1. Introduction
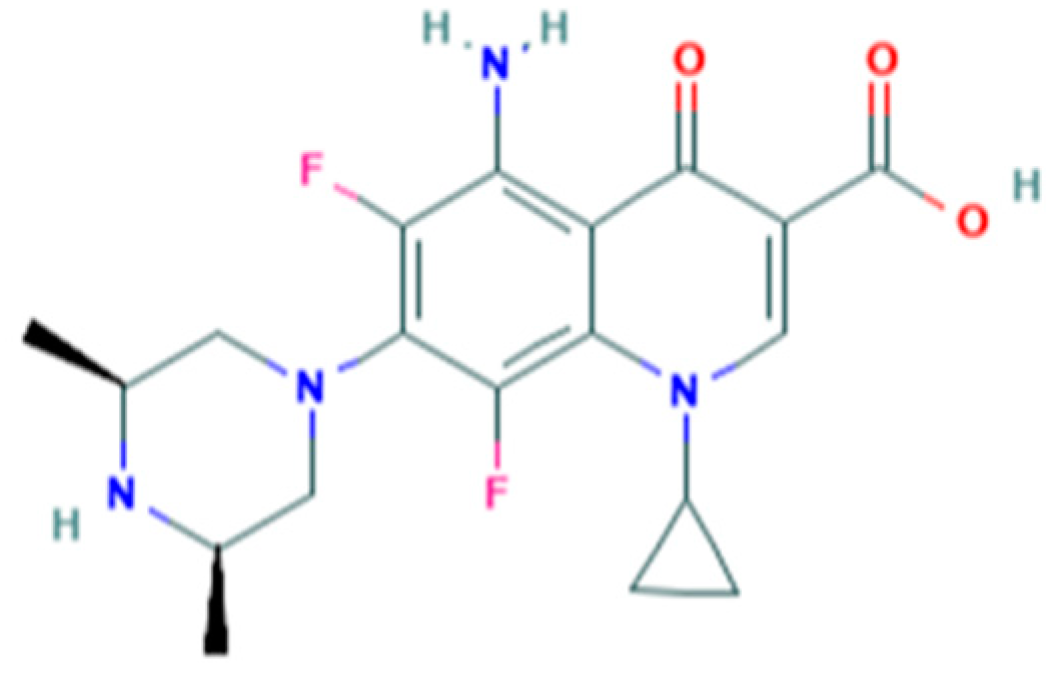
Sparfloxacin (SPAR), 5-amino-1-cyclopropyl-7-[(3
R,5
S)-3,5-dimethylpiperazin-1-yl]-6,8-difluoro-4-oxoquinoline-3-carboxylic acid (IUPAC), is a synthetic bactericidal antibiotic belonging to the third-generation fluoroquinolones (
Figure 1) [
1].
SPAR is a difluorinated quinolone active against a wide range of common and atypical Gram-negative and Gram-positive pathogenic microorganisms, and its spectrum embraces anaerobes,
Chlamydia spp.,
Mycoplasma spp. and
Mycobacterium spp. [
2,
3]. Methicillin-resistant
Staphylococcus aureus (MRSA) is also susceptible to SPAR [
4]. The molecular target of SPAR is considered the inhibition of DNA gyrase or topoisomerase II and topoisomerase IV. Both are essential enzymes that control DNA topology and assist in DNA replication, repair, deactivation and transcription [
2,
5,
6].
This fluoroquinolone exhibits very good bioavailability following oral administration, with values of about 61.7 to 80% for broiler chicken [
7], rats [
8,
9] and humans [
10,
11]. SPAR binds weakly to plasma protein (mainly albumin) (37–46%) and exhibits excellent tissue distribution, with the exception of the central nervous system and adipose tissue, and a long elimination half-life. Furthermore, it shows an effective penetration into extracellular fluids in all species tested, including humans. Concentrations of the drug in most tissues are similar to, or higher than, concomitant plasma concentrations. SPAR is distributed slightly into the cerebrospinal fluid, and only one inactive metabolite, the acylglucuronide of SPAR, has been found [
4,
8,
12]. SPAR and its metabolite are excreted by renal and extrarenal processes including biliary excretion and probably transintestinal secretion [
10].
According to a literature survey, there are few methodologies for determining SPAR in various liquids, plasma and dosage forms in humans [
13,
14,
15,
16]. In addition, there are few reports regarding pharmacokinetic analysis of SPAR in animals [
2,
3,
8,
17,
18], and none of them have been carried out in sheep, although an HPLC method using it as an internal standard was validated in sheep plasma [
19]. The numerous microbiological and pharmacokinetic advantages of SPAR compared to other members of its group could make it an excellent candidate to treat infections caused by susceptible pathogenic species in sheep.
SPAR has been temporarily used in human medicine with good results; however, its application in human health is currently banned in Europe due to phototoxicity problems, making its use in veterinary medicine a valuable market. Following the recommendations of the regulatory agencies, its exclusive use in veterinary medicine would contribute to reducing the risk of the development of bacterial resistance.
Thus, the objective of this study was to develop and validate an analytical HPLC method to quantify SPAR in sheep plasma, following the recommendations set out in the Guideline on Bioanalytical Method Validation of the European Union (EMEA/CHMP/EWP/192217/2009) [
20].
2. Materials and Methods
2.1. Chemicals and Reagents
SPAR was purchased from Sigma-Aldrich (Schenelldorf, Germany) with a purity of 98%. The internal standard (IS) used was genabilic acid (purity 98.5%, Sigma-Aldrich, Schenelldof, Germany). All reagents and solvents used were of HPLC grade: methanol (LiChrosolv. Merck, Madrid, Spain), acetonitrile (HiPerSolv CHROMANORM, Radnor, PA, USA), sodium hydroxide 1N (Panreac. Quimica S.A., Barcelona, Spain), monopotassium phosphate (AnalaR NORMAPUR, Radnor, PA, USA) and acetic acid 10% (Chromanorm VWR Chemicals, Radnor, PA, USA). HPLC-grade water was used for the extraction and quantification procedures. This was produced in our laboratory by using an Ultramatic system by Wasserlab. For the solid-phase extraction process, a Manifold with 20 positions (Waters Corporation, Milford, MA, USA) and Oasis HLB 1cc 30 mg cartridge (Waters Corporation, Milford, MA, USA) were employed.
2.2. Animals and Experimental Procedures
Six non-lactating healthy female Spanish Churra sheep (4–5 years old) weighing 70 ± 7 kg were used. The study was carried out in the experimental farm of the Veterinary Faculty of the University of Leon. Animals’ health was closely monitored before and throughout the experimental period by a veterinarian. Sheep were allowed to acclimatize to their environment before the experiment was started, and they were maintained in an adequately ventilated building. They were provided a diet of hay and pelleted feed concentrate twice a day with water and saltlick ad libitum. The study was approved by the Ethics Committee of the University of León (OEBA-ULE-004-2019).
Blood samples were collected from the jugular veins into heparinized tubes (Vacutainer, BD, Plymouth, UK). Samples were centrifuged at 1500 rpm for 20 min, and plasma was stored at −20 °C until analysis.
2.3. Preparations of Stock, Calibration and Quality Working Solutions
Stock solutions, calibration working solutions and quality control working solutions (QC) were prepared daily.
Stock solution (1 mg/mL) was prepared in HPLC-grade water, also adding 100 µL NaOH 1N. IS (1 mg/mL) was also dissolved in HPLC-grade water and 100 µL NaOH 1N.
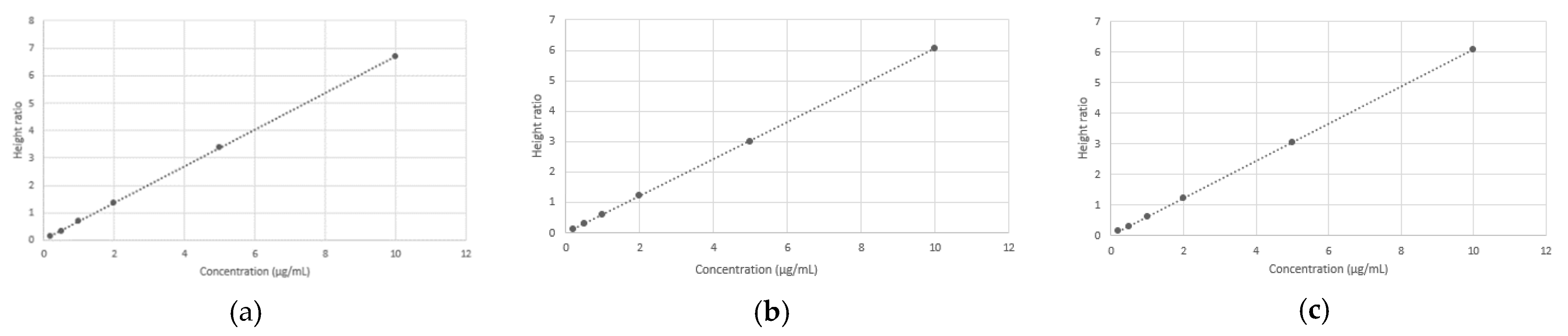
Calibration working solutions containing both SPAR (2, 5, 10, 20, 50 and 100 µg/mL) and IS (20 µg/mL) were then prepared by diluting an appropriate volume of the stock solution in 10 mL of HPLC-grade water.
Quality control working solutions (QC) were obtained by adding an aliquot of each stock solution to obtain final concentrations of 0.2 µg/mL (QC1: LLOQ, lower limit of quantitation), 0.6 µg/mL (QC2: LOW, three times the LLOQ), 3 µg/mL (QC3: MED, between 30 and 50% of the calibration curve range) and 8 µg/mL (QC4: HIGH, 75% of the upper calibration curve range).
2.4. Preparation of Analysis Samples
Blank samples: biological matrix without SPAR and IS (1 mL).
Zero samples: biological matrix (0.9 mL) with 0.1 mL IS (20 µg/mL).
Calibration standards: 0.9 mL plasma was spiked with 100 µL of each calibration working solution to obtain calibration curves. Thus, concentrations of calibration samples were 0.2, 0.5, 1, 2, 5 and 10 µg/mL for SPAR and 2 µg/mL for IS.
Quality control samples were also prepared in plasma (0.9 mL) at concentrations of 0.2, 0.6, 3 and 8 µg/mL for SPAR (0.1 mL) and 2 µg/mL for IS (0.1 mL).
All samples were fully thawed at room temperature.
2.5. Extraction Method
Plasma samples were deproteinized with 1 mL 10% acetic acid, shaken for 1 min and centrifuged at 1620 g for 10 min. The supernatant was then transferred into the SPE cartridge. Cartridges were previously conditioned with 1 mL methanol and then with 1 mL HPLC-grade water. After washing twice with 1 mL HPLC-grade water, the cartridge was properly dried and eluted with 1 mL mobile phase. Finally, 20 µL of eluate was injected into the HPLC system. All procedures were performed at room temperature.
2.6. HPLC System and Conditions
The samples were analyzed by reverse-phase high-performance liquid chromatography (HPLC) in a Waters Alliance e2695 HPLC system equipped with a photodiode array detector (model 2998) (Waters Corporation, Milford, MA, USA).
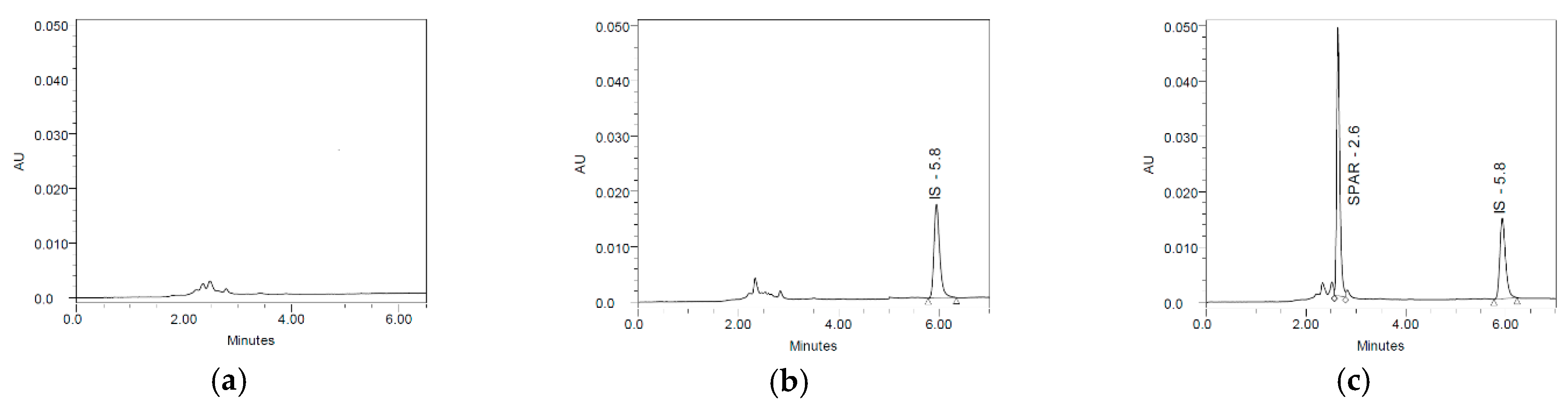
Chromatographic separation was performed at room temperature (21 °C) with an Xbridge BEH C18 column (5 µm, 4.6 × 250 mm) (Waters Corporation, Milford, MA, USA). The mobile phase consisted of a mixture of acetonitrile and monopotassium phosphate buffer (1.36 g/L) 49:51 (v/v). The flow rate was 1 mL/min, and the wavelength was set at 236 and 298 nm. The injection volume was 10 µL. Genabilic acid was used as internal standard. The limit of detection (LOD) was estimated by integrating the baseline noise of the HPLC system in the height covering the mean retention time of SPAR in six plasma samples spiked with the IS and defined as the mean baseline noise/IS peak height ratio plus three standard derivations.
Photodiode array detectors provide three-dimensional information that allows an accurate assessment of peak identification, purity and quantitation in a single run. We used our spectral library to establish peaks’ homogeneity and identity. The study was conducted under the Good Laboratory Practice (GLP) regulations at our GLP-compliant laboratory LAFARLE (University of Leon, Leon, Spain), certified by the Spanish Agency of Medicines and Medical Devices (AEMPS) [
21].
2.7. Method Validation Procedure
The validation of the method was carried out by using the following parameters: selectivity, carry-over, lower limit of quantification, calibration curve, precision, accuracy and stability, in accordance with the Guideline on Bioanalytical Method Validation of the European Medicines Agency (EMA/CHMP/EWP/192217/2009) [
20].
2.8. Method Application
SPAR concentrations were measured in plasma samples obtained from 2 sheep to explore the applicability of the method in clinical practice. SPAR was administered intravenously at a dose of 5 mg/kg. Blood samples were alternately collected from the jugular veins into heparinized tubes (Vacutainer, BD, Plymouth, UK) at 15 and 30 min and 2, 4, 8, 12 and 24 h. Samples were centrifuged for 20 min at 406 g, and then plasma was stored at −20 °C until analysis. Before analysis, plasma samples (1 mL) were spiked with 20 µL IS.
Animal procedures and management protocols were authorized in advance by both the Ethics Committee of the University of Leon and the regional authorities (OEBA-ULE-004-2019). No invasive procedure was involved beyond blood sampling.
2.9. Data Analysis
For data acquisition and processing, HPLC Empower 3 (Waters Corporation, USA) software was employed. A descriptive statistical analysis (mean and standard deviation) was carried out on data values using SPSS Statistical Software V. 26.0 (IBM Corporation, Armonk, NY, USA).
4. Conclusions
A new, sensitive, simple and fast HPLC method for sparfloxacin quantification in sheep plasma was successfully developed. It was validated according to the Guideline on Bioanalytical Method Validation of the European Union (EMEA/CHMP/EWP/192217/2009) [
20]. This method can be used to perform pharmacokinetic studies with sparfloxacin in this animal species.
Selectivity, carry-over, lower limit of quantification (LLOQ), calibration range, accuracy, precision and stability always fulfilled the criteria established. Its low cost, the low volume of sample needed and the simple instrumentation required should also be considered as advantages of this method. Moreover, the chromatographic runtime is about 6 min, which allows a large number of samples to be processed in a short time.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}