
Screening and Experimental Validation for Selection of Open Metal Sites Metal-Organic Framework (M-CPO-27, M = Co, Mg, Ni and Zn) to Capture CO2

 and
and
Abstract
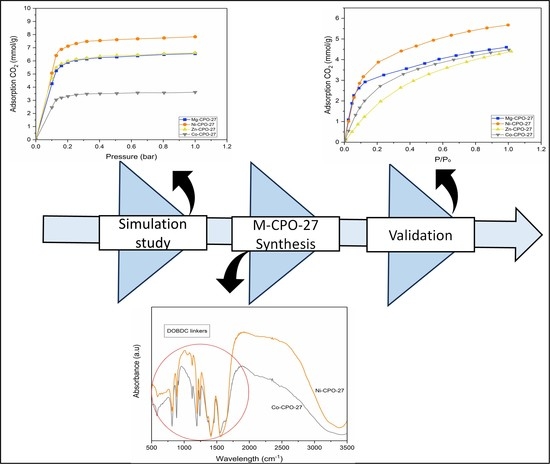
1. Introduction
2. Materials and Methods
2.1. Screening of M-CPO-27
2.2. Synthesis and Characterization of M-CPO-27
3. Results
3.1. Screening of M-CPO-27 (M = Mg, Ni, Co, and Zn)
3.2. Physical Properties of M-CPO-27 (M = Co, Mg, Ni, and Zn)
3.2.1. X-ray Diffraction
3.2.2. Fourier Transform IR (FTIR) Spectroscopy
3.2.3. Adsorption CO2 of Synthesis M-CPO-27 (M = Co, Mg, Ni and Zn)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, S.; Zhu, M.; Tang, Y.; Fu, Y.; Li, W.; Xiao, B. Molecular simulation and experimental investigation of CO2 capture in a polymetallic cation-exchanged 13X zeolite. J. Mater. Chem. A 2018, 6, 19570–19583. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, M.; Majeed, Z.; Xie, Y.; Zheng, K.; Luo, Z.; Li, C.; Zhao, C.J.S. Application of Hdrogen-Bonded Organic Frameworks in Environmental Remediation: Recent Advances and Future Trends. Separations 2023, 10, 196. [Google Scholar] [CrossRef]
- Andirova, D.; Cogswell, C.F.; Lei, Y.; Choi, S. Effect of the structural constituents of metal organic frameworks on carbon dioxide capture. Microporous Mesoporous Mater. 2016, 219, 276–305. [Google Scholar] [CrossRef]
- Mercado, R.; Vlaisavljevich, B.; Lin, L.-C.; Lee, K.; Lee, L.; Mason, J.A.; Xiao, D.J.; Gonzalez, M.I.; Kapelewski, M.T.; Neaton, B.; et al. Force Field Development from Periodic Density Functional Theory Calculations for Gas Separation Applications Using Metal–Organic Frameworks. J. Phys. Chem. 2016, 23, 12590–12604. [Google Scholar] [CrossRef]
- Hassan, R.Z. Adsorption of Gases (CO2, CH4) Using Novel Porous Materials (MOFs). Ph.D. Thesis, Curtin University, Bentley, WA, Australia, 2016. [Google Scholar]
- Xiang, S.; He, Y.; Zhang, Z.; Wu, H.; Zhou, W.; Krishna, R.; Chen, B. Microporous metal-organic framework with potential for carbon dioxide capture at ambient conditions. Nat. Commun. 2012, 3, 954. [Google Scholar] [CrossRef]
- Bolotov, V.A.; Kovalenko, K.A.; Samsonenko, D.G.; Han, X.; Zhang, X.; Smith, G.L.; McCormick, L.J.; Teat, S.J.; Yang, S.; Lennox, M.J.; et al. Enhancement of CO2 Uptake and Selectivity in a Metal-Organic Framework by the Incorporation of Thiophene Functionality. Inorg. Chem. 2018, 57, 5074–5082. [Google Scholar] [CrossRef] [PubMed]
- Trickett, C.A.; Helal, A.; Al-Maythalony, B.A.; Yamani, Z.H.; Cordova, K.E.; Yaghi, O.M. The chemistry of metal–organic frameworks for CO2 capture, regeneration and conversion. Nat. Rev. Mater. 2017, 2, 17045. [Google Scholar] [CrossRef]
- Gutiérrez-Serpa, A.; Fernandez, I.P.; Pasan, J.; Pino, V.J. Metal-Organic Frameworks as Key Materials for soild-Phase Microextraction Devices-A Review. Separations 2019, 6, 47. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. A review on solid adsorbents for carbon dioxide capture. J. Ind. Eng. Chem. 2015, 23, 1–11. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Assiri, M.A.; Al-Sehemi, A.G.; Sagir, M.; Kareem, F.A.A.; Elkhalifah, A.E.; Mukhtar, A.; Gonfa, G. Synthesis, and characterization of metal-organic frameworks-177 for static and dynamic adsorption behavior of CO2 and CH4. Microporous Mesoporous Mater. 2019, 288, 109569. [Google Scholar] [CrossRef]
- Mahdipoor, H.R.; Halladj, R.; Babakhani, E.G.; Amjad-Iranagh, S.; Ahari, J.S. Adsorption of CO2, N2 and CH4 on a Fe-based metal organic framework, MIL-101 (Fe)-NH2. Colloids Surf. A Physicochem. Eng. Asp. 2021, 619, 126554. [Google Scholar] [CrossRef]
- Ismail, M.; Bustam, M.A.; Kari, N.E.F. Screening of Gallate-Based Metal-Organic Frameworks for Single-Component CO2 and CH4 Gas. In E3S Web of Conferences; EDP Sciences: Les Ulis, France, 2021; p. 02005. [Google Scholar]
- Krishna, R. Screening metal–organic frameworks for mixture separations in fixed-bed adsorbers using a combined selectivity/capacity metric. RSC Adv. 2017, 7, 35724–35737. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Assiri, M.A.; Al-Sehemi, A.G.; Gonfa, G.; Mukhtar, A.; Kareem, F.A.A.; Ayoub, M.; Saqib, S.; Mellon, N.B. Synthesis and characterization of mesoporous MOF UMCM-1 for CO2/CH4 adsorption; an experimental, isotherm modeling and thermodynamic study. Microporous Mesoporous Mater. 2020, 294, 109844. [Google Scholar] [CrossRef]
- Sunder, N.; Fong, Y.-Y.; Bustam, M.A.; Lau, W.-J. Study on the Performance of Cellulose Triacetate Hollow Fiber Mixed Matrix Membrane Incorporated with Amine-Functionalized NH2-MIL-125(Ti) for CO2 and CH4 Separation. Separations 2023, 10, 41. [Google Scholar] [CrossRef]
- Alonso, G.; Bahamon, D.; Keshavarz, F.; Giménez, X.; Gamallo, P.; Sayós, R. Density Functional Therory-Based Adsorption Isotherm for Pure and Flue Gas Mixtures om Mg-MOF-74. Application in CO2 Capture Swing Adsorption Processes. J. Phys. Chem. C 2018, 122, 3945. [Google Scholar] [CrossRef]
- de Oliveira, A.; de Lima, G.F.; De Abreu, H.A. Structural and electronic properties of M-MOF-74 (M=Mg, Co or Mn). Chem. Phys. Lett. 2018, 691, 283. [Google Scholar]
- Maurin, G. Role of molecular simulations in the structure exoplaration of Metal Organic Frameworks Illustrations through recent advances in the field. CR Chim. 2016, 19, 207. [Google Scholar] [CrossRef]
- Wilmer, C.E.; Leaf, M.; Lee, C.Y.; Farha, O.; Hauser, B.; Hupp, J.T.; Snurr, R.Q. Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 2011, 171, 83–89. [Google Scholar]
- Bao, Z.; Yu, L.; Ren, Q.; Lu, X.; Deng, S. Adsorption of CO2 and CH4 on a magnesium-based metal organic framework. J. Colloid. Interface. Sci. 2011, 353, 549–556. [Google Scholar] [CrossRef]
- Bautista, P.R.; Mancera, I.T.; Pasan, J.; Pino, V. Metal-Organic Frameworks in Green Analytical Chemistry. Separations 2019, 6, 33. [Google Scholar] [CrossRef]
- Caskey, S.R.; Adam, J. Dramatic Tuning of Carbon Dioxide Uptake via Metal Substitution in a Coordination Polymer with Cylindrical Pores. J. Am. Chem. Soc. 2008, 130, 10870–10871. [Google Scholar] [CrossRef] [PubMed]
- Becker, T.M.; Lin, L.C.; Dubbeldam, D.; Vlugt, T.J. Polarizable Force Field for CO2 in M-MOF-74 Derived from Quantum Mechanics. J. Phys Chem. C 2018, 122, 24488. [Google Scholar] [CrossRef]
- Wang, X.; Ramirez-Hinestrosa, S.; Dobnikar, J.; Frenkel, D. The Lennard-Jones potential: When (not) to use it. Phys. Chem. Chem. Phys. 2020, 22, 10624–10633. [Google Scholar] [CrossRef] [PubMed]
- Konstantakou, M.; Gotzias, A.; Kainourgiakis, M.; Stubos, A.K.; Steriotis, T.A. GCMC Simulation of Gas Adsorption in Carbon Pore Structure. In Applications of Monte Carlo Method in Science and Engineering; Intechopen: London, UK, 2011. [Google Scholar]
- Fairen, D.F.; Seaton, N.A.; Düren, T. Unusual Adsorption Behavior on Metal-Organic Frameworks. Langmuir 2010, 26, 14694–14699. [Google Scholar] [CrossRef] [PubMed]
- Yazaydın, A.O.; Snurr, R.Q.; Park, T.H.; Koh, K.; Liu, J.; LeVan, M.D.; Benin, A.I.; Jakubczak, P.; Lanuza, M.; Galloway, D.B.; et al. Screening of Metal-Organic Frameworks for Carbon Dioxide from Fuel Gas Using a Combined Experimental and Modeling Approcah. J. Am. Chem. Soc. 2009, 131, 18189–18199. [Google Scholar] [CrossRef] [PubMed]
- Remy, T.P.; Sunil, A.; Perre, A.; Valvekens, P.; Vos, P.; Baron, G.; Joeri, J. Selective Dynamic CO2 Seperation on Mg-MOF-74 at Low Pressures: A Detailed Comparison with 13X. J. Phys. Chem. C 2013, 117, 9301–9310. [Google Scholar] [CrossRef]
- Xin, J.H.; He, P.; Li, H.; Wang, X. Understanding the Adsorption of C2H2, CO2, and CH4 in Isostructural Metal-Organic Frameworks with Cooridinatively Unsaturated Metal Sites. J. Phys. Chem. C 2013, 117, 2824–2834. [Google Scholar]
- Keskin, S.; Altintas, C. Computational screening of MOFs for C2H6/C2H4 and C2H6/CH4 separations. Chem Eng. Sci. 2016, 139, 49–60. [Google Scholar]
- Tim, M.B.; Heinen, J.; Dubbeldam, D.; Lin, L.; Vlugt, T.H. Polarized Force Field for CO2 and CH4 Adsorption in M-MOF-74. J. Phys. Chem. C 2017, 121, 4659–4673. [Google Scholar]
- Jung, D.H.; Kim, D.; Lee, T.B.; Choi, S.B.; Yoon, J.H.; Kim, J.; Choi, K.; Choi, S.-H. Grand Canonical Monte Carlo Simulation Study on the Catenation Effect on Hydrogen Adsorption onto the Interpenetrating Metal−Organic Frameworks. J. Phys. Chem. B 2006, 100, 22987–22990. [Google Scholar] [CrossRef]
- García, M.D.; Sanchez, M. Synthesis and characterization of a new Cd-based metal-organic framework isostructural with MOF-74/CPO-27 material. Micropor. Mesopor. Mat. 2014, 190, 248–254. [Google Scholar] [CrossRef]
- Biovia, D.S. Modules Tutorials Materials Studio; Dassault Systèmes: San Diego, CA, USA, 2017; Volume 470, pp. 226–233. [Google Scholar]
- Agrawal, A.; Agrawal, M.; Suh, D.; Fei, S.; Alizadeh, A.; Ma, Y.; Matsuda, R.; Hsu, W.; Daiguji, H. Augmenting the Carbon Dioxide Uptake and Selectivity of Metal-Organic Frameworks by Metal Substitution: Molecular Simulations of LMOF-202. ACS Omega 2020, 5, 17193–17198. [Google Scholar] [CrossRef]
- Sladekova, K.; Campbell, C.; Grant, C.; Fletcher, A.J.; Gomes, J.R.; Jorge, M. The Effect of Atomic Point Charges on Adsorption Isotherms of CO2 and Water in Metal Organic Frameworks. Adsorption 2020, 26, 663–685. [Google Scholar] [CrossRef]
- Deng, J.; Zhao, G.; Zhang, L.; Ma, H.; Rong, Y. CO2 Adsorption and Separation Properties of M-MOF-74 Materials Determined by Molecular Simulation. Capillarity 2023, 6, 13–18. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, J.; Ma, X.; Liu, J.; Lin, Y. Mechanism of CO2 adsorption on Mg/DOBDC with elevated CO2 loading. Fuel 2016, 181, 340–346. [Google Scholar] [CrossRef]
- Queen, W.L.; Hudson, M.R.; Bloch, E.D.; Mason, J.A.; Gonzalez, M.I.; Lee, J.S.; Gygi, D.; Howe, J.D.; Lee, K.; Darwish, T.A.; et al. Comprehensive study of carbon dioxide adsorption in the metal–organic frameworks M2(dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci. 2014, 5, 4569. [Google Scholar] [CrossRef]
- Canivet, J.; Fateeva, A.; Coasne, B.; Farrusseng, D. Water adsorption in MOFs: Fundamentals and applications. Chem. Soc. Rev. 2014, 43, 5594. [Google Scholar] [CrossRef]
- Liu, J.; Thallapally, P.K.; McGrail, B.P. Selective CO2 Capture from Flue Gas Using Metal Organic Frameworks—A Fixed Bed Study. J. Phys. Chem. C 2012, 116, 9575–9581. [Google Scholar] [CrossRef]
- Gonzalez, M.I.; Mason, J.A.; Bloch, E.D.; Teat, S.J.; Gagnon, K.J.; Morrison, G.Y.; Queen, W.L.; Long, J.R. Structural characterization of framework–gas interactions in the metal–organic framework Co2(dobdc) by in situ single-crystal X.-ray diffraction. R. Soc. Chem. 2017, 9, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Botas, J.A.; Calleja, G.; Sánchez-Sánchez, M.; Orcajo, M.G. Effect of Zn/Co ratio in MOF-74 type materials containing exposed metal sites on their hydrogen adsorption behaviour and on their band gap energy. Int. J. Hydrogen Energy 2011, 36, 10834–10844. [Google Scholar] [CrossRef]
- Naeem, R. Lennard Jones Potential; UC Davis Chem Wiki: Davis, CA, USA, 2015. [Google Scholar]
- Zhao, Z.; Zuhra, Z.; Qin, L.; Zhou, Y.; Zhang, L.; Tang, F.; Mu, C. Confinement of microporous MOF-74(Ni) within mesoporous γ-Al2O3 beads for excellent ultra-deep and selective adsorptive desulfurization performance. Fuel Process. Technol. 2018, 176, 276–282. [Google Scholar] [CrossRef]
- Thakkar, H.; Eastman, S.; Al-Naddaf, Q.; Rownaghi, A.A.; Rezaei, F. 3D-Printed Metal–Organic Framework Monoliths for Gas Adsorption Processes. ACS Appl. Mater. Interfaces 2017, 9, 35908–35916. [Google Scholar] [CrossRef]
- Lee, K.; Howe, J.D.; Lin, L.C.; Smit, B.; Neaton, J.B. Small-Molecule Adsorption in Open-Site Metal−Organic Frameworks: A Systematic Density Functional Theory Study for Rational Design Chemistry of Materials. Chem. Mater. 2015, 3, 668–678. [Google Scholar] [CrossRef]
- Koh, H.S.; Rana, M.K.; Wong-Foy, A.G.; Siegel, D.J. Predicting Methane Storage in Open-Metal-Site Metal–Organic Frameworks. J. Phys. Chem. C 2015, 119, 13451–13458. [Google Scholar] [CrossRef]
- Concepcih, P.; Mifsud, A. Preparation and Characterization of Mg-Containing AFI and Chabazite-Type Material. Zeolites 1996, 16, 56–64. [Google Scholar] [CrossRef]
- Valenzano, L.; Civalleri, B.; Chavan, S.; Palomino, G.T.; Areán, C.O.; Bordiga, S. Computational and Experimental Studies on the Adsorption of CO, N2, and CO2 on Mg-MOF-74. J. Phys. Chem. C 2010, 114, 11185–11191. [Google Scholar] [CrossRef]
- Ullah, S.; Bustam, M.A.; Assiri, M.A.; Al-Sehemi, A.G.; Sagir, M.; Kareem, F.A.A.; Elkhalifah, E.I.; Mukhtar, A.; Gonfa, G. Influence of post-synthetic graphene oxide (GO) functionalization on the selective CO2/CH4 adsorption behavior of MOF-200 at different temperatures; an experimental and adsorption isotherms study. Microporous Mesoporous Mater. 2019, 296, 110002. [Google Scholar] [CrossRef]
- Kizzie, A.C.; Wong-Foy, A.G.; Matzger, A.J. Effect of Humidity on the Performance of Microporous Coordination Polymers as Adsorbents for CO2 Capture. Langmuir 2011, 27, 6368–6373. [Google Scholar] [CrossRef]
- Lopez, M.G.; Canepa, P.; Thonhauser, T. NMR study of small molecule adsorption in MOF-74-Mg. J. Chem. Phys. 2013, 138, 154704. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metal | Lattice Constant (Å), a = b | Lattice Constant (Å), c | Bond Length (M-O) (Å) |
|---|---|---|---|
| Mg-CPO-27 | 25.94 | 6.72 | 2.61 |
| Ni-CPO-27 | 25.86 | 6.71 | 2.04 |
| Co-CPO | 26.13 | 6.72 | 2.71 |
| Zn-CPO | 26.18 | 6.65 | 2.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kari, N.E.F.; Ismail, M.; Ahmad, A.; Kamal, K.; Chew, T.L.; Bustam, M.A. Screening and Experimental Validation for Selection of Open Metal Sites Metal-Organic Framework (M-CPO-27, M = Co, Mg, Ni and Zn) to Capture CO2. Separations 2023, 10, 434. https://doi.org/10.3390/separations10080434
Kari NEF, Ismail M, Ahmad A, Kamal K, Chew TL, Bustam MA. Screening and Experimental Validation for Selection of Open Metal Sites Metal-Organic Framework (M-CPO-27, M = Co, Mg, Ni and Zn) to Capture CO2. Separations. 2023; 10(8):434. https://doi.org/10.3390/separations10080434
Chicago/Turabian StyleKari, Nor Ernie Fatriyah, Marhaina Ismail, Aqeel Ahmad, Khaliesah Kamal, Thiam Leng Chew, and Mohamad Azmi Bustam. 2023. "Screening and Experimental Validation for Selection of Open Metal Sites Metal-Organic Framework (M-CPO-27, M = Co, Mg, Ni and Zn) to Capture CO2" Separations 10, no. 8: 434. https://doi.org/10.3390/separations10080434
APA StyleKari, N. E. F., Ismail, M., Ahmad, A., Kamal, K., Chew, T. L., & Bustam, M. A. (2023). Screening and Experimental Validation for Selection of Open Metal Sites Metal-Organic Framework (M-CPO-27, M = Co, Mg, Ni and Zn) to Capture CO2. Separations, 10(8), 434. https://doi.org/10.3390/separations10080434