Abstract
The rigid-backbone bidentate ligands Indoline-2-carboxylic acid (IndoliH) and Indole-2-carboxylic acid (IndolH) were evaluated for rhodium(I). IndoliH formed [Rh(Indoli)(CO)(PPh3)] (A2), while IndolH yielded the novel dinuclear [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) complex (Indol’ = Indol2−), which were characterized by SCXRD. In B2, the Rh1(I) fragment [Rh1(Indol’)(CO)(PPh3)] (bidentate N,O-Indol) exhibits a square-planar geometry, while Rh2(I) shows a ‘Vaska’-type trans-[O-Rh2(PPh3)2(CO)] configuration (bridging the carboxylate ‘oxo’ O atom of Indol2−). The oxidative addition of MeI to A2 and B2 via time-resolved FT-IR, NMR, and UV/Vis analyses indicated only Rh(III)-alkyl species (A3/B3) as products (no migratory insertion). Variable temperature kinetics confirmed an associative mechanism for A2 via an equilibrium-based pathway (ΔH≠ = (21 ± 1) kJ mol−1; ΔS≠ = (−209 ± 4) J K−1mol−1), with a smaller contribution from a reverse reductive elimination/solvent pathway. The dinuclear complex B2 showed the oxidative addition of MeI only at Rh1(I), which formed a Rh(III)-alkyl, but cleaved the bridged Rh2(I) site, yielding trans-[RhI(PPh3)2(I)(CO)] (5B) as a secondary product. A significantly smaller negative activation entropy [ΔH≠ = (73.0 ± 1.2) kJ mol−1; ΔS≠ = (−21 ± 4) J K−1mol−1] via a more complex/potential interchange mechanism (the contribution of ΔS≠ to the Gibbs free energy of activation, ΔG≠, only ±10%) was inferred, contrary to the entropy-driven oxidative addition of MeI to A2 (the contribution of ΔS≠ to ΔG≠ ± 75%).
1. Introduction
Rhodium complexes represent some of the most widely propagated industrial homogeneous catalysts, and classic examples include olefin hydrogenation by Wilkinson’s catalyst [RhCl(PPh3)3], the carbonylation of methanol to produce acetic acid by using the catalyst precursor [Rh(CO)2I2]– in the Monsanto process, and the hydroformylation of alkenes to produce aldehydes by Union Carbide using [RhHCO(PPh3)2] [1,2]. The catalytic cycles are defined by several fundamental reactions, of which oxidative addition is a prime example, and are prominent in the mechanistic scheme of a number of these applied processes. Thus, the formations of RhIII-alkyl and RhIII-acyl entities are important reaction products in a number of these prominent catalytic processes and has prompted much research aiming to optimize systems.
The Rh catalyst precursor in the Monsanto process, [RhI2(CO)2]– contains only ‘simple’ iodido ancillary ligands [1,2]. Since the catalytic reactivity of rhodium(I) complexes is controlled by the nature of the ligands around the metal center, manipulating the electro-steric properties of these groups allows the reaction rates of two essential steps in homogeneous catalysts, such as oxidative addition and migratory insertion, to be tuned. The catalyst precursor [RhI(I)2(CO)2]– in the Monsanto process is relatively unstable and may thus be manipulated by introducing different bidentate ligands, generating, for example, [RhI(L,L’-Bid)(CO)2] complexes (L,L’-Bid−, a monocharged bidentate ligand, and L and L’, coordinating donor atoms).
In addition, depending on the reaction conditions, monodentate (typically tertiary phosphine entities) and non-symmetric bidentate (L,L’-Bid) ancillary non-labile ligands with different donor atoms (e.g., O, N, S, P, Se) may be visualized to engineer pre-selected additional characteristics in new catalyst analogues. Thus, one of the carbonyl ligands in the precursor [RhI(L,L’-Bid)(CO)2] may additionally be replaced by a monodentate tertiary phosphine, arsine, or stibine ligand to form [RhI(L,L’-Bid)(CO)(AX3)] complexes (where A = P, As, and Sb, and X3 = Ph3, Ph2Cy, PhCy2, Cy3, etc.). Moreover, tertiary phosphines, when employed in conjunction with the bidentate ligands, allows for further synergistic tuning of the electronic and steric accessibility of the metal center to a model catalyst [3,4]. In this regard, platinum group metals [5,6], particularly rhodium(I) complexes, and their oxidative addition (RhIII-alkyl) and migratory insertion products (which form RhIII-acyl entities) have been extensively studied in the past few decades [4,7,8,9,10,11,12,13,14].
Our group focuses on structure/reactivity relationships in model catalytic systems; therefore, we extensively use X-ray crystallography as an integral research component, which is supplemented by IR and multi-nuclear NMR spectroscopy (including time-resolved) to study applicable systems. Different examples have been presented in the past, which include crystallographic studies on [RhI(L,L’-Bid)(CO)(PPh3)] complexes that revealed that the reaction between [RhI(L,L’-Bid)(CO)2] with PPh3 yields only one isomer that crystallizes from solution [4,15,16,17]. The only example wherein both isomers crystallized in the same unit cell has been reported for the [RhI(BA)(CO)(PPh3)] complex (BA = benzoylacetonate) [18].
Thus, we present herein examples of rigid-backbone bidentate ligands based on the Indole framework, such as Indole-2-carboxylic acid and Indoline-2-carboxylic acid, which, due to its electronic and relative robust structure, warrants additional and extended investigation. It produced surprising new compounds and provide us with insight into both a monomeric (A2) and a dinuclear (B2) reactant and the kinetic behavior during the iodomethane (MeI) oxidative addition, including the associated energies therewith. An added advantage resulting from these ligand systems is that migratory insertion is eliminated, which significantly simplifies the interpretation of the reaction mechanism without the interference of any side isomerization reactions.
2. Results
2.1. Synthesis of Compounds
Indole-2-carboxylate (IndolH = Indole-2-carboxylic acid) and Indoline-2-carboxylate (IndoliH = Indoline-2-carboxylic acid) are known ligands for transition metals. Both act as bidentate ligands by undergoing a mono-dissociation of the carboxylic acid proton, as shown in Scheme 1. However, Indole-2-carboxylic acid showed a two-proton dissociation, as illustrated in Scheme 1b.
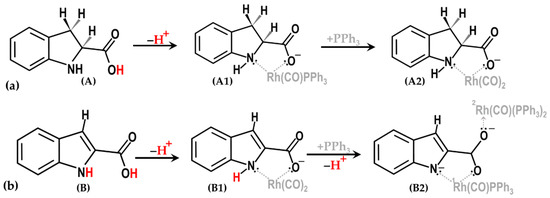
Scheme 1.
General scheme for the proton dissociation of Indoline-2-carboxylic acid (a) and Indole-2-carboxylic acid (b) in order to enable the formation of complexes A1, A2, B1, and B2, respectively (see Scheme 2).
The two seemingly very similar ligands, Indoline-2-carboxylate and Indole-2-carboxylate (IndoliH = Indoline-2-carboxylic acid (A); IndolH=Indole-2-carboxylic acid (B)), were thus explored as N,O-donor systems for rhodium(I) carbonyl systems. Dicarbonyl complexes, [Rh(Indoli)(CO)2] (A1) and [Rh(Indol)(CO)2] (B1), were obtained directly from RhCl3∙xH2O via the in situ synthesis of μ-[RhCl(CO)2]2/[RhICl2(CO)2]− [7,19,20], as illustrated in Scheme 2. From these, triphenylphosphine complexes could be obtained (A2, B2), which were utilized as reactants for the oxidative addition of iodomethane thereto, yielding A3 and B3/B5).

Scheme 2.
General scheme for the synthesis of the compounds in this study (starting from the μ-[RhICl(CO)2]2/[RhICl2(CO)2]–, generated in situ from RhCl3·xH2O) and associated ligands utilized [A1–A4: Indoline-2-carboxylic acid (IndoliH; A); B1–B4: Indole-2-carboxylic acid (IndolH; B)]. Note that (i) the dinuclear species’ Rh(I) metal centers are distinguished by 1 and 2, respectively; (ii) Indol’H2 = IndolH = Indole-2-carboxylic acid (thus, Indole-2-carboxylic acid can be effectively doubly deprotonated, losing both its carboxylic and pyrone-nitrogen protons, such as in B2); (iii) the absolute geometry of the methyl and iodido entities following the oxidative addition of iodomethane (MeI) is not 100% defined, although current information in hand from the literature indicates that, due to steric effects, five-membered metallocycles such as those used in this study primarily yield a cis-alkyl product, which typically results from a three-centered transition state [9,11,17,21].
Scheme 2.
General scheme for the synthesis of the compounds in this study (starting from the μ-[RhICl(CO)2]2/[RhICl2(CO)2]–, generated in situ from RhCl3·xH2O) and associated ligands utilized [A1–A4: Indoline-2-carboxylic acid (IndoliH; A); B1–B4: Indole-2-carboxylic acid (IndolH; B)]. Note that (i) the dinuclear species’ Rh(I) metal centers are distinguished by 1 and 2, respectively; (ii) Indol’H2 = IndolH = Indole-2-carboxylic acid (thus, Indole-2-carboxylic acid can be effectively doubly deprotonated, losing both its carboxylic and pyrone-nitrogen protons, such as in B2); (iii) the absolute geometry of the methyl and iodido entities following the oxidative addition of iodomethane (MeI) is not 100% defined, although current information in hand from the literature indicates that, due to steric effects, five-membered metallocycles such as those used in this study primarily yield a cis-alkyl product, which typically results from a three-centered transition state [9,11,17,21].

2.2. X-ray Crystallography
This study presents the solid state structure of an Indol rhodium(I) complex of the type [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2]∙(CH3COCH3) (B2a), where Indol’H2: = IndolH and Indol’= Indole2–, i.e., the dianion of Indole-2-carboxylic acid following the double deprotonation of the carboxylic acid entity, as well as the nitrogen on the pyrrole. Complex B2a was isolated following the addition of triphenylphosphine to [Rh(Indol’)(CO)2], which results in a carbonyl substitution from the rhodium coordination sphere, and which is isolated as an acetone adduct. The Vaska-type complex trans-[Rh(CO)(I)(PPh3)2]∙(CH3COCH3) (B5a) is produced following the oxidative addition of iodomethane to [Rh1(Indol’)(CO)(PPh3)Rh1(CO)(PPh3)2]∙(CH3COCH3) (B2a) and recrystallization from acetone. These two structures were characterized by means of single-crystal X-ray crystallography and are described herein, with basic data given in Table 1 and Figure 1.

Table 1.
Crystal data and refinement parameters of [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2]- ∙(CH3COCH3) (B2a) and trans-[Rh(CO)(I)(PPh3)2]∙(CH3COCH3) (B5a).
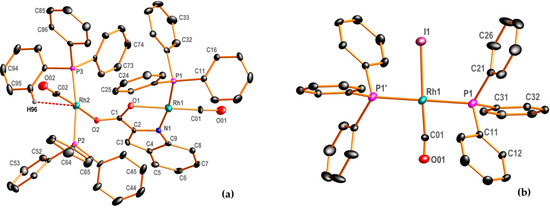
Figure 1.
DIAMOND perspective view of (a) [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2), where Indol’ = Indole2−, i.e., the dianion of Indole-2-carboxylic acid, generated following double deprotonation of the carboxylic acid entity and the pyrrole-nitrogen; (b) trans-[Rh(CO)(I)(PPh3)2] (B5), with a 50/50% positional disorder (not shown) occurring between the CO and Iodido ligands, with the CO oxygen situated exactly on the iodide (50% probability displacement ellipsoids). For the phenyl rings, the first digit indicates ring number, and the second digit indicates the position of the carbon atom in the ring. Apart from H96, all other hydrogen atoms, as well as the acetone solvates, have been omitted for clarity.
The crystal structures for B2a and B5a were determined at 100 K and the selected bond distances and angles are reported in Table 2 and Table 3, respectively.
Table 2.
Bond lengths [Å] and angles [°] for [Rh1(Indol′)(CO)(PPh3)Rh2(CO)(PPh3)2]·(CH3COCH3) (B2a).
Table 3.
Bond lengths [Å] and angles [°] for trans-[Rh(CO)(I)(PPh3)2]·(CH3COCH3) (B5a).
2.3. Spectroscopic Evaluation of the Oxidative Addition of Iodomethane to Rh(Indoli)(CO)(PPh3)] (A2) and [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2)
The solutional behavior of the complexes in the presence of iodomethane as a substrate was next evaluated by FT-IR, 13P NMR, and UV/vis spectroscopies. The monomeric complex [Rh(Indoli)(CO)(PPh3)] (A2) was first investigated, followed by the dinuclear complex [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) to determine the solution characteristics and, eventually, the reactivities of the three different Rh(I) metal centers.
2.3.1. Rh(Indoli)(CO)(PPh3)] (A2)
- IR Results
Figure 2 illustrates the dynamics [22] observed within the [Rh(Indoli)(CO)(PPh3)] (A2) complex when reacted with iodomethane. A clear decrease in the reactant is observed, coinciding with the formation of an IR band at 2054 cm−1, which is typical of a Rh(III)-alkyl species (A3) [4].
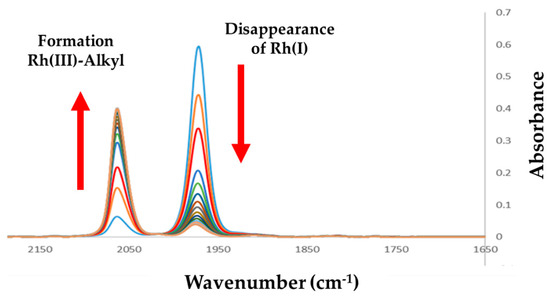
Figure 2.
Successive IR spectra illustrating the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)] (A2) (ν(CO) =1976 cm–1) and the formation of the RhIII-alkyl A3 (ν(CO) =2054 cm–1); [Rh] = 0.01 M, [MeI] = 1.0 M, Δt = 120 s; Total time = 30 min.
A notable observation is the seemingly non-complete disappearance of the Rh(I) species as illustrated in Figure 2, and the absence of the formation of a Rh(III)-acyl species (with a typical ν(CO) of around 1700–1720 cm−1).
- 31P NMR study
Figure 3 illustrates the corresponding dynamics observed via 31P NMR within [Rh(Indoli)(CO)(PPh3)] (A2) when reacted with iodomethane compared to those noted in the infrared spectrum (Figure 2). Again, a clear systematic decrease in the reactant is observed (δ = 41.7 ppm; 1J(Rh-P) = 169 Hz), coinciding with the formation of two doublets (δ=30.5 ppm, 1J(Rh-P) = 124 Hz, and δ = 29.5 ppm; 1J(Rh-P) = 124 Hz), typical of alkyl species [4], with the species represented by δ = 30.5 ppm being the dominant and the final Rh(III)-alkyl complex.
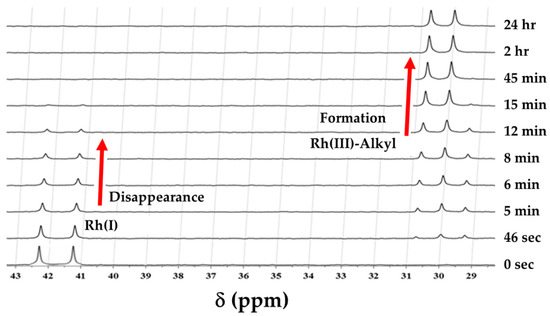
Figure 3.
Successive 31P NMR spectra illustrating the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)] (A2) in dichloromethane at 25 °C. [Rh] = 0.02 M and [MeI] = 1.0 M. A minor Rh(III)-alkyl isomer (doublet at 29.5 ppm) is observed, which disappears after ca. 15 min, never exceeds ca 5–10% of the major isomer, and does not noticeably contribute to the reaction; thus, it has been ignored further in this study. Total time = 24 h.
2.3.2. [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2)
- IR Study
Contrary to that observed for the Indoline complex A2 reported in Section 2.3.1, a completely different behavior was noted for [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2). The typical reaction progress is shown in Figure 4 below.
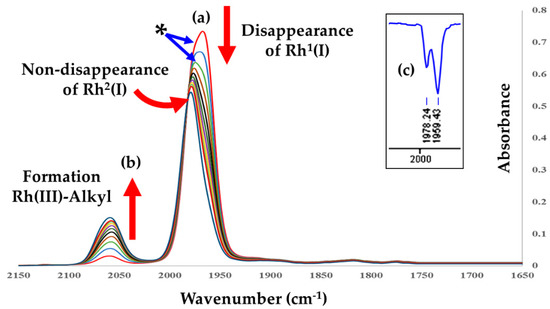
Figure 4.
Successive infrared spectra illustrating the oxidative addition of iodomethane to [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2), showing (a) the disappearance of the signal at ν(CO) = 1967 cm−1 (representing the ‘sum’ of the two Rh fragments, Rh1(I) and Rh2(I); also note the shoulder at the asterisk as indicated) and (b) the simultaneous definition of (i) the Vaska fragment at 1980 cm−1 and (ii) the appearance of the Rh(III)-alkyl species at (ν(CO) = 2060 cm−1), in dichloromethane at 25 °C. [B2] = 0.0058 M, [MeI] = 0.064 M, and Δt = 120 s; Total time = 20 min. Insert (c) illustrates the similar wavenumber range in the solid state IR ATR spectrum, and the two ν(CO) peaks associated with Rh1(I) and Rh2(I) are more clearly defined at ν = 1959 and 1978 cm−1, respectively (see also Figure S4 in Supplementary Materials).
In Figure 4, a new signal of the Vaska fragment at ca. 1980 cm−1 is seemingly formed. However, the deconvolution of the original spectrum before the addition of iodomethane clearly shows a shoulder in B2, which may be attributed to a Vaska fragment (Rh2(I)) from the start, when the complete molecule [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] was still intact. This is additionally confirmed by the solid-state ATR IR as illustrated in Figure 4c.
- NMR study
Repetitive 31P{1H} NMR scans (Figure 5) of the oxidative addition of iodomethane observed from IR spectroscopy in Figure 4 confirmed the behavior noted by the latter, where the rate of disappearance of Rh1(I) (δ = 30.09 ppm; 1JRh-P = 132 Hz) and the rate of appearance of RhIII-alkyl (δ = 27.2 ppm; 1JRh-P = 122 Hz) are equal within the experimental error. The rate of disappearance of Rh1(I) is relatively fast and difficult to monitor reproducibly by quantitative 31P{1H} NMR spectroscopy, but it was still successfully evaluated.
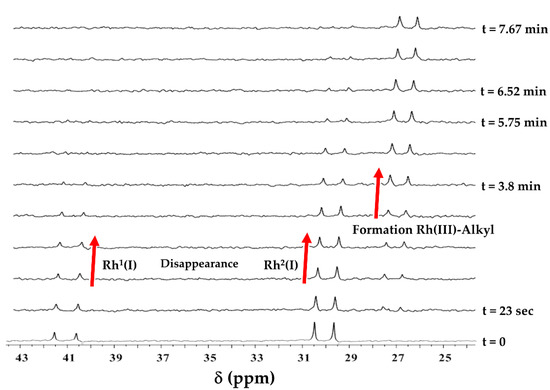
Figure 5.
Observed 31P{1H} NMR spectra illustrating the oxidative addition of iodomethane to [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) in dichloromethane at 25 °C. Spectra show the disappearance of the Rh1(I) and Rh2(I) sites and the formation of the RhIII-alkyl (progress as per red arrows). Total peak integrals were obtained against time for 98 spectra. However, only selected spectra are plotted here to illustrate the decrease/increase in peaks (the number of scans = 12), kobs = (5.6 ± 0.2) × 10−3s−1 for Rh1(I)/Rh2(I), and (5.62 ± 0.17) × 10−3s−1 for RhIII-alkyl. The spectra were referenced externally to 85% H3PO4 (δ = 0 ppm). [B2] = 0.0058 M; [MeI] = 0.064 M.
A summary of chemical shifts, the first-order coupling constants, and kobs for B2 is given in Table 4 for this reaction.
2.3.3. Summary of the Oxidative Addition of Iodomethane to Rh(Indoli)(CO)(PPh3)] (2A) and [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) as Monitored by Different Spectroscopies
- Rh(Indoli)(CO)(PPh3)] (A2)
The formation of Rh(III)-alkyl was monitored by both IR and 31P NMR time-resolved spectroscopy, wherein the species’ resonances were respectively analyzed. The relationship regarding the absorbance values/NMR peak integrals vs. time of the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)] in dichloromethane at 25 °C was now fitted to the first-order exponential given in Equation (1) (Par 4.2), and the relative reaction progress is illustrated by the four different data set fits in Figure 6.
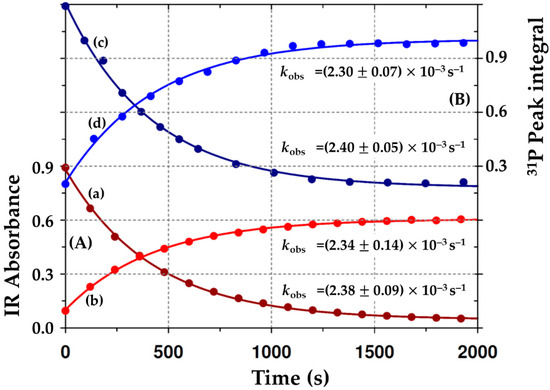
Figure 6.
First-order fits to Equation (1) (Section 4.2) of the kinetic process at 25 °C in dichloromethane defined by the disappearance of the [Rh(Indoli)(CO)(PPh3)] (A2) species, and the concurrent formation of the Rh(III)-alkyl species (A3). (A) In Red: IR Absorbance changes vs. time from Figure 2: (a) Disappearance of Rh(I); (b) formation of Rh(III)-acyl; ([A2] = 0.01 M; [MeI] = 1.0 M). (B) In blue: 31P{1H} peak integral changes vs. time from Figure 3: (c) Disappearance of Rh(I); (d) formation of Rh(III)-acyl; ([A2] = 0.02 M; [MeI] = 1.0 M). The data points represent the observed IR absorbance values/31P NMR peak integrals, while the lines give the least-squares data fits to yield the pseudo first-order rate constants.
The good fits obtained by all the data sets illustrated in Figure 6 enabled the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)] (A2) to be reliably studied (i) as a function of [MeI] and (ii) with respect to temperature. A summary of the chemical shifts, the first-order coupling constants, and the kobs of the oxidative addition of iodomethane to A2 is given in Table 4.
Both the FT-IR and 31P-NMR studies agree and confirm the oxidative addition of iodomethane to A2, with the first reaction step being the formation of the Rh(III)-alkyl species. The 31P-NMR results show that the Rh(I) species is completely depleted at 45 min, while the Rh(III)-alkyl signal grew at the same rate.
Table 4.
Summary of chemical shifts, coupling constants, and preliminary kinetic data in dichloromethane for the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)] (A2) and [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2).
Table 4.
Summary of chemical shifts, coupling constants, and preliminary kinetic data in dichloromethane for the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)] (A2) and [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2).
| Species | IR Data | 31P{1H} NMR Data | |||
|---|---|---|---|---|---|
| νCO (cm−1) | kobs (s−1) | δ (ppm) | 1J(Rh-P) (Hz) | kobs (s–1) | |
| A2(a,b) | |||||
| Rh(I) | 1967 | (2.38 ± 0.09) × 10−3 (c) | 41.7 | 169 | (2.40 ± 0.05) × 10–3 (c) |
| RhIII-alkyl | 2054 | (2.34 ± 0.14) × 10−3 (c) | 30.3 | 124 | (2.30 ± 0.07) × 10–3 (c) |
| B2(d,e) | |||||
| Rh1(I) | 1967 | (5.56 ± 0.12) × 10−3 (f) | 41.0 | 150 | (5.50 ± 0.02) × 10–3 (f) |
| Rh2(I) | 1980 (d) | ” | 30.0 | 133 | ” |
| Rh1(III)-Alkyl | 2060 | (5.50 ± 0.04) × 10−3 (f) | 27.2 | 122 | (5.52 ± 0.02) × 10–3 (f) |
| B5 | 1968 | - | 29.0 | 126 | - |
- [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2)
Again, as for the Indoli complex A2, the time-resolved experiments via IR and 31P{1H} NMR spectroscopy facilitated the kinetic evaluation of the Rh(III)-alkyl formation from the [Rh1(Indol’)(CO)(PPh3)-Rh2(CO)(PPh3)2] (B2) dinuclear species, wherein the relative species’ resonances were respectively analyzed. The time-resolved IR data for the oxidative addition of iodomethane to (B2) in dichloromethane at 25 °C, as illustrated in Figure 4, were fitted to the first-order exponential given in Equation (1) (Section 4.2), and the relative reaction progress is illustrated in Figure 7A. Thus, well-behaved kinetics were also observed for B2 when monitored by infrared spectroscopy.
Moreover, by monitoring the 31P{1H} NMR signal versus time as shown in Figure 5, well-behaved first-order kinetics predicted by Equation (1) for the formation of the Rh(III)-alkyl product were also observed as shown in Figure 7B, yielding virtually identical rate constants for the disappearance of the reactant, and the corresponding formation of the Rh(III)-alkyl product.
Finally, it should be noted that both IR and 31P NMR spectroscopy require fairly large concentrations (tens of millimolar) to allow for reproducible spectra, particularly when the reactions are fast. Thus, UV/vis spectroscopy, which allows for much lower concentrations to be used (a few millimolar, or even fractions thereof) is preferred when large numbers of kinetic runs are required, e.g., in detailed temperature and iodomethane concentration evaluations. Therefore, UV/vis spectroscopy was utilized, but always, when appropriate, in conjunction with IR and 31P NMR, as illustrated in Figure 7.

Figure 7.
Typical first-order fits (kobs values given on graph) to Equation (1) of: (A) Absorbance changes as a function of time (see Figure 4): (a) IR: disappearance of B2; the Rh1(I) fragment; (b) IR: formation of the RhIII-alkyl species in dichloromethane at 25 °C ([B2] = 0.0058 M and [MeI] = 0.064 M); (c) UV/Vis time trace in dichloromethane at 25 °C, λ = 450 nm, [B2] = 8.35 × 10−4 M, and [MeI] = 0.088 M (when kobs normalized to [MeI] = 0.064 M, kobs = 5.00(3) × 10−3 s−1). (B) 31P{1H} NMR Integrals as a function of time (see Figure 5) for the (d) disappearance of the Rh1(I) species and (e) formation of the RhIII-alkyl species and of B5 ([B2] = 0.0058 M; in dichloromethane at 25 °C; [MeI] = 0.064 M).
Figure 7.
Typical first-order fits (kobs values given on graph) to Equation (1) of: (A) Absorbance changes as a function of time (see Figure 4): (a) IR: disappearance of B2; the Rh1(I) fragment; (b) IR: formation of the RhIII-alkyl species in dichloromethane at 25 °C ([B2] = 0.0058 M and [MeI] = 0.064 M); (c) UV/Vis time trace in dichloromethane at 25 °C, λ = 450 nm, [B2] = 8.35 × 10−4 M, and [MeI] = 0.088 M (when kobs normalized to [MeI] = 0.064 M, kobs = 5.00(3) × 10−3 s−1). (B) 31P{1H} NMR Integrals as a function of time (see Figure 5) for the (d) disappearance of the Rh1(I) species and (e) formation of the RhIII-alkyl species and of B5 ([B2] = 0.0058 M; in dichloromethane at 25 °C; [MeI] = 0.064 M).

A summary of the chemical shifts, the first-order coupling constants, and kobs values for the [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) complex is given in Table 4. A low concentration of iodomethane was necessitated due to the rapid reaction, and only a small amount of RhIII-alkyl was formed from the two Rh(I) reactants (Figure 5). Nevertheless, even at low concentrations of MeI and Rh(I) starting materials, the data from the IR and NMR spectroscopy still correlate very well.
Interestingly, the [Rh(Indoli)(CO)(PPh3)] (A2) complex showed no signal for a Rh(III)-acyl species as a final product from the potential migratory CO-insertion of the coordinated CO into the Rh-Me bond, even after prolonged experimental time frames (>24 h), despite being a common feature following many oxidative addition reactions of typical Rh(L,L’-Bid)(CO)(PR3)] entities (see Scheme 2) [7,12,13]. The same observation was made for B2. Thus, the kinetics are significantly simplified, as illustrated in Scheme 3. Moreover, the exploratory results obtained from the IR/31P{1H} NMR experiments, as described in Section 2.3.3 for the kinetic process described in Scheme 3, could thus be used to analyze the kinetics further in more expanded detail.
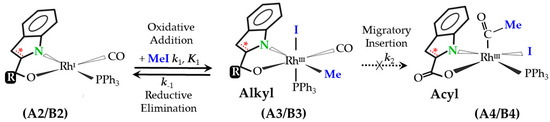
Scheme 3.
General scheme [7] for the kinetic study of the oxidative addition of iodomethane to [RhI(N,O-Bid)(CO)(PPh3)] complexes, showing the potential successive migratory insertion step. Note: (i) A2–A4: N,O-BidR = Indoline-2-carboxylate (without olefinic bond (asterisked) in pyrrole ring, and R = carboxylate ketonic oxygen ‘O==’); (ii) B2–B4: N,O-BidR = Indole-2-carboxylate (with olefinic bond in pyrrole ring, and with R = Vaska-type fragment—[ORh2(CO)(PPh3)2]). A solvent pathway is not considered since it could not be formally verified in this study, although solvent effects are always present [4]. Additionally, note that although the migratory insertion step is indicated, it was not detected in the current study (shown with dotted arrow), as was observed on some occasions [23].
Notably, the formation of the Rh(III)-alkyl species in both A2 and B2 seems to be quite fast when compared to the literature and reactions with different bidentate systems [21,24,25,26,27,28].
3. Discussion
3.1. Synthesis
The synthesis of these rhodium complexes generally emanates from using the Rh(I) precursor(s) μ-[RhICl(CO)2]2/[RhICl2(CO)2]−, which, upon the addition of the mono-charged bidentate ligand, generate the corresponding dicarbonyl rhodium(I) complexes [4,11,19,20,29,30,31]. The formation of these dicarbonyl species can easily be traced by the two prominent IR stretching frequency signals at 2083 and 1987 cm−1 in response to the two coordinated carbonyl ligands (see, e.g., Supplementary Materials Figure S1). The stoichiometric reaction of a variety of tertiary phosphine derivatives often facilitates the production of asymmetric square planar complexes, sometimes forming two isomers in solution. In the current study, triphenylphosphine was used as a monodentate ligand to substitute (only) one carbonyl ligand, yielding only one product due to the highly asymmetric Indol and Indoli bidentate ligands. The obtained light yellow precipitates were easily confirmed by one prime IR stretching frequency of one coordinated carbonyl at 1960 cm−1 and the phosphine resonance frequency of 40.9 ppm, confirming the product to be that of the [RhI(Indoli)(CO)(PPh3)] (A2) complex (see Scheme 2, Figure 2 and Figure 3). The other product and reactant complexes (A3, B1–B3) have been successfully synthesized and confirmed as characterized by FT-IR, NMR, UV/Vis, and elemental analysis.
3.2. X-ray Crystallography
3.2.1. Comparison of B2a Data with the Literature
The surprising and novel dinuclear configuration, showing the Indol ligand to act ambidentately as a bridging entity, is very rare, only being identified once in the past for a completely different functionalized thiourea-type bidentate ligand [32].
The Indole ligand coordinates with metal ions as a bidentate ligand via the carboxylate oxygen and pyrrole nitrogen atoms to form a five-membered metallocycle, as well as a monodentate ligand coordinated with the oxygen atom [33,34,35]. There are a few crystal-structural reports of Indole-2-carboxylic acid ligands coordinated with different metal complexes in the literature (nine structures) [36], but not with Rh(I), and all these structures contain Indole-2-carboxylic acid coordinated as a monodentate ligand (only in this study is it coordinated as a bidentate as well as a monodentate ligand). The dianion formed by the double deprotonation of the Indole-2-carboxylic acid (Indol2−) ligand displays an unprecedented and unexpected mode of coordination with Rh(I) by forming a bridging ligand to two rhodium centers [complex B2, Figure 1a].
The dinuclear complex is simplified as [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2), where the Rh1 center exhibits the expected κ2N,O coordination by the Indole-2-carboxylate ligand. However, although N,O atoms bind to the Rh1 atom in the more familiar bidentate κ2N,O fashion, the carboxylate keto-oxygen atom coordinated (κO’) to the Rh2 atom. Hence, the Rh2 center can be considered as forming a ‘Rh(I) Vaska type analogue’ of the common type trans-[Rh2(PPh3)2(CO)(R)], where R = Cl− in the typical Ir(I) complex [37].
Selected bond distances and angles for [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2]-∙(CH3COCH3) (B2a) are given in Table 2 and Table 3 and Table S1 (Supplementary Materials). Kemp et al. [32] had determined the structure of a complex similar to B2, i.e., [Rh(nbnpt)(CO)(PPh3)Rh(CO)(PPh3)2]∙(CH3COCH3), which differs with respect to its bidentate ligand, utilizing N-benzoyl-N’-phenylthiourea, nbnptH [32]. Table S1 presents some crystallographic data of the two structures.
It is clear from Table 2 that there is a distortion from the ideal square planar geometry in complex B2, as well as in the complex published by Kemp et al. as evidenced by the angles around the coordination polyhedron, which differ from 90° and 180°. The P2-Rh2-P3 bond angle of 171.78(4)° for complex B2 and the 174.78(1)° for the complex described by Kemp et al. are both smaller than 180°. The Rh2-P2 and Rh2-P3 bond distances of 2.3356(1) and 2.3419(1) Å for complex B2, and the 2.328(3) Å and 2.330(3) Å, respectively, for the complex by Kemp et al., are relatively long, but normal for a trans-P-Rh-P moiety [8]. The Rh1-P1 bond distance of 2.2623(1) Å in B2 is slightly shorter than the corresponding distance of 2.275(3) Å for the complex described by Kemp et al.
Interestingly, the H14–H94 distance between the outermost H-atoms on the periphery of B2 (and upon including the H-atom radii) is >1.7 nm and indicates that 2B is a ‘molecular nanomaterial’.
3.2.2. Comparison of B5a Data with the Literature
The structure of [Rh(CO)(I)(PPh3)2] (B5) has previously been reported in a study by Basson et al. [38] at 298 K, without the acetone solvate. Thus, B5a as isolated in this study is a polymorph of Basson’s structure. A comparison between the structure of this study’s complex B5 and the structure by Basson et al. is given in Table S2 (Supplementary Materials).
A distortion from the ideal square planar geometry was observed in complex B5, as evidenced by the small I-Rh-P angle of 87.59(3)°, C01-Rh-P of 85.99(18)°, and the I-Rh-C01 angle of 176.2(2)°.
Rh-P1 and Rh-P1i have the same bond distance of 2.3178(9) Å (i refers to the symmetry-generated atom (1−x, 1−y, and 1−z) in complex B5), while for the complex published by Basson et al., 2.336 (2) Å and 2.316 (2) Å, respectively, were reported. These distances are relatively longer than those of the [Rh(L,L’-Bid)(CO)(PPh3)] complexes [39], (where L,L’-Bid = different monocharged bidentate ligands) but normal for a trans-P-Rh-P moiety [4,40]. Nevertheless, the Rh-C bond distances of 1.725(5) Å for complex B5, as well as the C=O bond of 1.00(1) Å (shorter; overshadowed by the massive iodide), are in reasonable agreement with the bond distances of related compounds [7,41]. The Rh-I bond distances of 2.7103(7) Å for complex B5 and 2.683(1) Å for Basson et al.’s complex are in good agreement with the related literature complex [42].
Basson et al. showed that the Rh complex crystallized in the monoclinic P21/n system (Z = 4) and did so without a disorder along the I-Rh-CO axis [38]. The current study determined the space group of the structure of B5 as monoclinic P21/c with a disorder along the I-Rh-CO axis. The cell parameters were different to those reported by Basson et al. and may be attributed to the non-standard P21/n space group and/or due to the high temperature at which the complex published by Basson et al. was collected (ambient temperature). As indicated, B5a is, therefore, a polymorph of that published by Basson et al. [38].
Crucially, B5a was obtained directly from the oxidative addition of iodomethane to B2, albeit after >24 h, which is clearly confirmed as (i) is a subsequent product of the reaction and since (ii) the Rh2(I)-Vaska type fragment in B2 undergoes substitution, but not oxidative addition, even after an extended period.
3.3. Spectroscopic Study
3.3.1. [Rh(Indoli)(CO)(PPh3)] (A2)
As indicated in Section 2, illustrated in Figure 2 and Figure 3, and summarized in Figure 6 and Table 4, typical first-order fits to Equation (1) for the time-resolved IR and 31P{1H} NMR data were obtained and allowed for a detailed analysis of the oxidative addition to A2. The kinetics are straight-forward, as previously observed in a number of other analogous Rh(I) systems in the literature [4,7,11,13,23], and will be further discussed in context in Section 3.4.
3.3.2. [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2)
The IR data show that the reaction consists of a fairly rapid oxidative addition step on the [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) complex to form a Rh1(III)-alkyl species (Figure 4; kinetic traces in Figure 7). Figure 4 adequately illustrates how the oxidative addition of iodomethane proceeds, and it is observed that the total dynamics are clearly different from those exhibited by A2.
Firstly, the IR vibrations corresponding to both the Rh1 and the Rh2 fragments’ decrease ((a) ν(Rh1)CO = 1967 cm−1: completely disappearing; (b) ν(Rh2)CO = 1975–1980 cm−1: shifting only a few cm−1 to a higher wavenumber).
Secondly, this coincides exactly with the appearance of a Rh(III)-alkyl fragment (ν(RhIII-alkyl)CO = 2060 cm−1). The largest effect is observed on the Rh1-site, while the change in absorbance in the Rh2(I)-fragment is much less pronounced.
The observations in the IR spectra (Figure 4) are exactly mimicked in the 31P{1H} spectra (Figure 5), and the entire process can be summarized as follows:
- A fairly rapid oxidative addition equilibrium step causes the Rh1(I) fragment to form an oxidized RhIII-alkyl entity at the Rh1(N,O-Indol) part, i.e., [Rh1(N,O-Indol)(CO)(PPh3)(Me)(I)] (B3).
- This is usually followed by the much slower carbonyl insertion still at Rh1, i.e., the conversion of the corresponding Rh1(III)-alkyl fragment to a corresponding [Rh1(N,O-Idol)(COMe)-(PPh3)(I)] (B5) Rh1(III)-acyl species. However, this is not observed on the time scale utilized in the current study.
- The Rh2(I)-Vaska-type fragment, on the other hand, is cleaved off, and does not undergo appreciable oxidative addition, as shown in the literature [43]. Additional evidence of the effect wherein the Rh2(I) fragment does not undergo appreciable oxidative addition comes from the fact that the corresponding trans-[Rh(PPh3)2(I)(CO)] (B5) has been isolated from the final solution even after 1 week (see Section 4.3.7).
- There is some uncertainty as to exactly when the Rh2(I)-Vaska type fragment is cleaved off, since the product Rh(III)-alkyl (B3 in Scheme 2 and Scheme 3) with a signal at δ = 27.2 ppm (d, 1J(Rh-P) = 122 Hz) is close yet slightly different from the final signal of B5 at δ 28.9 ppm (d, 1J(Rh-P) = 126.8 Hz), although the integral value of the Rh(III)-alkyl is close to three equivalents of PPh3 (see the 31P NMR traces in Figure 6 pointing to an average, potentially dynamic equilibrium that might exist between the three different PPh3 sites in the ‘Rh(III)-alkyl’ (B3) species.
3.4. Detailed Kinetic Study: Variation of [MeI] and Temperature
Once the dynamics within B2 had been properly defined, as given in Section 3.3, the oxidative addition of iodomethane to [Rh(Indoli)(CO)(PPh3)2] (A2) and [Rh1(Indol’)(CO)(PPh3)-Rh2(CO)(PPh3)2] (B2) could be further reliably analyzed in more detail.
Both complexes (that is, A2 as such, and then the Rh1(I) fragment of B2) essentially follow the same simplified pathway as illustrated in Scheme 2 and as confirmed by FT-IR, 31P{1H} NMR, and UV/Vis spectroscopy. However, as indicated, migratory insertion (Rh(III)-acyl formation) was detected for neither A2 or B2.
UV/vis spectroscopy requires the use of far fewer of the reactants A2 and B2 and thus enables a more extended variation of the different experimental parameters; consequently, it was primarily used for the [MeI] variation and temperature study. The observed pseudo first-order rate constant data (absorbance vs. time), used to determine the effect of [MeI] and temperature on the oxidative addition reaction, as well as the second order rate constants for the oxidative addition k1, were fitted to Equations (1)–(3) (Section 4.2).
3.4.1. [Rh(Indoli)(CO)(PPh3)] (A2)
The oxidative addition of iodomethane to Rh(I) complexes frequently progresses as an equilibrium reaction, as presented in Scheme 3, wherein the rate of Rh(III)-alkyl species formation is dependent on the relative concentration of MeI. For example, the oxidative addition of [MeI] to the [Rh(acac)(CO)(PR3)] (acacH = general β-diketones and analogs, with PR3 different tertiary phosphine ligands) complexes regularly leads to linear relationship plots with non-zero intercepts [8,11,12,13,14].
In the current study, Figure 8 illustrates a similar typical linear relationship plot of the observed pseudo first-order rate constant against [MeI], corresponding to the formation of Rh(III)-alkyl species. The solid lines in Figure 8 represent the least-squares fits of the kobs data versus [MeI] at three different temperatures—4.5, 14.7, and 26.1 °C—and the corresponding values obtained from a global fit [23,44]. Prominent intercepts are also observed.
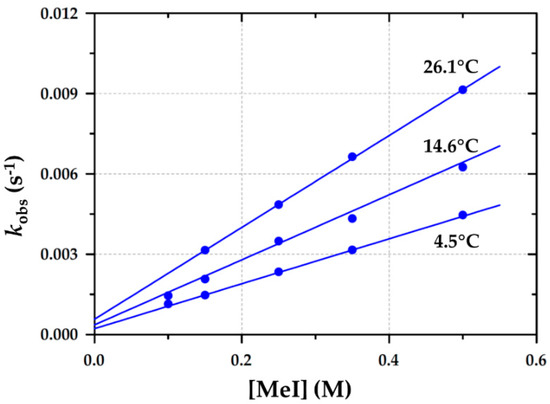
Figure 8.
Temperature and [MeI] dependence of the pseudo first-order rate constant for the formation of the RhIII-alkyl from [Rh(Indoli)(CO)(PPh3)] (A2) via time-resolved UV/vis spectroscopy in dichloromethane; (λ = 450 nm) and [A2] = 3.0 × 10−3 M.
As illustrated in Figure 8, the results are consistent with the rate expression of a simple two-step reversible reaction as given in Equation (2) (Section 4.2) and Scheme 3, which is in agreement with iodomethane’s oxidative addition to different Rh(I) systems in the literature [7,8,17].
The disappearance of A2 and the formation of the Rh(III)-alkyl product A3 was thus monitored with IR, NMR, and UV/vis spectroscopy as illustrated in Section 2.3, which gave virtually identical observed rate constants within the experimental error. The results obtained from Figure 8 as fitted to Equation (2) yielded the second-order forward rate constant, k1, and are summarized in Table 5.
Table 5.
Rate and equilibrium constants and activation parameters at different temperatures for the oxidative addition of iodomethane in dichloromethane to (a) [Rh(Indoli)(CO)(PPh3)] (A2) and (b) [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2).
If the oxidative addition to A2 proceeds to completion, it, in principle, excludes the possibility that the intercept could represent a significant reverse reaction, i.e., reductive elimination (k−1). This implies large positive values for the equilibrium constant as denoted by K1 if the Rh(III)-alkyl species is quite stable, indicating that the non-zero intercept is indicative of a solvent pathway. This is supported by Figure 3, which indicates the complete disappearance of the Rh(I) reactant (no reverse reaction).
However, the opposite is suggested by Figure 3, where there is clearly some unreacted Rh(I) at the end of the reaction, suggesting that not all the Rh(I) reactant has been consumed. This seemingly contradictory evidence left us assuming the presence of a potential solvent pathway but did not provide sufficient proof of whether the intercept indicates reductive elimination or a solvent pathway.
The experimental temperature variation study of the oxidative addition of iodomethane to A2 (as illustrated in Figure 8) enabled the determination of the activation parameters (Table 5) from which an Eyring plot [11] was constructed (Supplementary Materials: Figure S18) or via a global fit of all the data points in Figure 8 [23,44].
3.4.2. [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2)
UV/Vis spectroscopy was further used to analyze the oxidative addition of iodomethane to B2 and produced virtually identical results to those found by IR and NMR spectroscopy in dichloromethane at 25 °C (Figure 7 and Table 4). The temperature dependence of the reaction is illustrated in Figure 9, while the rate constants obtained are reported Table 5.
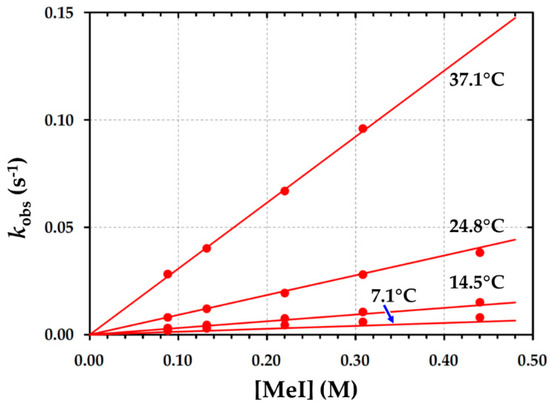
Figure 9.
Temperature and [MeI] dependence of the pseudo first-order rate constant for the formation of RhIII-alkyl in [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) via time-resolved UV/vis spectroscopy in dichloromethane; (λ = 450 nm) and [B2] = 8.35 × 10−4 M.
This complex follows the same mechanism for oxidative addition as described in Scheme 3, and forms only one major isomer, B3, negating any formation of isomerization Rh(III)-acyl products during the oxidative addition of iodomethane. Still, the formation of B5 took place as indicated, but did not prevent the detailed study of the oxidative addition of iodomethane at the Rh1(I) site in B2.
The small intercept value (near zero) of k−1 suggests that neither reductive elimination nor a solvent pathway are operative in the overarching oxidative addition of iodomethane to B2. Therefore, this implies a large value for the equilibrium (K1 = k1/k−1), which significantly favors a forward reaction. As a result of some spectral cross-interference in the Rh(I) and RhIII-alkyl signals, a combined effect was observed at a high concentration of iodomethane, which, in turn, led to low absorption readings at advanced reaction times. The equilibrium constant (K1) was not determined precisely because of the uncertainty in most of the values of k−1. A reasonable estimate, however, is that K1 > 100 M−1, which is typical of less steric complexes and more readily accessible by the entering MeI.
The experimental temperature variation study of the oxidative addition of iodomethane to B2, as illustrated in Figure 9, again enabled the determination of the activation parameters from which an Eyring plot could be constructed [11] (Supplementary Material: Figure S19) or via a global fit of the data in Figure 9 [23,44]. As given in Table 5, the current iodomethane oxidative addition study yielded a significantly smaller negative activation entropy [ΔH≠ = (73.0 ± 1.2) kJ mol−1 and ΔS≠ = (−21 ± 4) J K−1mol−1], wherein the ΔS≠ contribution to the free energy of activation, ΔG≠, at 25 °C is only ca. 10%. This might be due to the bulkiness of B2, which results in a more complex transition state and may be interpreted as indicating a more interchange-associated type of mechanism.
3.4.3. Kinetic Correlation of [Rh1(Indoli)(CO)(PPh3)] (A2) with [Rh1(Indol’)(CO)(PPh3)-Rh2(CO)(PPh3)2] (B2)
The oxidative addition of iodomethane to both A2 and B2 shows a direct conversion of the rhodium(I) starting material to the rhodium(III) alkyl species as the final product, without suggesting a Rh(III)-acyl species as a final product. Thus, a migratory CO-insertion of the coordinated methyl into the Rh-CO bond, even after prolonged experimental time frames > 24 h, is not favored, as indicated by the IR and 31P NMR data.
Table 5 illustrates the activation parameters obtained from the Eyring equation as well as a global fit of all the kobs vs. [MeI] vs. Temperature data to the exponential form of the Eyring equation. It presents the forward second-order rate constant k1 for both A2 and B2 at different temperatures, as well as the free energy of activation, ΔG≠, at 25 °C.
It is evident that a slower reaction is observed for A2 when compared to B2, wherein a larger contribution of the activation entropy [ΔH≠ = (21 ± 1) kJ mol−1 and ΔS≠ = (−209 ± 4) J K−1mol−1] to the free energy of activation, ΔG≠, is obtained (the contribution of ΔS≠ to ΔG≠ of ca. 75%) [45]. The significant negative value of ΔS≠ for A2 is indicative of an associative mechanism for the oxidative addition step and, subsequently, an effective entropy-controlled reaction, as found in many [Rh(L,L’-Bid)(CO)(PPh3)] complexes [4,7,8,9,23].
Although the oxidative addition of iodomethane to [Rh1(Indol’)(CO)(PPh3)-Rh2(CO)(PPh3)2] (B2) is expected to be influenced by the increased complexity within the molecule and potentially the added large steric effect augmented by the two trans triphenylphosphine ligands coordinated to Rh2, the rate of the reaction for B2 is actually ca. five times faster at 25 °C than A2. Thus, the net electronic effect of the three groups of triphenylphosphine surrounding the two rhodium metals seems to overshadow the steric effect. It also yielded a significantly smaller negative activation entropy [ΔH≠ = (73.0 ± 1.2) kJ mol−1 and ΔS≠ = (−21 ± 4) J K−1mol−1], wherein the contribution of ΔS≠ to the free energy of activation, ΔG≠, at 25 °C was only ca. 10%. This is contrary to the entropy-driven oxidative addition of iodomethane to A2 (ΔS≠ contribution to ΔG≠ of ca. 75%) (see Table 5). The literature assumes typical normal associative processes for the oxidative addition of iodomethane to these types of complexes and displays large negative values for ΔS≠ [4,7,10,11].
These seemingly different results regarding the processes applied to the Indoline-2-carboxylate and Indole-2-carboxylate bidentate ligands explored in this study, although structurally very similar, are very interesting, notable, and illustrate the fact that small changes in ligand systems (main group elements-based) may induce significant effects in the behavior of transition metals—in this case, rhodium(I).
4. Materials and Methods
4.1. General Procedure and Instrumentation
All chemical reagents used for the syntheses were of analytical grade and obtained from Sigma-Aldrich, South Africa. Solvents were of analytical grade and were used without additional purification, and all syntheses were performed under aerobic conditions. 1H, 13C{1H}, and 31P{1H} FT-NMR solution state spectra for the complexes were recorded at 25 °C on a Bruker Avance II 600 MHz spectrometer (1H = 600.28 MHz, 13C = 150.95 MHz, and 31P = 242.99 MHz), Bruker Avance III 400 MHz spectrometer (1H = 400.13 MHz,13C = 100.61 MHz, and 31P = 161.97 MHz), and Bruker Fourier NMR 300 MHz spectrometer (1H = 300.18 MHz, and 13C = 75.48 MHz). Chemical shifts are reported in ppm and coupling constants in Hz. For all complexes, the deuterated solvents used for analysis are mentioned with the spectral results. A Bruker Digilab FTS 2000 Fourier-transform spectrometer (ATR) was used to record all infrared spectra of complexes in the range 600–3000 cm−1.
A Bruker ApexII 4K CCD diffractometer was used for crystal data collection via Mo Κα (0.71073 Å) and ω-scans at 100(2) K. All reflections were merged and integrated with SAINT-PLUS [46], and SADABS [46] was used to correct Lorentz, polarization, and absorption effects. The heavy atom method was used to determine the structures and was refined through full-matrix least squares cycles using SHELX-97 [47] as part of the WinGX [48] package, with Σ(‖Foǀ − ǀFc‖)2 and Olex2 [49] being minimized. All non-H atoms were refined with anisotropic displacement parameters, while H atoms were constrained to parent atom sites using a riding model (aromatic C–H = 0.95 Å; aliphatic C–H = 0.98 Å). The graphics were created using DIAMOND Visual Crystal Structure Information System software [50] with 50% probability displacement ellipsoids for non-hydrogen atoms. Elemental analyses for CHN were performed by Atlantic Microlab Inc., 6180 Atlantic Blvd., Suite M, Norcross, GA 30071 (USA).
All reagents used for the syntheses and characterization were of analytical grade and were purchased from Sigma-Aldrich, South Africa. Some reagents were used as received without further purification. All organic solvents were purified and dried according to accepted procedures.
4.2. Kinetic Measurements
All experiments were performed under an air atmosphere, and only pure organic solvents were used for the kinetic measurements. All 31P NMR spectra were collected on a Bruker Avance III 400 MHz NMR spectrometer in CD2Cl2 at 161.97 MHz. For the collection of data, a pre-selected number of scans were applied to ensure quantitative spectra, (typically 12–84 scans) using a 90° pulse for 31P (25 μs), decoupling 90° pulse for 1H (8 μs), 64 k data points, delay time of 2 s, and broadband H-decoupling. 31P chemical shifts are reported relative to 85% H3PO4 (0 ppm); positive shifts are downfield. The kinetic data reported were calibrated using CD2Cl2 as standard. The FT-IR spectra were recorded as liquid samples in dry organic solvents (DCM) in a NaCl cell on a Bruker Tensor 27 spectrometer in the range of 3000–600 cm−1, equipped with a temperature cell regulator accurate within 0.3 °C. UV/Vis absorbance spectra were collected in 1.000 ± 0.001 cm tandem quartz cuvettes on a Varian Cary 50 Conc spectrophotometer, equipped with a temperature cell regulator, accurate within 0.1 °C.
The kinetic data were analyzed with the Scientist software package [51] by fitting the UV/vis-absorbance/NMR/IR data to the first-order exponential given in Equation (1), as reported previously [24,25,52] based on the simple reaction shown in Scheme 3.
The kobs values were calculated from plots of absorbance against time graphs using the following first-order equation with parameters Aobs, A0, A∞, and t as the overall observed absorbance, initial absorbance at the start of the reaction, final absorbance, and time, respectively.
Aobs = A∞ − (A∞ – A0)e(−kobs. t)
The oxidative addition steps were studied using different iodomethane concentrations at different temperatures. The observed pseudo first-order rate constant data were plotted against [MeI] in order to determine the rate constant for oxidative addition k1 and reductive elimination or solvent pathway as intercept k−1, fitted to Equation (2) [24,25,52].
kobs = k1[MeI] + k−1
Under equilibrium conditions and in absence of an appreciable solvent pathway, the equilibrium constant for the reaction in Scheme 3 is given by Equation (3).
K1 = k1/k−1
The Indoline and Indole Rh(I) systems [A2 and B2] were studied at different temperatures to determine the effect of temperature on the iodomethane oxidative addition reaction using the Eyring equation (Equation (4); logarithmic form [53,54,55,56,57] and Equation (5); exponential form (for global fit) [23,44,58]).
ln(k1/T) = ln(k/h) – ΔH≠/RT + ΔS≠/R
k1= (kBT/h)·e(ΔS≠/R)·e(−ΔH≠/RT)
4.3. Synthesis
4.3.1. Synthesis of Dicarbonyl(Indoline-2-Carboxylato-κO,N)rhodium(I), [Rh(Indoli)(CO)2] (A1)
RhCl3∙xH2O (0.100 g; 0.380 mmol) was dissolved in 5 cm3 of DMF and refluxed for 30 min. The solution was cooled to room temperature. [Rh(Indoli)(CO)2] was synthesized by addition of Indoline-2-carboxylic acid (A; 0.093 g and 0.570 mmol) to the cooled solution. An equivalent amount (0.0470 g; 0.570 mmol) of sodium acetate was added for deprotonation. A light-yellow precipitate was formed after the addition of ice water. The precipitate was filtered off and dried (Yield: 0.070 g; 59.0%). IR: νCO 2083, and 1987 cm−1. 1H NMR (400 MHz, dimethyl sulfoxide-d6): 8.88 (d, J = 53.4 Hz, 1H), 7.37 – 7.23 (m, 2H), 7.17 (d, J = 19.7 Hz, 1H), 4.24 (s, 1H), and 3.33 (s, 2H). 13C{1H} NMR (101 MHz, dichloromethane-d6): 188.9 (m), 181.9 (m), 180.6 (s), 148.2 (w), 146.1 (s), 133.1 (d, 1J = 30 Hz),128.3 (s), 126.2 (d, 1J = 30 Hz), 118.4 (d, 1J = 30 Hz), 62.1 (s), and 35.0 (m).
4.3.2. Synthesis of carbonyl(indoline-2-caboxylato-κO,N)(triphenylphosphine-κP)-rhodium(I), [Rh(Indoli)(CO)(PPh3)] (A2)
[Rh(Indoli)(CO)(PPh3)2] was synthesized by dissolving [Rh(Indoline)(CO)2] (0.062 g, 0.193 mmol) in 5 cm3 of acetone. Triphenylphosphine (PPh3) (0.050 g and 0.193 mmol) was added to the solution with stirring applied, resulting in the immediate evolution of CO gas. After a few minutes, the reaction was completed, as determined by IR spectroscopy. Ice water was added dropwise to the solution. A yellow precipitate was filtered off and dried. (Yield: 0.077 g; 61.0%). IR: νCO 1960 cm−1. 1H NMR (300 MHz, dichloromethane-d2): 7.80 – 7.37 (m, 15H), 7.22 (d, 1J = 26.2 Hz, 2H), 6.33 (t, 1H), 4.46 (t, 1H), 3.50 (d, 1J = 23.6 Hz, 2H), and 2.19 (t, 1J = 21.2 Hz, 1H). 13C{1H} NMR (101 MHz; acetonitrile-d6): 190.2 (m), 179.2 (s), 145.0 (w), 140.5 (s), 134.3 (s), 132.6 (s), 131.9 (s), 130.3 (s), 128.5 (s) 124.5 (s), 120.0 (s), 116.3 (d, 1J = 50 Hz), 103.3 (s), 101.3 (s), 61.1 (m), and 32.5 (m). 31P{1H} NMR (243 MHz, dichloromethane-d2): 40.94 (d, 1J = 167.6 Hz).
4.3.3. In Situ Synthesis/Characterization of carbonyl(indoline-2-caboxylato-κO,N)(iodido)(methyl)(triphenylphosphine-κP)-rhodium(III)] [Rh(Indoli)(I)(Me)(CO)(PPh3)] (A3)
Typical in situ characterization of the Rh(III)-alkyl species was primarily performed by 31P NMR and Infrared spectroscopy in dichloromethane as follows: [MeI] = 0.10 M was added to a solution of A2 (0.010 M) in dichloromethane at ca. 25 °C. Successive infrared spectra illustrating the oxidative addition of iodomethane to [Rh1(Indoli)(CO)(PPh3)] (A2) indicated the disappearance of the signal at ν(CO) = 1976 cm−1 and yielded the Rh(III)-alkyl, [Rh(Indoli)(I)(Me)(CO)(PPh3)] (A3) (ν(CO) = 2054 cm−1; Figure 2).
A similar solution when monitored by 31P{1H} NMR spectroscopy showed the synchronized disappearance of A2 (δ[Rh(I)] = 41.9 ppm; Figure 3) and appearance of [Rh(Idoli)(I)(Me)-(CO)(PPh3)] (A3) (δ[Rh(III)-alkyl] = 27.4 ppm).
4.3.4. Synthesis of dicarbonyl(Indole-2-carboxylato-κO,N)rhodium(I), [Rh(Indol)(CO)2] (B1)
RhCl3∙xH2O (0.0500 g; 0.1898 mmol) was dissolved in 5 cm3 of DMF and refluxed for 30 min. The solution was cooled to room temperature. Indol-2-carboxylic acid (B; 0.037 g and 0.228 mmol) was added to the cooled solution. An equivalent amount (0.0187 g; 0.2277 mmol) of sodium acetate was added for deprotonation. A pink precipitate was formed after the addition of ice water. The precipitate was filtered off and dried. (Yield: 0.0415 g; 68.60%). IR: νCO 2075 and 1995 cm−1. 1H NMR (300 MHz; acetone-d6): 7.99 (s, 1H), 7.53 (d, 1J = 7.5 Hz, 1H), 7.44 (d, 1J = 8.1 Hz, 1H), 7.13 (t, 1H), 6.90 (t, 1H). 13C{1H} NMR (75 MHz, acetone-d6): 191.1 (w), 190.1 (s), 176.0 (s), 138.2(s), 131.4 (m), 129.2 (m), 123.5 (s), 119.2(s), 116.1(s), 108.6 (s), and 105.6 (m). Anal. Calcd for C11H11NO7Rh (%): C, 35.50; H, 2.98; N, 3.76. Found: C, 35.15; H, 3.09; N, 6.03.
4.3.5. Synthesis of [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2)
The dinuclear species [Rh(Indol’)(CO)(PPh3)Rh(CO)(PPh3)2] (B2) was synthesized by dissolving [Rh(Indol)(CO)2] (0.0412g; 0.129 mmol) in 5 cm3 of acetone. Triphenylphosphine (PPh3) (0.0339 g; 0.1292 mmol) was added to the solution with stirring, resulting in the immediate evolution of CO gas. After a few minutes, the reaction was completed as determined by IR spectroscopy. Ice water was added dropwise to the solution. A yellow precipitate was filtered off and dried. (Yield: 0.097 g; 61.8%) Yellow cuboid crystals suitable for X-ray diffraction were obtained from recrystallization in acetone and a few drops of water.
IR: νCO 1978, 1959 cm−1. 1H NMR (600 MHz, dichloromethane-d2): 7.74–7.68 (m, 1H), 7.52 (dd, 1J = 1J = 12.2, 5.8 Hz, 2H), 7.45 (d, 1J = 6.0 Hz, 2H), 7.40–7.37 (m, 1H), 7.35 (t, 1J = 7.4 Hz, 1H), 7.25 (t, 1J = 7.5 Hz, 2H), 6.98 (t, 1J = 7.4 Hz, 1H), 6.80 (t, 1J = 7.3 Hz, 1H), 6.16 (s, 1H).13C{1H} NMR (75 MHz, CDCl2): 190.4 (m), 189.6 (m), 177.9 (s), 144.3 (s), 140.3(s), 133.7 (s), 133.2 (s), 131.2 (s), 130.4 (s), 129.1 (s), 127.1 (s), 120.1 (s), 115.5 (d 1J = 76 Hz), and 102.6 (s). 31P{1H} NMR (243 MHz, dichloromethane-d2): 41.08 (d, 1J = 150.2 Hz), 30.09 (d, 1J = 132.9 Hz). Anal. Calcd for C68H56NO5P3Rh2 (%): C, 64.52; H, 4.46; N, 1.11. Found: C, 63.85; H, 4.36; N, 1.13.
4.3.6. In Situ Synthesis/Characterization of carbonyl(Indole-2-carboxylato-κO,N)(iodido)(methyl)(triphenylphosphine-κP)rhodium(I), [Rh(Indol)(I)(Me)(CO)(PPh3)] (B3)
Typical in situ characterization of the Rh(III)-akyl species was primarily performed by 31P NMR and Infrared spectroscopy in dichloromethane as follows: [MeI] = 0.064 M was added to a solution of B2 (0.010 M) in dichloromethane at ca. 25 °C. Successive infrared spectra illustrating the oxidative addition of iodomethane to [Rh1(Indol’)(CO)(PPh3)-Rh2(CO)(PPh3)2] indicated the disappearance of the signal at ν(CO) = 1967 cm−1 (representing the ‘sum’ of the two Rh fragments, Rh1(I) and Rh2(I)) and yielded the Rh(III)-alkyl, [Rh(Indol)(I)(Me)(CO)(PPh3)] (B3) (ν(CO) = 2060 cm−1; Figure 4). However, the simultaneous appearance of the Vaska fragment at 1980 cm−1 was also prominent.
A similar solution when monitored by 31P{1H} NMR spectroscopy showed the synchronized disappearance of B2 (Rh fragments: δ[Rh1(I)] = 40.1 ppm, and δ[Rh2(I)] = 30.1 ppm; Figure 5) and appearance of [Rh(Indol)(I)(Me)(CO)(PPh3)] (B3), (δ[Rh(III)-alkyl] ca. 28 ppm.
4.3.7. Synthesis of the Vaska-Type Complex trans-[Rh(CO)(I)(PPh3)2] (B5)
The Vaska-type complex trans-[Rh(CO)(I)(PPh3)2] (B5) was obtained from the kinetic reaction of iodomethane’s oxidative addition to [Rh(Indol’)(CO)(PPh3)Rh(CO)(PPh3)2] (B2). It was synthesized by dissolving B2 (0.0121 g; 0.0100 mmol) in 1 cm3 of acetone. Iodomethane (0.0227 g; 0.1600 mmol) was added to the aforementioned solution and the solution was kept for one week. (Yield: 0.0032 g; 40.8%.) Yellow cuboid crystals, suitable for X-ray diffraction, were obtained.
Trans-[Rh(CO)(I)(PPh3)2] (B5) was also independently synthesized by dissolving [Rh(CO)(Cl)(PPh3)2] (0.0050 g; 0.0072 mmol) in 1 cm3 of acetone. Tetra-n-butylammonium iodide (CH3CH2CH2CH2)4N(I) (0.0027g; 0.0072 mmol) was added to the solution with stirring applied. Yellow powder was obtained after evaporating the acetone. (Yield: 0.0039 g; 70.00%.) IR: νCO 1968 cm−1. 31P{1H} NMR (162 MHz, dichloromethane-d2) δ 28.9.0 (d, J = 126.8 Hz).
5. Conclusions
An important contribution to the coordination chemistry of rhodium(I) Indole/Indoline complexes has been presented herein. It underlines the delicate interplay between appropriately selected main group bidentate ligand systems such as Indoline-2-carboxylic acid (B, IndoliH) and Indole-2-carboxylic acid (IndolH, B) to platinum group metals, which is further manifested by a detailed study of the oxidative addition of iodomethane to rhodium(I) carbonyl triphenylphosphine complexes.
IndoliH formed [Rh(Indoli)(CO)(PPh3)] (A2), while IndolH yielded the novel dinuclear [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) complex (Indol’ = Indol2−) as characterized by SCXRD. In B2, the Rh1(I) fragment [Rh1(Indol’)(CO)(PPh3)] (bidentate N,O-Indol) exhibits a distorted square-planar geometry, while Rh2(I) shows a ‘Vaska’-type distorted square-planar trans-[O-Rh2(PPh3)2(CO)] configuration (bridging the carboxylate ‘oxo’ O atom of Indol2−).
A detailed kinetic study of the oxidative addition of iodomethane to A2 and B2 via time-resolved FT-IR, NMR, and UV/Vis spectroscopy indicated only Rh(III)-alkyl species (A3/B3) as products; thus, no migratory insertion occurred. A variable temperature kinetic study confirmed an associative mechanism for A2 via the general equilibrium-based pathway (ΔH≠ = (21 ± 1) kJ mol−1; ΔS≠ = (−209 ± 4) J K−1mol−1), with a smaller contribution from a reverse reductive elimination/solvent pathway. The dinuclear complex B2 showed MeI oxidative addition only at Rh1(I), forming a Rh(III)-alkyl, but cleaved the bridged Rh2(I) site, yielding trans-[RhI(PPh3)2(I)(CO)] (5B) as a secondary product (characterized by SCXRD).
The significantly smaller negative activation entropy [ΔH≠ = (73.0 ± 1.2) kJ mol−1; ΔS≠ = (−21 ± 4) J K−1mol−1] suggests a more complex and potential interchange mechanism for the dinuclear complex B2 (the contribution of ΔS≠ to the Gibbs free energy of activation, ΔG≠, is only ca. 10%), contrary to the entropy-driven and purely associative oxidative addition of MeI to A2 (ΔS≠ contribution to ΔG≠ ± 75%).
Finally, this study allowed for important comparisons to be drawn with similar structures from the literature and illustrated the significant chemical and geometric differences between the complexes A2 and B2. Consequently, these findings enabled further understanding of the physical and chemical behaviors of potential larger, dinuclear Rh(I) model catalysts and their structure/reactivity relationships and may prompt further investigation into their future applications as, e.g., applied catalysts.
Supplementary Materials
The complete list of supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics10120251/s1, Figure S1: ATR IR spectrum of [Rh(indoli)(CO)2] (A1); Figure S2: ATR IR spectrum of [Rh(indoli)(CO)(PPh3)] (A2); Figure S3: ATR IR spectrum of [Rh(indol)(CO)2] (B1); Figure S4: ATR IR spectrum of [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2); Figure S5: 1H NMR spectrum (DCM) of [Rh(indoli)(CO)2] (A1); Figure S6: 13C NMR spectrum of the IndoliH ligand in Acetonitrile d3; Figure S7: 13C NMR spectrum (DCM) of [Rh(indoli)(CO)2] (A1); Figure S8: 1H NMR spectrum (DCM) of [Rh(indoli)(CO)(PPh3)] (A2); Figure S9: 13C NMR spectrum (Acetone-d6) of [Rh(indoli)(CO)(PPh3)] (A2); Figure S10: 31P NMR spectrum (DCM) of [Rh(indoli)(CO)(PPh3)] (A2); Figure S11: 1H NMR spectrum (DCM) of [Rh(indol)(CO)2] (B1); Figure S12: 1H NMR spectrum (DCM) of [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2); Figure S13: 13C NMR spectrum (Acetone-d6) of IndolH; Figure S14: 13C NMR spectrum (Acetone-d6) of [Rh(indol)(CO)2] (B1); Figure S15: 13C NMR spectrum (CDCl3) of [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2); Figure S16: 31P NMR spectrum (DCM) of [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2); Figure S17: 31P NMR spectrum (DCM) of (a) trans-[RhI(CO)(PPh3)2] (B4) and (b) trans-[RhClCO)(PPh3)2]; Figure S18: Eyring plot: k1 rate constant (DCM): iodomethane oxidative addition to [Rh(indoli)(CO)(PPh3)] (A2); Figure S19: Eyring plot: OA k1 rate constant to [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2] (B2) in dichloromethane.; Table S1: Comparison of selected crystallographic data in [Rh1(Indol’)(CO)(PPh3)Rh2(CO)(PPh3)2]-·(CH3COCH3) (B2a) with an isostructural thiourea molecule reported by Kemp et al. [Ref see ms]; Table S2: Comparison of geometric parameters and crystal data of trans-[Rh(CO)(I)(PPh3)2]-·(CH3COCH3) (B5a) and similar structure found in the literature.
Author Contributions
Conceptualization, A.R. and J.A.V.; methodology, A.R. and J.A.V.; software, A.R. and O.T.A.; validation, M.A.E.E., A.R. and J.A.V.; formal analysis, M.A.E.E.; investigation, M.A.E.E.; resources, G.J.S.V., A.R. and J.A.V.; data curation, M.A.E.E.; writing—original draft preparation, M.A.E.E. and O.T.A.; writing—review and editing, M.A.E.E., A.R., J.A.V. and O.T.A.; visualization, M.A.E.E., A.R. and O.T.A.; supervision, J.A.V. and A.R.; project administration, G.J.S.V., J.A.V. and A.R.; funding acquisition, G.J.S.V., J.A.V. and A.R. All authors have read and agreed to the published version of the manuscript.
Funding
We acknowledge the University of the Free State, Department of Chemistry, the South African National Research Foundation (SA NRF) (UID 99782) for its financial support. This includes funding under the Swiss–South Africa joint research program (SSAJRP) from the SA NRF (AR: UID: 107802), the Competitive Program for Rated Researchers of the SA NRF (AR: UID 111698), and the Competitive Program for Unrated Researchers of the SA NRF (J.A.V.: 116302).
Data Availability Statement
The crystallographic data sets for compounds B2a and B5a are available as Supplementary Material and from the Cambridge Structural Data Center with the following codes: CCDC 2214777 (B2a) 2214778 (B5a). Other data and spectroscopy are available as Supplementary Information [Figures S1–S16].
Acknowledgments
Sincere thanks are also due to Linette Twigge for the NMR data analysis. Additional acknowledgement further goes to the crystallographic data collection team at the University of the Free State in Bloemfontein, South Africa.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Maitlis, P.M.; Haynes, A.; Sunley, G.J.; Howard, M.J. Methanol carbonylation revisited thirty years on. J. Chem. Soc. Dalton Trans. 1996, 2187–2196. [Google Scholar] [CrossRef]
- Bohnen, H.W.; Cornils, B. Hydroformylation of Alkenes: An Industrial View of the Status and Importance. Adv. Catal. 2002, 47, 1–64. [Google Scholar]
- van Leeuwen, P.W.N.M. Homogeneous Catalysis Understanding the Art; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; pp. 109–124. [Google Scholar]
- Roodt, A.; Visser, H.G.; Brink, A. Structure/reactivity relationships and mechanism from X-ray data and spectroscopic kinetic analysis. Cryst. Rev. 2011, 17, 241–280. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Crous, R.; Bennie, L.; Meij, A.M.; Blann, K.; Bezuidenhoudt, B.C.B.; Young, D.A.; Green, M.J.; Roodt, A. Borate Esters as Alternative Acid Promoters in the Palladium-Catalyzed Methoxycarbonylation of Ethylene. Angew. Chem. Int. Ed. 2007, 119, 2323–2325. [Google Scholar] [CrossRef]
- Jackman, K.M.K.; Liang, G.; Boyle, P.D.; Zimmerman, P.M.; Blacquiere, J.M. Changes in Ligand Coordination Mode Induce Bimetallic C-C Reductive Elimination. Dalton Trans. 2022, 51, 3977–3991. [Google Scholar] [CrossRef]
- Warsink, S.; Kotze, P.D.R.; van Rensburg, J.M.J.; Venter, J.A.; Otto, S.; Botha, E.; Roodt, A. Kinetic-Mechanistic and Solid-State Study of the Oxidative Addition and Migratory Insertion of Iodomethane to [Rhodium(S,O-BdiPT or N,O-ox)(CO)(PR1R2R3)] Complexes. Eur. J. Inorg. Chem. 2018, 2018, 3615–3625. [Google Scholar] [CrossRef]
- Roodt, A.; Otto, S.; Steyl, G. Structure and solution behaviour of rhodium(I) Vaska-type complexes for correlation of steric and electronic properties of tertiary phosphine ligands. Coord. Chem. Rev. 2003, 245, 121–137. [Google Scholar] [CrossRef]
- Basson, S.S.; Leipoldt, J.G.; Roodt, A.; Venter, J.A. Mechanism for the Oxidative Addition of Iodomethane to Carbonyl(N-hydroxy-Nitrosobenzenaminato-κ2O,O′)-triarylphosphine-rhodium(I) Complexes and Crystal Structure of [Rh(cupf)(CO)(CH)(I)(PPh3)]. Inorg. Chim. Acta 1987, 128, 31–37. [Google Scholar] [CrossRef]
- Venter, J.A.; Leipoldt, J.G.; van Eldik, R. Solvent, Temperature, and Pressure Dependence of the Oxidative Addition of Iodomethane to Complexes of the Type Rhl(β-diketonate)(CO)( PPh3). Inorg. Chem. 1991, 30, 2207–2209. [Google Scholar] [CrossRef]
- Elmakki, M.A.E.; Alexander, O.T.; Venter, G.J.S.; Venter, J.A.; Roodt, A. Synthesis and structural determination of [Rh(opo)(CO)(PR3)] complexes (opo− = 2-oxopyridin-1-olate) and in situ isomeric behavior from preliminary kinetic study of iodomethane oxidative addition. J. Coord. Chem. 2021, 74, 444–466. [Google Scholar] [CrossRef]
- Conradie, J.; Swarts, J.C. Oxidative Addition of CH3I and CO Migratory Insertion in a Series of Ferrocene-Containing Carbonyl Phosphine β-Diketonato Rhodium(I) Complexes. Organometallics 2009, 28, 1018–1026. [Google Scholar] [CrossRef]
- Conradie, J.; Lamprecht, G.J.; Roodt, A.; Swarts, J.C. Kinetic study of the oxidative addition reaction between methyl iodide and [Rh(FcCOCHCOCF3)(CO)(PPh3)]: Structure of [Rh(FcCOCHCOCF3)(CO)(PPh3)(CH3)(I)]. Polyhedron 2007, 26, 5075–5087. [Google Scholar] [CrossRef]
- Conradie, M.M.; Conradie, J. Methyl iodide oxidative addition to [Rh(acac)(CO)(PPh3)]: An experimental and theoretical study of the stereochemistry of the products and the reaction mechanism. Dalton Trans. 2011, 40, 8226–8237. [Google Scholar] [CrossRef]
- Steyn, G.J.J.; Roodt, A.; Poletaeva, I.; Varshavsky, Y.S. Structural correlation between Rh-P and Rh-C bond distances vs. 31P and 13C NMR parameters in monocarbonyl-phosphinerhodium(I) complexes: Crystal structure of (methyl 2-(amino)-1-cyclopentene-1-dithiocarboxylato-κN,κS)-carbonyl(triphenylphosphine) rhodium(I). J. Organomet. Chem. 1997, 536–537, 197–205. [Google Scholar]
- Varshavsky, Y.S.; Galding, M.R.; Cherkasova, T.G.; Podkorytov, I.S.; Nikol’skii, A.B.; Trzeciak, A.M.; Olejnik, Z.; Lis, T.; Zio´łkowski, J.J. 31P-NMR and X-ray studies of new rhodium(I) β-ketoiminato complexes Rh(R1C(O)CHC(NH)R2)(CO)(PZ3) where PZ3=PPh3, PCy3, P(OPh)3 or P(NC4H4)3. J. Organomet. Chem. 2001, 628, 195–210. [Google Scholar] [CrossRef]
- Warsink, S.; Fessha, F.G.; Purcell, W.; Venter, J.A. Synthesis and characterization of rhodium(I) 2-methylcupferrate complexes and their kinetic behaviour in iodomethane oxidative addition. J. Organomet. Chem. 2013, 726, 14–20. [Google Scholar] [CrossRef]
- Purcell, W.; Basson, S.S.; Leipoldt, J.G.; Roodt, A.; Preston, H. First structural confirmation of different geometrical isomers in the same crystal lattice: The crystal structure of benzoylacetonatocarbonyltriphenylphosphinerhodium(I). Inorg. Chim. Acta 1995, 234, 153–156. [Google Scholar] [CrossRef]
- McCleverty, J.A.; Wilkinson, G. 20. Tetracarbonyldichlorodirhodium. In Inorganic Syntheses; Angelici, R.J., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1990; Volume 28, pp. 84–86. [Google Scholar]
- Serp, P.; Hernandez, M.; Kalck, P. Dimethylformamide as a convenient CO source for the facile preparation of rhodium-, iridium- or rutheniumchlorocarbonyl complexes directly from RhCI3-3H2O, IrCI3-3H2O or RuCI3-3H2O. In Chimie des Surfaces et Catalysel/Surface Chemistry and Catalysis; Serie II; C.R. Acad. Sci.: Paris, France, 1999; pp. 267–272. [Google Scholar]
- Preston, H. Spesifieke Isomeervorming en Oksidatiewe Addisiegedrag van Rodium(I)-Tiolatokomplekse. Ph.D. Thesis, University of the Free State, Bloemfontein, South Africa, 1993. [Google Scholar]
- Roodt, A.; Leipoldt, J.G.; Helm, L.; Merbach, A.E. Equilibrium behavior and proton transfer kinetics of the dioxotetracyanometalate complexes of molybdenum(IV), tungsten(IV), technetium(V), and rhenium(V): Carbon-13 and Oxygen-17 NMR Study. Inorg. Chem. 1994, 33, 140–147. [Google Scholar] [CrossRef]
- Mokolokolo, P.P.; Brink, A.; Roodt, A.; Schutte-Smith, M. Subtle variation of stereo-electronic effects in rhodium(I) carbonyl Schiff base complexes and their iodomethane oxidative addition kinetics. J. Coord. Chem. 2020, 73, 2740–2762. [Google Scholar] [CrossRef]
- Otto, S. Structural and Reactivity Relationships in Platinum(II) and Rhodium(I) Complexes. Ph.D. Thesis, University of the Free State, Boemfontein, South Africa, 1999. [Google Scholar]
- Pretorius, C. Structural and Reactivity Study of Rhodium(I) Carbonyl Complexes as Model Nano Assemblies. Ph.D. Thesis, University of the Free State, Bloemfontein, South Africa, 2015. [Google Scholar]
- Shestakova, E.P.; Cherkasova, T.G.; Osetrova, L.V.; Varshavsky, Y.S.; Leipoldt, J.G.; Roodt, A. NMR Study of the Octahedral Rhodium(III) Complexes formed by the Iodomethane Oxidative Addition to the Rhodium(I)-Diketonato bis-triphenylphosphine Complexes. Rhodium Express 1994, 7–8, 24–29. [Google Scholar]
- Damoense, L. Fundamental Aspects of Selected Rhodium Complexes in Homogeneous Catalytic Acetic Acid Production. Ph.D. Thesis, University of the Free State, Bloemfontein, South Africa, 2001. [Google Scholar]
- Basson, S.S.; Leipoldt, J.G.; Nel, J.T. The Oxidative Addtion of Methyl Iodide to Betadiketonecarbonyltriphenylphosphine-rhodium(I) Complexes. Inorg. Chim. Acta 1984, 84, 167–172. [Google Scholar] [CrossRef]
- Goswami, K.; Singh, M.M. Di and Monocarbonyl Complexes of Rhodium(I) containing Singly Charged Bidentate Ligands. Transit. Met. Chem. 1980, 5, 83–85. [Google Scholar] [CrossRef]
- Singh, M.M.; Szafran, Z.; Pike, R.M. Microscale Chemistry and Green Chemistry: Complementary Pedagogies. J. Chem. Educ. 1999, 76, 1684–1686. [Google Scholar] [CrossRef]
- Goswami, K.; Singh, M.M. Few carbonyl oximato compounds of rhodium. J. Inorg. Nucl. Chem. 1977, 39, 1718–1719. [Google Scholar] [CrossRef]
- Kemp, G.; Roodt, A.; Purcell, W.; Koch, K.R. Unprecedented N,S,O co-ordination of the doubly deprotonated anion of N-benzoyl-N9-phenylthiourea (H2L2) bridging two rhodium(I) centres: Crystal structure of the acetone solvate of [Rh(nbnpt)(CO)(PPh3)Rh(CO)(PPh3)2].(CH3COCH3). J. Chem. Soc. Dalton Trans. 1997, 4481–4483. [Google Scholar] [CrossRef]
- Morzyk-Ociepa, B.; Michalska, D.; Pietraszko, A. Structures and vibrational spectra of Indole carboxylic acids. Part I. Indole-2-carboxylic acid. J. Mol. Struct. 2004, 688, 79–86. [Google Scholar] [CrossRef]
- Viossat, V.; Lemoine, P.; Dayan, E.; Dung, N.-H.; Viossat, B. Synthesis, crystal structure and IR spectroscopy of MnII(2-IC)2(NC)(DMSO) and [MnII(2-IC)2(phen)(H2O)]·DMA; (2-HIC, Indole-2-carboxylic acid; phen, 1,10-phenanthroline; NC, 2,9-dimethyl-1,10-phenanthroline; DMSO, dimethyl sulfoxide; DMA, dimethyl acetamide); catalysts for the disproportionation of hydrogen peroxide. Polyhedron 2003, 22, 1461–1470. [Google Scholar]
- Li, Y.X.; Zhang, B.S.; Zheng, M. Bis(2,2′-bipyridine-κ2N,N′)bis-(1H-Indole-2-carboxyl-ato- κ2O,O)cadmium-2,2′-bipyridine-water (1/0.5/2). Acta Crystallogr. Sect. E Struct. Rep. 2011, 67, m879–m880. [Google Scholar] [CrossRef]
- Allen, F.H. (Ed.) Cambridge Structural Database (CSD), Version 5.38, November 2016 Update. In Acta Crystallographica; Wiley Online Library: Hoboken, NJ, USA, 2002; pp. 380–388. [Google Scholar]
- Kemp, G.; Roodt, A.; Purcell, W. A New Crystalline Form of the Rhodium(I) Analogue of Vaska’s Complex: Crystal Structure of trans-Chlorocarbonyl-bis(triphenylphosphine)rhodium(I). Rhodium Express 1995, 12, 21–26. [Google Scholar]
- Basson, S.S.; Leipoldt, J.G.; Roodt, A. trans-Carbonyliodo bis(triphenyl-phosphine)rhodium(I). Acta Cryst. 1990, C46, 142–143. [Google Scholar]
- Botha, L.J.; Basson, S.S.; Leipoldt, J.G. The crystal structure of thioacetylacetonatocarbonyltriphenylphosphinerhodium(I). Inorg. Chim. Acta 1987, 126, 25–28. [Google Scholar] [CrossRef]
- Koshevoy, I.O.; Sizova, O.V.; Tunik, S.P.; Lough, A.; Poë, A.J. A Novel Five-Coordinate Rhodium(I) Complex. Eur. J. Inorg. Chem. 2005, 2005, 4516–4520. [Google Scholar] [CrossRef]
- Dilworth, J.R.; Morales, D.; Zheng, Y. Rhodium and iridium complexes with thiolate and tertiary phosphine ligands. The synthesis and structures of trans-[Ir(SC6H3Cl2-2,6)(CO)(PPh3)2], [Rh2(μ-SC6H3Pri3-2,4,6)2(CO)2(PPh3)2], [Rh2H2(μ-SC6H4PPh2-2)2(CO)2(PPh3)2][BF4]2, and [Rh2I6(MeSC6H4PPh2-2)2]. J. Chem. Soc. Dalton Trans. 2000, 17, 3007–3015. [Google Scholar] [CrossRef]
- Pruchnik, F.P.; Smoleñski, P.; Gałdecka, E.; Gałdecki, Z. New water-soluble rhodium(I) complexes containing 1-methyl-1-azonia-3,5-diaza-7-phosphaadamantane iodide. New J. Chem. 1998, 22, 1395–1398. [Google Scholar] [CrossRef]
- Otto, S.; Mzamane, S.N.; Roodt, A. Tuning rhodium(I) metal center accessibility in iodomethane oxidative addition to Vaska-type complexes by interchanging tertiary phosphine for arsine and stibine. In Legacy of Joseph Chatt; Winterton, N., Leigh, G., Eds.; Royal Society of Chemistry: Cambridge, UK, 2001; pp. 675–683. [Google Scholar]
- Schutte-Smith, M.; Roodt, A.; Visser, H.G. Ambient and high-pressure kinetic investigation of methanol substitution in fac-[Re(Trop)(CO)3(MeOH)] by different monodentate nucleophiles. Dalton Trans. 2019, 48, 9984–9997. [Google Scholar] [CrossRef]
- Otto, S.; Roodt, A.; Elding, L.I. Bridge-splitting kinetics, equilibria and structures of trans-bis-cyclooctene complexes of platinum(II). Dalton Trans. 2003, 2519–2525. [Google Scholar] [CrossRef]
- Bruker SAINT-Plus (Version 7.12) and SADABS, Bruker AXS Inc., Madison: Fitchburg, WI, USA, 2004.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Brandenburg, K. DIAMOND. Visual Crystal Structure Information System; Version 3.0c Crystal Impact; IUCr: Bonn, Germany, 2005. [Google Scholar]
- Scientist for Windows (32). Version 2.01: 1986-1995. Micromath Inc.: Salt Lake City, UT, USA.
- Romeo, R.; Arena, G.; Scolara, L.M.; Plutino, M.R. Ion-pair mechanism in square planar substitution. Reactivity of cationic platinum (II) complexes with negatively charged nucleophiles in solvents of high, medium and low polarity. Inorg. Chim. Acta 1995, 240, 81–92. [Google Scholar] [CrossRef]
- Purcell, W.; Roodt, A.; Basson, S.S.; Leipoldt, J.G. Kinetic study of the reaction between trans-tetracyanodioxorhenate(V) and Thiocyanate ions. Transit. Met. Chem. 1989, 14, 224–226. [Google Scholar] [CrossRef]
- Leipoldt, J.G.; Basson, S.S.; Roodt, A.; Potgieter, I.M. Kinetics and mechanism of fluoride ion substitution in trans-dioxotetracyanotungstate(IV) ions and the crystal structure of K3WOF(CN)4. S. Afr. J. Chem. 1986, 39, 179–183. [Google Scholar]
- Leipoldt, J.G.; Van Eldik, R.; Basson, S.S.; Roodt, A. Kinetics and mechanism of the reaction between trans-dioxotetracyanotungstate (IV) and azide in aqueous solution. Inorg. Chem. 1986, 25, 4639–4642. [Google Scholar] [CrossRef]
- Smit, J.P.; Purcell, W.; Roodt, A.; Leipoldt, J.G. Kinetics of the substitution reaction between aquaoxotetracyanomolybdate (IV) and cyanide/hydrogen cyanide. Polyhedron 1993, 12, 2271–2277. [Google Scholar] [CrossRef]
- Muller, A.J.; Otto, S.; Roodt, A. Rapid phosphorus (III) ligand evaluation utilising potassium selenocyanate. Dalton Trans. 2008, 650–657. [Google Scholar] [CrossRef]
- Atkins, P.; Paula, J.D. Physical Chemistry, 8th ed.; Oxford University Press: Oxford, UK, 2006; pp. 880–883. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).