
Prediction of the Iron–Sulfur Binding Sites in Proteins Using the Highly Accurate Three-Dimensional Models Calculated by AlphaFold and RoseTTAFold


Abstract
:1. Introduction
1.1. AlphaFold and RoseTTAFold
1.2. Does a Given Protein Bind an Fe–S Cluster?
1.3. Biochemical and Structural Characterization of the Fe–S-Binding Protein
2. Results and Discussion
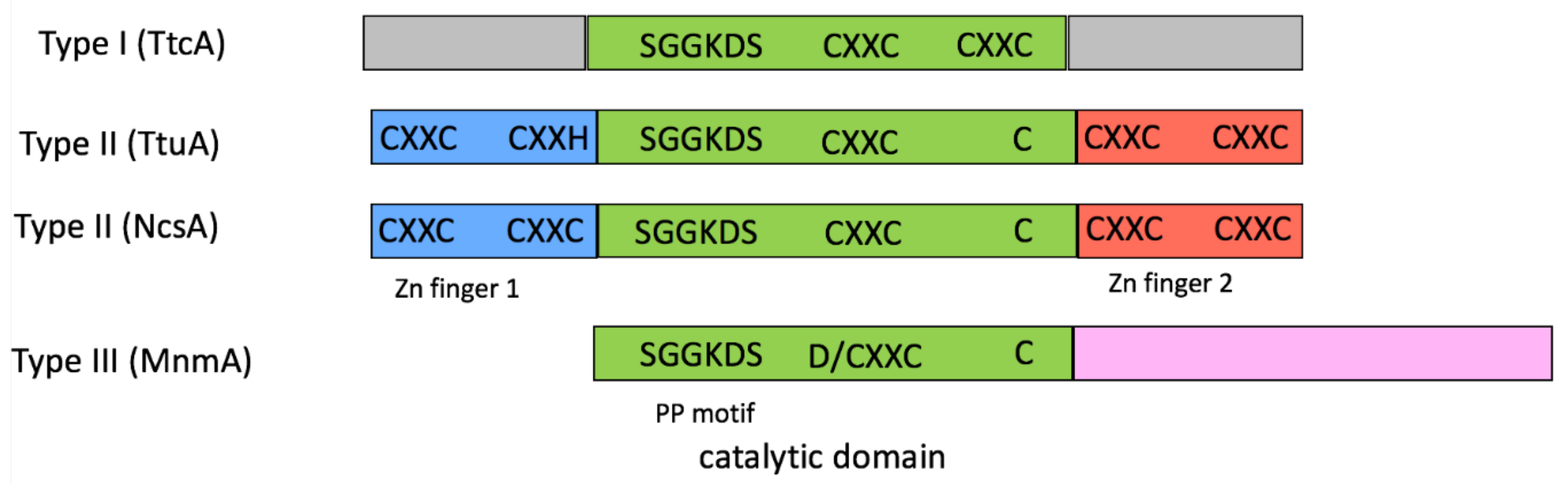
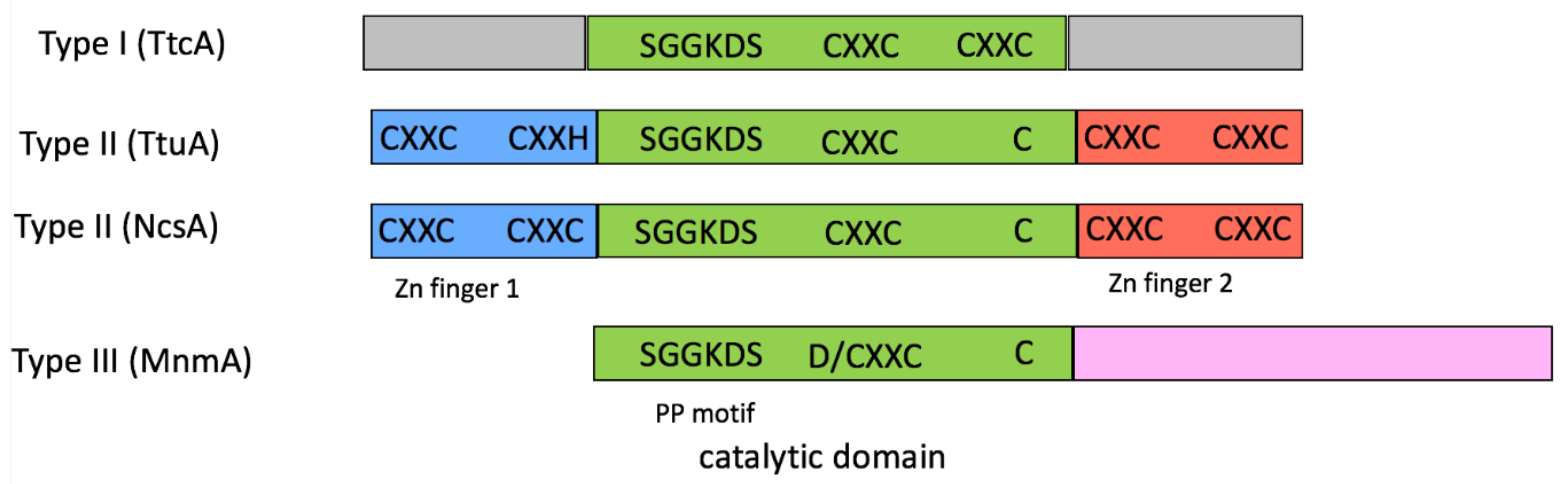
2.1. tRNA Sulfuration Enzymes
2.2. The Cluster Binding Site Is Pre-Formed in the Crystal Structure of the Apo-Form of U54–tRNA Sulfuration Enzyme TtuA
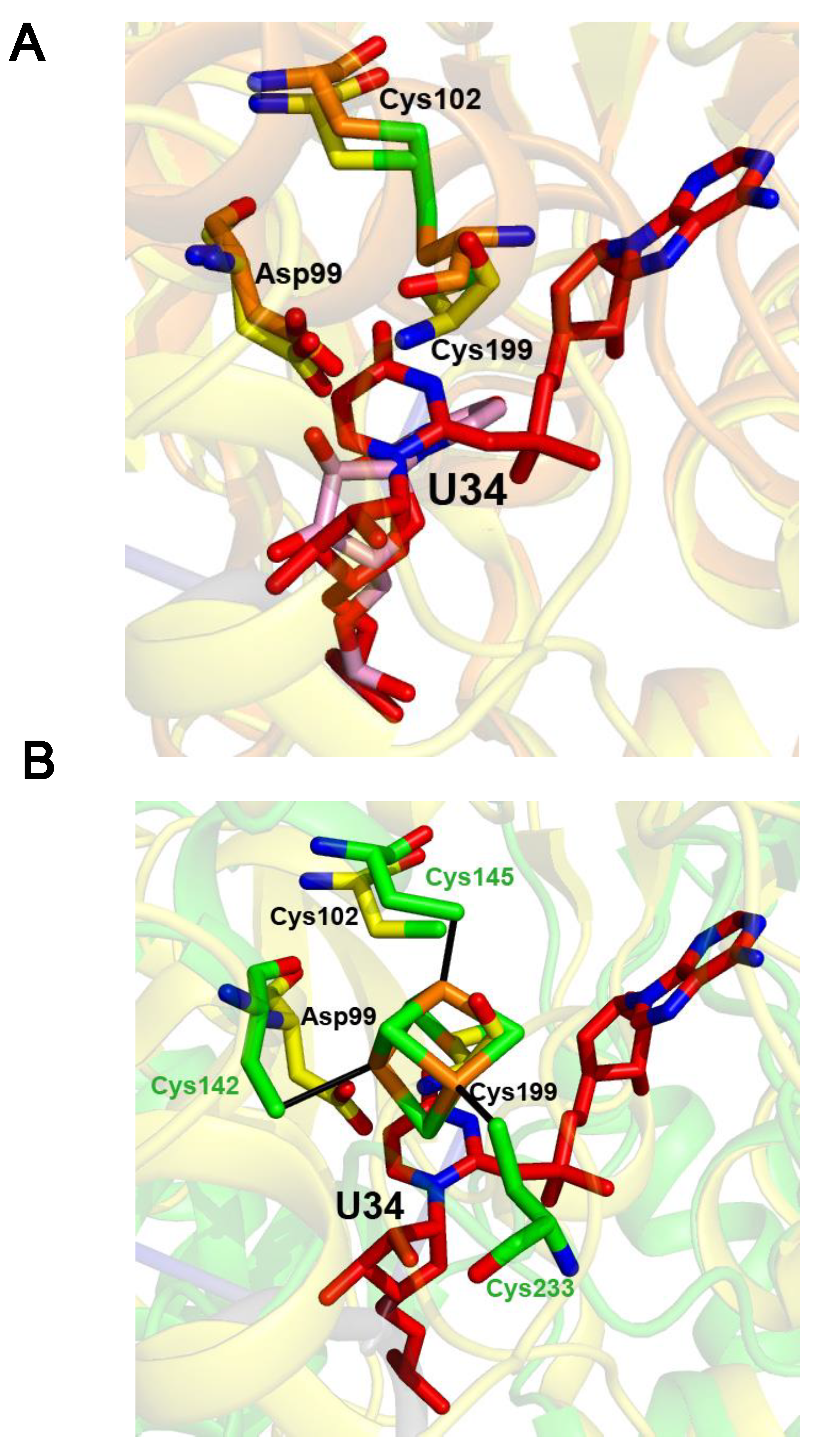
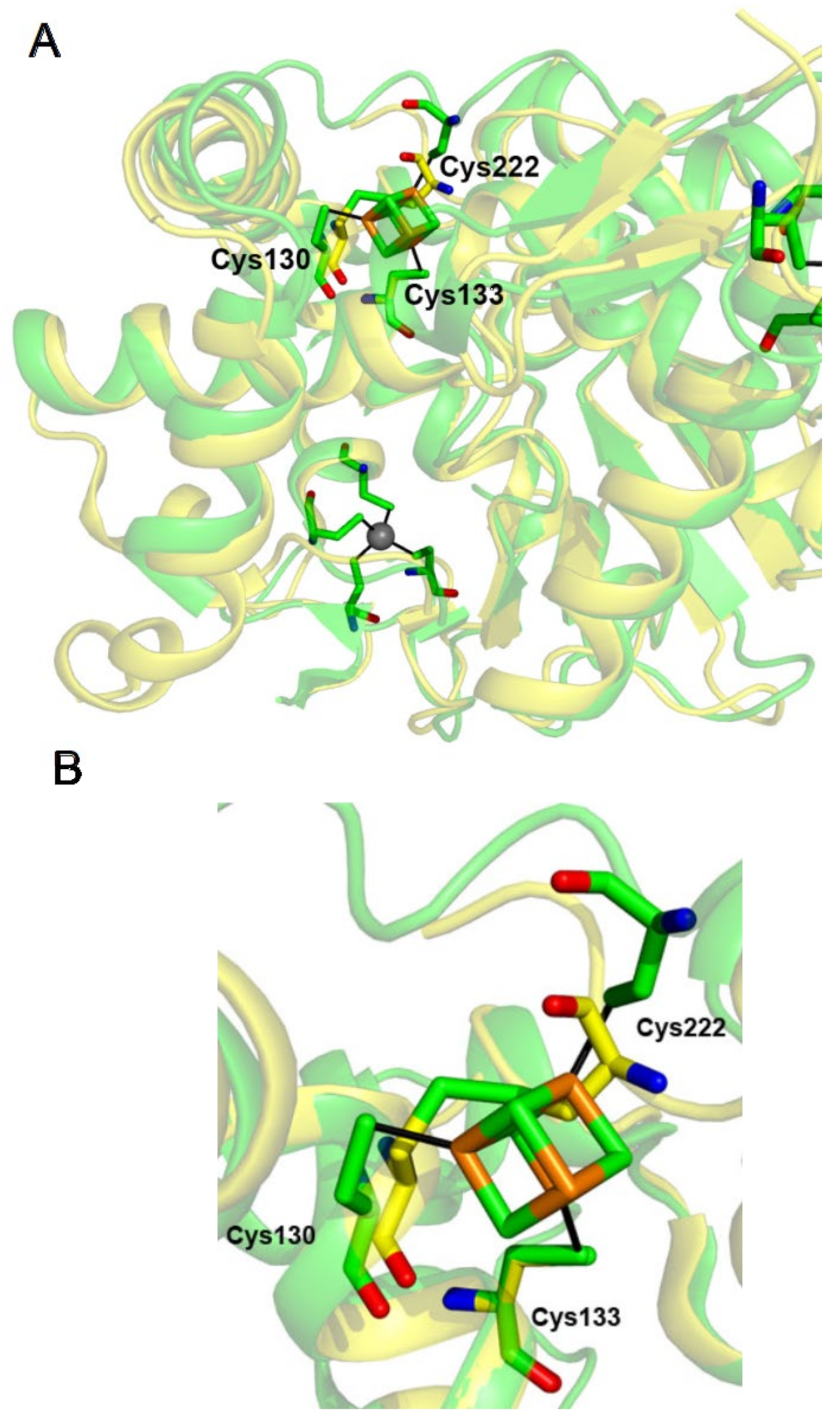
2.3. The [4Fe-4S] Cluster of TtuA Is Bound by Three Protein Ligands Only
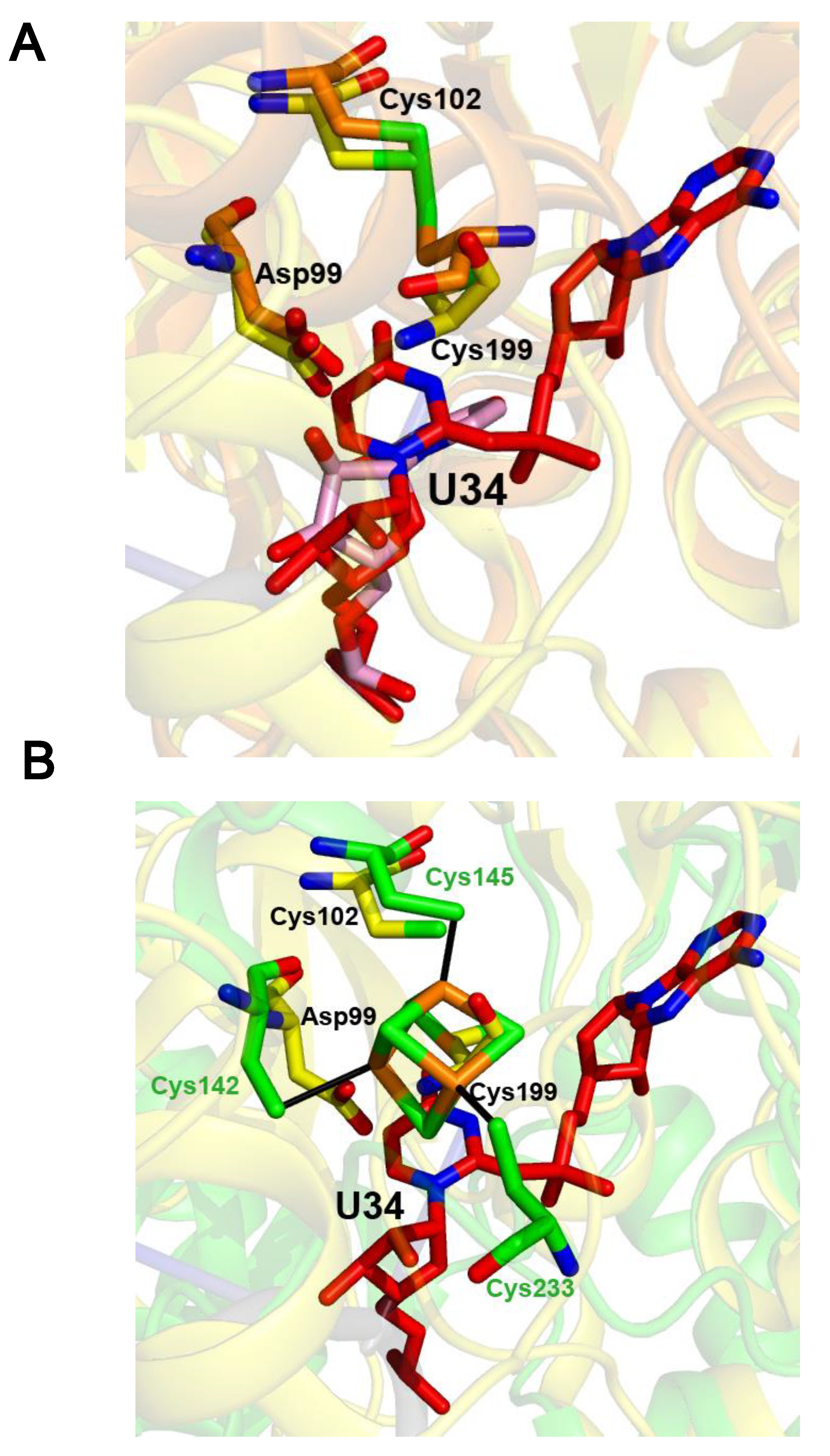
2.4. The Crystal Structure of the U34–tRNA Sulfuration Enzyme MnmA, in the Apo-Form, Suggested That a Cluster Could Bind in the Active Site
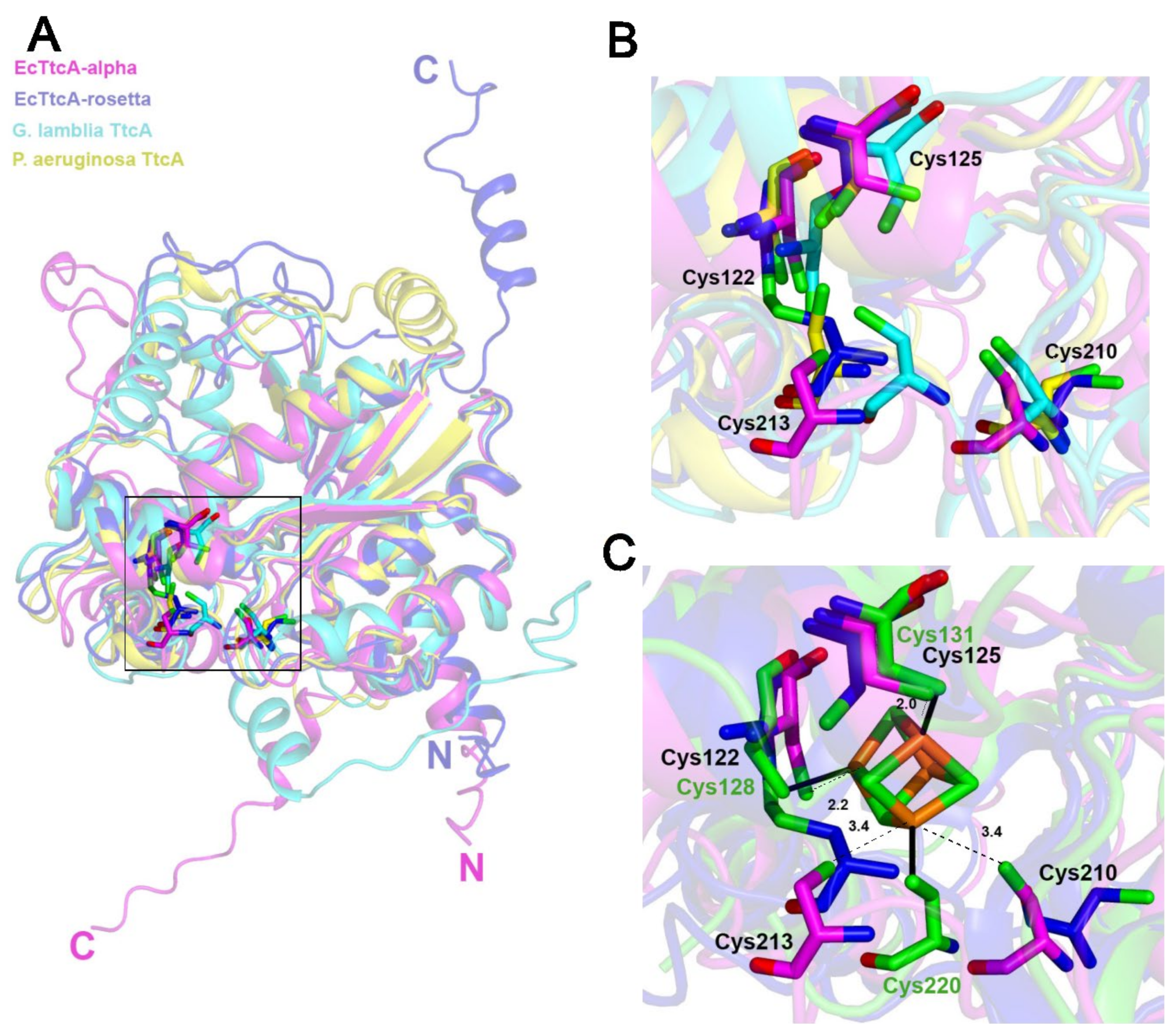
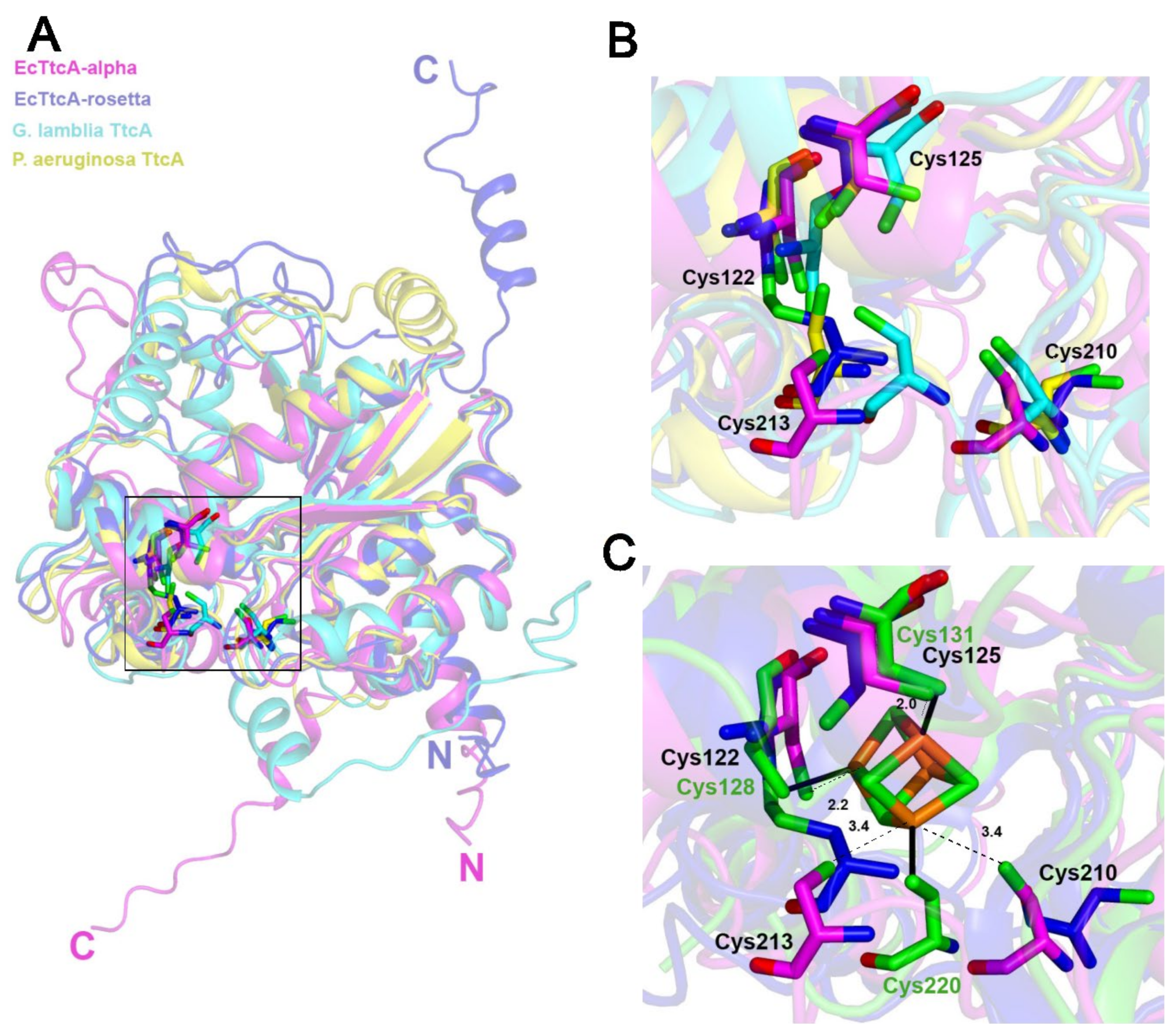
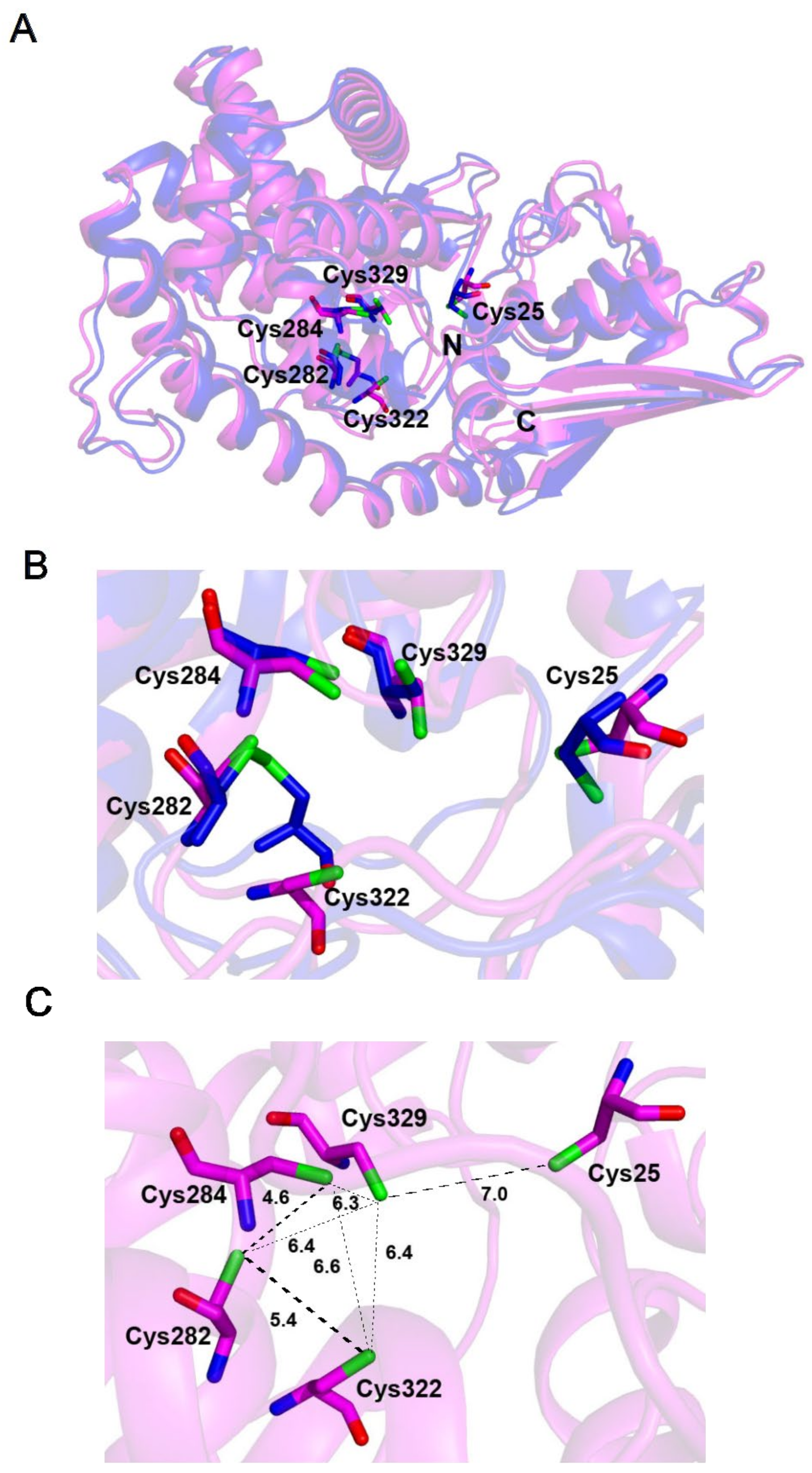
2.5. The Fe–S-Binding Site Is Pre-Formed in the 3D Model of the C32–tRNA Sulfuration Enzyme TtcA, but the Cluster Coordination Is Ambiguous
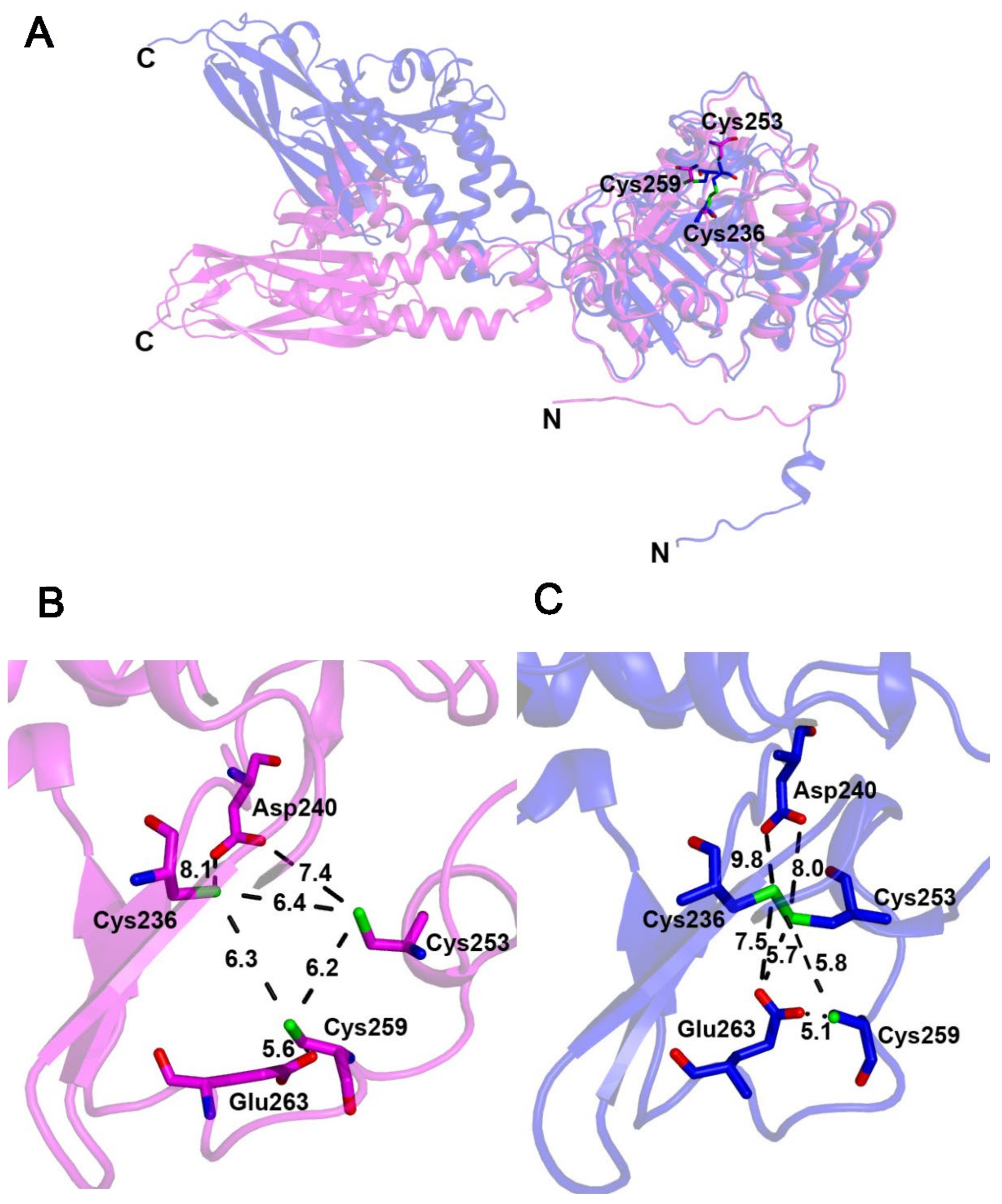
2.6. The Fe–S-Binding Site Is Pre-Formed in the 3D Model of Cysteine Desulfidase CyuA, but the Cluster Coordination Is Ambiguous
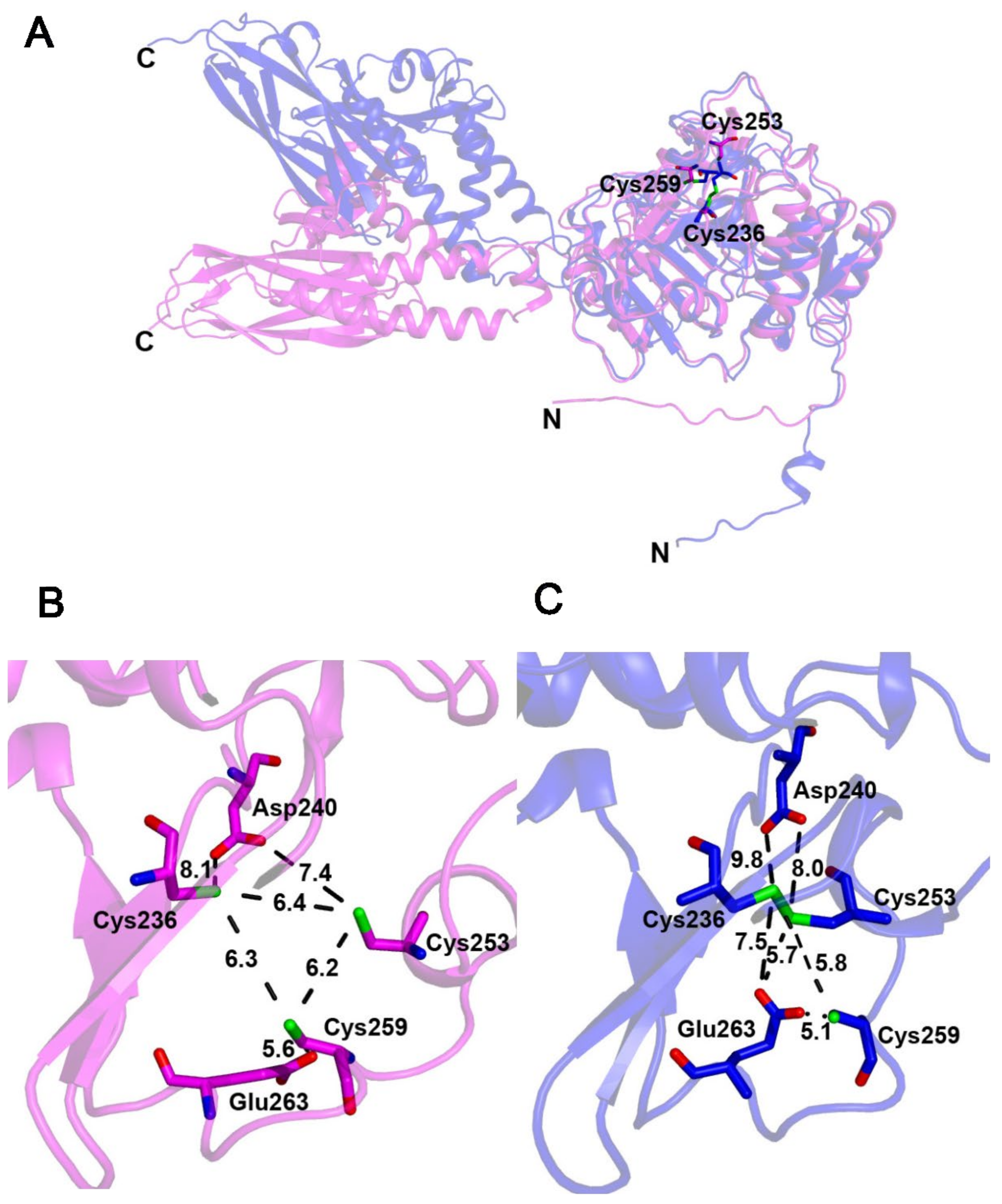
2.7. Blsk, a [3Fe-4S]-Dependent Enzyme Involved in Amide Bond Formation with an Unkown Mechanism
3. Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Cramer, P. AlphaFold2 and the future of structural biology. Nat. Struct. Mol. Biol. 2021, 28, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Baek, M.; Anishchenko, I.; Park, H.; Humphreys, I.R.; Baker, D. Protein oligomer modeling guided by predicted interchain contacts in CASP14. Proteins 2021, 89, 1824–1833. [Google Scholar] [CrossRef] [PubMed]
- Wehrspan, Z.J.; McDonnell, R.T.; Elcock, A.H. Identification of Iron-Sulfur (Fe-S) Cluster and Zinc (Zn) Binding Sites Within Proteomes Predicted by DeepMind’s AlphaFold2 Program Dramatically Expands the Metalloproteome. J. Mol. Biol. 2021, 434, 167377. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Cowan, J.A. Structural, mechanistic and coordination chemistry of relevance to the biosynthesis of iron-sulfur and related iron cofactors. Coord Chem Rev. 2011, 255, 688–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estellon, J.; Ollagnier De Choudens, S.; Smadja, M.; Fontecave, M.; Vandenbrouck, Y. An integrative computational model for large-scale identification of metalloproteins in microbial genomes: A focus on iron–sulfur cluster proteins. Metallomics 2014, 6, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Valasatava, Y.; Rosato, A.; Banci, L.; Andreini, C. MetalPredator: A web server to predict iron-sulfur cluster binding proteomes. Bioinformatics 2016, 32, 2850–2852. [Google Scholar] [CrossRef] [PubMed]
- Freibert, S.A.; Weiler, B.D.; Bill, E.; Pierik, A.J.; Mühlenhoff, U.; Lill, R. Biochemical Reconstitution and Spectroscopic Analysis of Iron–Sulfur Proteins. Methods Enzymol. 2018, 599, 197–226. [Google Scholar] [CrossRef]
- Stich, T.A. Characterization of Paramagnetic Iron-Sulfur Clusters Using Electron Paramagnetic Resonance Spectroscopy. Methods Mol. Biol. 2021, 2353, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.E.; Lanz, N.D.; Booker, S.J.; Krebs, C. Mössbauer spectroscopy of Fe/S proteins. Biochim. Biophys. Acta 2015, 1853, 1395–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, S.; Teixeira, M. Resonance Raman spectroscopy of Fe-S proteins and their redox properties. J. Biol. Inorg. Chem. 2018, 23, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, D.H.; Allen, R.M. Iron-Sulfur Proteins with Nonredox Functions. Chem Rev. 1996, 96, 2315–2334. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H.; Kennedy, M.C.; Stout, C.D. Aconitase as Iron-Sulfur Protein, Enzyme, and Iron-Regulatory Protein. Chem. Rev. 1996, 96, 2335–2374. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.; Tórtora, V.; Mansilla, S.; Radi, R. Aconitases: Non-redox Iron-Sulfur Proteins Sensitive to Reactive Species. Acc. Chem. Res. 2019, 52, 2609–2619. [Google Scholar] [CrossRef]
- Hirano, Y.; Takeda, K.; Miki, K. Charge-density analysis of an iron-sulfur protein at an ultra-high resolution of 0.48 Å. Nature 2016, 534, 281–284. [Google Scholar] [CrossRef]
- Glatt, S.; Zabel, R.; Kolaj-Robin, O.; Onuma, O.F.; Baudin, F.; Graziadei, A.; Taverniti, V.; Lin, T.Y.; Baymann, F.; Seraphin, B.; et al. Structural basis for tRNA modification by Elp3 from Dehalococcoides mccartyi. Nat. Struct. Mol. Biol. 2016, 23, 794–802. [Google Scholar] [CrossRef] [Green Version]
- El Yacoubi, B.; Bailly, M.; de Crecy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Shigi, N. Biosynthesis and functions of sulfur modifications in tRNA. Front. Genet. 2014, 5, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigi, N. Recent Advances in Our Understanding of the Biosynthesis of Sulfur Modifications in tRNAs. Front. Microbiol. 2018, 9, 2679. [Google Scholar] [CrossRef] [PubMed]
- Shigi, N. Biosynthesis and Degradation of Sulfur Modifications in tRNAs. Int. J. Mol. Sci. 2021, 22, 11937. [Google Scholar] [CrossRef]
- Pierrel, F.; Björk, G.R.; Fontecave, M.; Atta, M. Enzymatic modification of tRNAs—MiaB is an iron-sulfur protein. J. Biol. Chem. 2002, 277, 13367–13370. [Google Scholar] [CrossRef] [Green Version]
- Arragain, S.; Handelman, S.K.; Forouhar, F.; Wei, F.Y.; Tomizawa, K.; Hunt, J.F.; Douki, T.; Fontecave, M.; Mulliez, E.; Atta, M. Identification of Eukaryotic and Prokaryotic Methylthiotransferase for Biosynthesis of 2-Methylthio-N-6-threonylcarbamoyladenosine in tRNA. J. Biol. Chem. 2010, 285, 28425–28433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bimai, O.; Arragain, S.; Golinelli-Pimpaneau, B. Structure-based mechanistic insights into catalysis by tRNA thiolation enzymes. Curr. Opin. Struct. Biol. 2020, 65, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Nakai, M.; Lill, R.; Suzuki, T.; Hayashi, H. Thio modification of yeast cytosolic tRNA is an iron-sulfur protein-dependent pathway. Mol. Cell Biol. 2007, 27, 2841–2847. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Vinyard, D.J.; Reesbeck, M.E.; Suzuki, T.; Manakongtreecheep, K.; Holland, P.L.; Brudvig, G.W.; Söll, D. A [3Fe-4S] cluster is required for thiolation in archaea and eukaryotes. Proc. Natl. Acad. Sci. USA 2016, 113, 12703–12708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Asai, S.I.; Narai, S.; Nambu, S.; Omura, N.; Sakaguchi, Y.; Suzuki, T.; Ikeda-Saito, M.; Watanabe, K.; Yao, M.; et al. Biochemical and structural characterization of oxygen-sensitive 2-thiouridine synthesis catalyzed by an iron-sulfur protein TtuA. Proc. Natl. Acad. Sci. USA 2017, 114, 4954–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arragain, S.; Bimai, O.; Legrand, P.; Caillat, S.; Ravanat, J.L.; Touati, N.; Binet, L.; Atta, M.; Fontecave, M.; Golinelli-Pimpaneau, B. Nonredox thiolation in tRNA occurring via sulfur activation by a [4Fe-4S] cluster. Proc. Natl. Acad. Sci. USA 2017, 114, 7355–7360. [Google Scholar] [CrossRef] [Green Version]
- Shigi, N.; Horitani, M.; Miyauchi, K.; Suzuki, T.; Kuroki, M. An ancient type of MnmA protein is an iron-sulfur cluster-dependent sulfurtransferase for tRNA anticodons. RNA 2020, 26, 240–250. [Google Scholar] [CrossRef]
- Zhou, J.; Lénon, M.; Touati, N.; Ravanat, J.-L.; Velours, C.; Fontecave, M.; Barras, F.; Golinelli-Pimpaneau, B. Iron sulfur biology invades tRNA modification: The case of U34 sulfuration. Nucleic Acids Res. 2021, 49, 3997–4007. [Google Scholar] [CrossRef]
- Mulliez, E.; Duarte, V.; Arragain, S.; Fontecave, M.; Atta, M. On the Role of Additional [4Fe-4S] Clusters with a Free Coordination Site in Radical-SAM Enzymes. Front. Chem. 2017, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esakova, O.A.; Grove, T.L.; Yennawar, N.H.; Arcinas, A.J.; Wang, B.; Krebs, C.; Almo, S.C.; Booker, S.J. Structural basis for tRNA methylthiolation by the radical SAM enzyme MiaB. Nature 2021, 597, 566–570. [Google Scholar] [CrossRef]
- Chen, M.; Ishizaka, M.; Narai, S.; Horitani, M.; Shigi, N.; Yao, M.; Tanaka, Y. The [4Fe-4S] cluster of sulfurtransferase TtuA desulfurizes TtuB during tRNA modification in Thermus thermophilus. Commun. Biol. 2020, 3, 168. [Google Scholar] [CrossRef] [Green Version]
- Leipuviene, R.; Qian, Q.; Björk, G.R. Formation of thiolated nucleosides present in tRNA from Salmonella enterica serovar Typhimurium occurs in two principally distinct pathways. J. Bacteriol. 2004, 186, 758–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, G.; Leipuviene, R.; Pollard, M.G.; Qian, Q.; Björk, G.R. The conserved Cys-X1-X2-Cys motif present in the TtcA protein is required for the thiolation of cytidine in position 32 of tRNA from Salmonella enterica serovar Typhimurium. J. Bacteriol. 2004, 186, 750–757. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, H.; Kuratani, M.; Goto-Ito, S.; Ito, T.; Katsura, K.; Terada, T.; Shirouzu, M.; Sekine, S.; Shigi, N.; Yokoyama, S. Crystallographic and mutational studies on the tRNA thiouridine synthetase TtuA. Proteins 2013, 81, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Chavarria, N.E.; Hwang, S.; Cao, S.; Fu, X.; Holman, M.; Elbanna, D.; Rodriguez, S.; Arrington, D.; Englert, M.; Uthandi, S.; et al. Archaeal Tuc1/Ncs6 homolog required for wobble uridine tRNA thiolation is associated with ubiquitin-proteasome, translation, and RNA processing system homologs. PLoS ONE 2014, 9, e99104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armengod, M.E.; Meseguer, S.; Villarroya, M.; Prado, S.; Moukadiri, I.; Ruiz-Partida, R.; Garzón, M.J.; Navarro-González, C.; Martínez-Zamora, A. Modification of the wobble uridine in bacterial and mitochondrial tRNAs reading NNA/NNG triplets of 2-codon boxes. RNA Biol. 2014, 12, 1495–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouvier, D.; Labessan, N.; Clemancey, M.; Latour, J.M.; Ravanat, J.L.; Fontecave, M.; Atta, M. TtcA a new tRNA-thioltransferase with an Fe-S cluster. Nucleic Acids Res. 2014, 42, 7960–7970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, T.; Ikeuchi, Y.; Fukai, S.; Suzuki, T.; Nureki, O. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature 2006, 442, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Romsang, A.; Duang-Nkern, J.; Khemsom, K.; Wongsaroj, L.; Saninjuk, K.; Fuangthong, M.; Vattanaviboon, P.; Mongkolsuk, S. Pseudomonas aeruginosa ttcA encoding tRNA-thiolating protein requires an iron-sulfur cluster to participate in hydrogen peroxide-mediated stress protection and pathogenicity. Sci. Rep. 2018, 8, 11882. [Google Scholar] [CrossRef]
- Thoden, J.B.; Holden, H.M.; Grant, G.A. Structure of L-serine dehydratase from Legionella pneumophila: Novel use of the C-terminal cysteine as an intrinsic competitive inhibitor. Biochemistry 2014, 53, 7615–7624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchong, S.I.; Xu, H.; White, R.H. L-cysteine desulfidase: An [4Fe-4S] enzyme isolated from Methanocaldococcus jannaschii that catalyzes the breakdown of L-cysteine into pyruvate, ammonia, and sulfide. Biochemistry 2005, 44, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Pecqueur, L.; Aučynaitė, A.; Fuchs, J.; Rutkienė, R.; Vaitekūnas, J.; Meškys, R.; Boll, M.; Fontecave, M.; Urbonavičius, J.; et al. Structural evidence for a [4Fe-5S] intermediate in the non-redox desulfuration of thiouracil. Angew. Chem. Int. Ed. 2021, 60, 424–431. [Google Scholar] [CrossRef]
- White, R.H. The biosynthesis of cysteine and homocysteine in Methanococcus jannaschii. Biochim. Biophys. Acta 2003, 1624, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, Y.; Gao, Y.G.; Luo, X.; Du, A.; Deng, Z.; Zabriskie, T.M.; He, X.; Jiang, M. A [3Fe-4S] cluster and tRNA-dependent aminoacyltransferase BlsK in the biosynthesis of Blasticidin S. Proc. Natl. Acad. Sci. USA 2021, 118, e2102318118. [Google Scholar] [CrossRef] [PubMed]
- Bak, D.W.; Elliott, S.J. Alternative FeS cluster ligands: Tuning redox potentials and chemistry. Curr. Opin. Chem. Biol. 2014, 19, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouet, P.; Courcelle, E.; Stuart, D.I.; Metoz, F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 1999, 15, 305–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | RoseTTAFold (Confidence Score) | AlphaFold (pLDDT) | AlphaFold TM Score |
|---|---|---|---|
| EcTtcA | 0.74 | 85.8 | 0.83 |
| PaTtcA | 0.79 | - | - |
| GlTtcA | 0.71 | - | - |
| MjCyuA | 0.82 | 93.9 | 0.92 |
| BlsK | 0.80 | 92.9 | 0.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golinelli-Pimpaneau, B. Prediction of the Iron–Sulfur Binding Sites in Proteins Using the Highly Accurate Three-Dimensional Models Calculated by AlphaFold and RoseTTAFold. Inorganics 2022, 10, 2. https://doi.org/10.3390/inorganics10010002
Golinelli-Pimpaneau B. Prediction of the Iron–Sulfur Binding Sites in Proteins Using the Highly Accurate Three-Dimensional Models Calculated by AlphaFold and RoseTTAFold. Inorganics. 2022; 10(1):2. https://doi.org/10.3390/inorganics10010002
Chicago/Turabian StyleGolinelli-Pimpaneau, Béatrice. 2022. "Prediction of the Iron–Sulfur Binding Sites in Proteins Using the Highly Accurate Three-Dimensional Models Calculated by AlphaFold and RoseTTAFold" Inorganics 10, no. 1: 2. https://doi.org/10.3390/inorganics10010002
APA StyleGolinelli-Pimpaneau, B. (2022). Prediction of the Iron–Sulfur Binding Sites in Proteins Using the Highly Accurate Three-Dimensional Models Calculated by AlphaFold and RoseTTAFold. Inorganics, 10(1), 2. https://doi.org/10.3390/inorganics10010002