1-phenyl-2,2,2-trifluoroethanol in the solution state and their relevance to enantiomer self-disproportionation on achiral-phase chromatography (ESDAC). Org. Biomol. Chem. 2009, 7, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Storch, G.; Haas, M.; Trapp, O. Attracting Enantiomers: Chiral Analytes That Are Simultaneously Shift Reagents Allow Rapid Screening of Enantiomeric Ratios by NMR Spectroscopy. Chem. Eur. J. 2017, 23, 5414–5418. [Google Scholar] [CrossRef]
- Guetté, J.P.; Boucherot, D.; Horeau, A. Interactions diastereoisomeres d’enantiomeres en phase liquide—II. Tetrahedron Lett. 1973, 14, 465–468. [Google Scholar] [CrossRef]
- Guetté, J.P.; Horeau, A. Interactions diastereoisomeres d’antipodes en phase liquid. Tetrahedron 1974, 30, 1923–1931. [Google Scholar]
- Katagiri, T.; Takahashi, S.; Tsuboi, A.; Suzaki, M.; Uneyama, K. Discrimination of enantiomeric excess of optically active trifluorolactate by distillation: Evidence for a multi-center hydrogen bonding network in the liquid state. J. Fluor. Chem. 2010, 131, 517–520. [Google Scholar] [CrossRef]
- Katagiri, T.; Uneyama, K. Chiral recognition by multicenter single proton hydrogen bonding of trifluorolactates. Chem. Lett. 2001, 30, 1330–1331. [Google Scholar] [CrossRef]
- Wzorek, A.; Kamizela, A.; Sato, A.; Soloshonok, V.A. Self-Disproportionation of Enantiomers (SDE) via achiral gravity-driven column chromatography of N-fluoroacyl-1-phenylethylamines. J. Fluor. Chem. 2017, 196, 37–43. [Google Scholar] [CrossRef]
- Wzorek, A.; Klika, K.D.; Drabowicz, J.; Sato, A.; Aceña, J.L.; Soloshonok, V.A. The self-disproportionation of the enantiomers (SDE) of methyl n-pentyl sulfoxide via achiral, gravity-driven column chromatography: A case study. Org. Biomol. Chem. 2014, 12, 4738–4746. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Remarkable magnitude of the self-disproportionation of enantiomers (SDE) via achiral chromatography: Application to the practical-scale enantiopurification of β-amino acid esters. Amino Acids 2016, 48, 605–613. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A. Self-disproportionation of Enantiomers (SDE) of Chiral Nonracemic Amides via Achiral Chromatography. Isr. J. Chem. 2016, 56, 977–989. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A. Self-disproportionation of enantiomers via achiral gravity-driven column chromatography: A case study of N-acyl-α-phenylethylamines. J. Chromatgr. A 2016, 1467, 270–278. [Google Scholar] [CrossRef]
- Dutta, A.; Gellman, A.J. Enantiomer surface chemistry: Conglomerate versus racemate formation on surfaces. Chem. Soc. Rev. 2017, 46, 7787–7839. [Google Scholar] [CrossRef]
- Yun, Y.; Gellman, A.J. Competing Forces in Chiral Surface Chemistry: Enantiospecificity versus Enantiomer Aggregation. J. Phys. Chem. C 2016, 120, 27285–27295. [Google Scholar] [CrossRef]
- Yun, Y.; Gellman, A.J. Adsorption-induced auto-amplification of enantiomeric excess on an achiral surface. Nat. Chem. C 2015, 7, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, L.; Amabilino, D.B. Spontaneous resolution, whence and whither: From enantiomorphic solids to chiral liquid crystals, monolayers and macro- and supra-molecular polymers and assemblies. Chem. Soc. Rev. 2007, 36, 941–967. [Google Scholar] [CrossRef] [PubMed]
- Karger, B.L.; DeVivo, D.G. General Survey of Adsorptive Bubble Separation Processes. Sep. Sci. 1968, 3, 393–424. [Google Scholar] [CrossRef]
- Somasundaran, P. Foam Separation Methods. Sep. Purif. Methods 1972, 1, 117–198. [Google Scholar] [CrossRef]
- Somasundaran, P. Separation Using Foaming Techniques. Sep. Sci. 1975, 10, 93–109. [Google Scholar] [CrossRef]
- Pinfold, T.A. Adsorptive Bubble Separation Methods. Sep. Sci. 1970, 5, 379–384. [Google Scholar] [CrossRef]
- Tharapiwattananon, N.; Scamehorn, J.F.; Osuwan, S.; Harwell, J.H.; Haller, K.J. Surfactant Recovery from Water Using Foam Fractionation. Sep. Sci. Technol. 1996, 31, 1233–1258. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Zhou, E.Y.; Chen, S.; Le, K.; Tang, Y. Foam Floatation Enrichment of Enantiomers. Anal. Chem. 1994, 66, 4278–4282. [Google Scholar] [CrossRef]
- Armstrong, D.W.; Schneiderheinze, J.M.; Hwang, Y.-S. Bubble Fractionation of Enantiomers from Solution Using Molecularly Imprinted Polymers as Collectors. Anal. Chem. 1998, 70, 3717–3719. [Google Scholar] [CrossRef]
- Han, J.; Dembinski, R.; Soloshonok, V.A.; Klika, K.D. A Call for a Change in Policy Regarding the Necessity for SDE Tests to Validate the Veracity of the Outcome of Enantioselective Syntheses, the Inherent Chiral State of Natural Products, and Other Cases Involving Enantioenriched Samples. Molecules 2021, 26, 3994. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D. Recommended Tests for the Self-Disproportionation of Enantiomers (SDE) to Ensure Accurate Reporting of the Stereochemical Outcome of Enantioselective Reactions. Molecules 2021, 26, 2757. [Google Scholar] [CrossRef]
- Flynn, A.J.; Ford, A.; Maguire, A.R. Localized Partitioning of Enantiomers in Solid Samples of Sulfoxides: Importance of Sampling Method in Determination of Enantiopurity. J. Org. Chem. 2020, 85, 10216–10221. [Google Scholar] [CrossRef] [PubMed]
- Doucet, H.; Fernandez, E.; Layzell, T.P.; Brown, J.M. The Scope of Catalytic Asymmetric Hydroboration/Oxidation with Rhodium Complexes of 1,1′-(2-Diarylphosphino-1-naphthyl)isoquinolines. Chem. Eur. J. 1999, 5, 1320–1330. [Google Scholar] [CrossRef]
- Dasarathy, D.; Ito, Y. Foam separation of Rhodamine-G and Evans Blue using a simple separatory bottle system. J. Chromatogr. A 2017, 1517, 215–218. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wzorek, A.; Soloshonok, V.A.; Klika, K.D. The Self-Disproportionation of Enantiomers (SDE) of α-Pinene via Evaporation off Silica Gel and Foam Fractionation—Validation of the Plausibility of SDE via Gas Chromatography (GC) for α-Pinene. Separations 2023, 10, 382. https://doi.org/10.3390/separations10070382
Wzorek A, Soloshonok VA, Klika KD. The Self-Disproportionation of Enantiomers (SDE) of α-Pinene via Evaporation off Silica Gel and Foam Fractionation—Validation of the Plausibility of SDE via Gas Chromatography (GC) for α-Pinene. Separations. 2023; 10(7):382. https://doi.org/10.3390/separations10070382
Chicago/Turabian StyleWzorek, Alicja, Vadim A. Soloshonok, and Karel D. Klika. 2023. "The Self-Disproportionation of Enantiomers (SDE) of α-Pinene via Evaporation off Silica Gel and Foam Fractionation—Validation of the Plausibility of SDE via Gas Chromatography (GC) for α-Pinene" Separations 10, no. 7: 382. https://doi.org/10.3390/separations10070382
APA StyleWzorek, A., Soloshonok, V. A., & Klika, K. D. (2023). The Self-Disproportionation of Enantiomers (SDE) of α-Pinene via Evaporation off Silica Gel and Foam Fractionation—Validation of the Plausibility of SDE via Gas Chromatography (GC) for α-Pinene. Separations, 10(7), 382. https://doi.org/10.3390/separations10070382