Abstract
Phosphinines and donor-substituted phosphinines are of recent interest due to their use in homogeneous catalysis. In this article, a Pd(II) bis(phosphinine) complex was characterised and phosphorus–selenium coupling constants were used to assess the donor properties of the diphenylphosphine substituents of phosphinine ligands to promote their further use in catalysis. The selenation of 2,5-bis(diphenylphosphino)-3,6-dimethylphosphinine (5) and 2-diphenylphosphino-3-methyl-6-trimethylsilylphosphinine (6) gave the corresponding phosphine selenides 8 and 9, respectively, leaving the phosphinine ring intact. Multinuclear NMR spectroscopy, mass spectrometry and single crystal X-ray diffraction confirmed the oxidation of all the diphenylphosphine substituents with 1JP-Se coupling constants determined to be similar to SePPh3, indicating that the phosphinine rings were electronically similar to phenyl substituents. Solutions of 6 were found to react with oxygen slowly to produce the phosphine oxide 10 along with other by-products. The reaction of [bis{3-methyl-6-(trimethylsilyl)phosphinine-2-yl}dimethylsilane] (4) with [PdCl2(COD)] gave the chelating dichloropalladium(II) complex, as determined by multinuclear NMR spectroscopy, mass spectrometry and an elemental analysis. The molecular structure of the intermediate 2 in the formation of 4,6-di(tert-butyl)-1,3,2-diazaphosphinine (3) was also determined, which confirmed the structure of the diazaphosphacycle P(Cl){N=C(tBu)CH=C(tBu)-N(H)}.
1. Introduction
Phosphinines are the phosphorus analogue of pyridine. The initial discovery of 2,4,6-triphenylphosphinine by Märkl [1] and the parent, unsubstituted phosphinine by Ashe [2] are now landmarks in low-coordinate main group chemistry and continue to inspire new research in the field [3,4]. Recent developments have focused on the use of phosphinines as ligands [5,6,7,8,9,10], particularly in homogeneous catalysis [11,12,13,14,15] where they show interesting differences to conventional phosphine ligands. Examples from our group include the use of phosphinophosphinine complexes as small bite-angle ligands [16] for Ru-catalysed transfer hydrogenation, alcohol upgrading and acceptorless dehydrogenation reactions [17,18] as well as Cr catalysts for ethylene oligomerisation reactions producing unusual alkyl- and alkenylcyclopentane products [19] and Rh complexes for the hydroboration of carbonyls [20]. Others researchers have demonstrated the exceptional behaviour of Rh-phosphinine complexes in hydroformylation [21,22] and hydrogenation reactions [23], Au-phosphinine catalysts for cycloisomerisations [24] and cyclometalated iridium(III)–phosphinine precatalysts for water oxidation reactions [25] among many others [5,11].
The oxidation of phosphines with sulphur and selenium is well-known [26,27] with phosphine selenides being particularly useful in determining the electronic properties of the parent phosphines via the measurement of phosphorus–selenium coupling constants [28,29,30]. With respect to phosphinine chemistry, oxidation with sulfur and selenium is usually difficult with phosphinine sulfides initially only tentatively characterised as intermediates in the reaction of S with 2-Ph-4,5-Me2PC5H2 (via trapping reactions with 2,3-dimethylbutadiene) and S with 2,3-(PPh2)2-6-PhPC5H (ca. 20% yield before a continued reaction led to decomposition), principally identified by 31P NMR spectroscopy [31,32]. The analogous reaction with Se gave only weak evidence of a phosphinine selenide [31]. The first well-characterised phosphinine sulfides were formed from the reactions of 2,6-R2-3,5-Ph2PC5H (R = Ph, SiMe3) with S in toluene at 90 °C for 5–7 days and revealed 31P chemical shifts (159 and 194 ppm, respectively) greatly reduced from the starting materials (206 and 269 ppm, respectively) [33]. Their molecular structures were determined by X-ray crystallography; P=S bond lengths (1.916(1) and 1.929(1) Å, respectively) shorter than those in S=PPh3 (1.952 Å) were observed [33]. Additional phosphinine sulfides were observed as products in the reaction of S with either a pyridyl-substituted phosphinine or with 2,4,6-triphenylphosphinine in the presence of pyridine [34]. The first well-characterised and quantitatively formed phosphinine selenide was only synthesised recently using red selenium and 2,6-(SiMe3)2PC5H3; a 31P chemical shift of 170 ppm was observed [35].
Phosphinophosphinines are useful ligands for transition metal complexes [5,9] and form four-membered chelates that can be extended into five-membered chelates through oxidation of the phosphine with sulfur, which has been well-studied [10], or oxygen, selenium and organic azides [36], which are almost unknown. For example, phosphinines bearing two ortho-P(S)Ph2 groups were cleanly synthesised from analogous diphenylphosphine-substituted phosphinines by a reaction with elemental sulfur [37,38]. These species were utilised as ligands in their phosphahexadienyl anion form after an attack on the phosphorus by suitable nucleophiles, e.g., lithium alkyls, chloride and alkoxide [39,40,41,42,43,44]. Phosphinines with one phosphine-sulfide substituent have also been accessed via the reaction of an azaphosphinine with a phosphine-sulfide-substituted alkyne [45]. In this contribution, we describe the reaction of phosphinophosphinines with selenium to better characterise the donor properties of the PPh2 substituents, we establish that the phosphinine moiety is left untouched under these conditions and we compare the reactivity of phosphinophosphinines with oxygen from the air. We also describe the synthesis of a bis(phosphinine) palladium complex and crystallographic characterisation of the chlorodiazaphosphacyclic intermediate formed in the synthesis of the key starting material, 1,3,2-diazaphosphinine.
2. Results
The phosphinines in this work were all synthesised using the 1,3,2-diazaphosphinine methodology pioneered by Le Floch, Mathey and co-workers [46,47]. PCl3 was reacted with a titanocycle (1) [48] formed from the reaction of Cp2TiMe2 with two equivalents of NCtBu [49] followed by the addition of NEt3 (Scheme 1) [46,47]. The anticipated intermediate in this synthesis was a chlorodiazaphosphacycle (2). We were fortunate to crystallise this air- and moisture-sensitive compound in the course of attempting to isolate 3. NMR spectroscopy revealed a 31P resonance at 109 ppm and the same resonance was also observed upon the addition of HCl in ether to 1,3,2-diazaphosphinine (3).
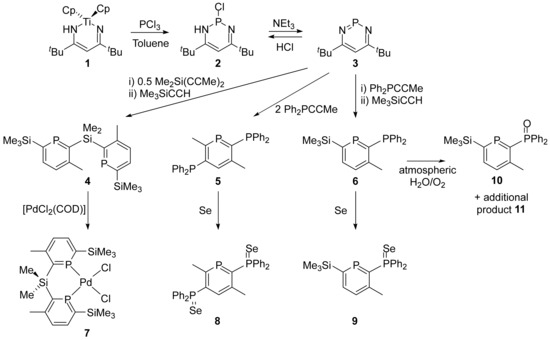
Scheme 1.
Formation and reactions of phosphinines.
Single crystal X-ray diffraction experiments revealed the molecular structure of 2 (Figure 1). The phosphorus atom was situated in an almost planar six-membered ring with single bonds to two nitrogen atoms (1.689(3) and 1.653(3) Å to N1 and N2, respectively). There was evidence of alternating single (C2-C3 1.432(4) Å) and double bonds (e.g., C1-C2 = 1.359(4) Å) around the ring albeit with the C-C single bond shorter than usual. The Cl atom was situated perpendicular to the plane of the ring and there was a stereochemically active lone pair on the phosphorus. The location of the double bonds was the same as in the parent titanocycle (1), with an N-H bond present rather than a methylene unit; the addition of HCl to 3 formed P-Cl and N-H bonds, as was expected from the higher electronegativity of nitrogen compared with phosphorus. Six-membered heterocycles featuring an N-P-N linkage are not uncommon in the Cambridge Structural Database but there are only a handful of examples with a P-Cl substituent and all feature an exocyclic substituent on both N atoms [50,51,52,53,54,55]. Other related examples are cationic and supported by a β-diketiminate framework [56,57].
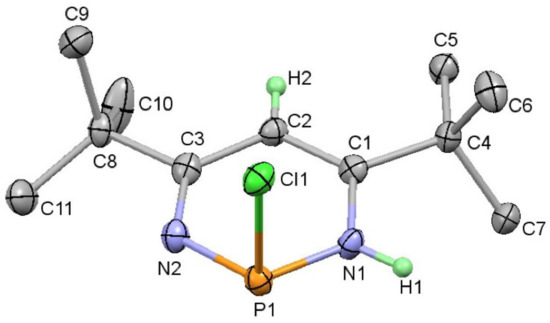
Figure 1.
Molecular structure of 2 (thermal ellipsoids at 50%; all H atoms except for those attached to the heterocyclic ring have been omitted for clarity). Selected bond lengths (Å) and angles (°): Cl1-P1 2.2417(11); P1-N1 1.689(3); P1-N2 1.653(3); N1-C1 1.377(4); C1-C2 1.359(4); C2-C3 1.432(4); N2-C3 1.320(4); N1-P1-Cl1 99.59(10); N2-P1-Cl1 98.29(10); N2-P1-N1 101.55(13).
The phosphinines 4 [58], 5 [36] and 6 [18] all featured additional P donors, either another phosphinine ring (4) or PPh2 moieties. They were synthesised by the reaction of diazaphosphinine (3) with alkynes in two sequential steps due to the slower reaction of the intermediate azaphosphinines with alkynes. This allowed two different alkynes to be used in the formation of 4 and 6. A reaction of the bis(phosphinine) (4) with [PdCl2(COD)] was attempted to ascertain whether the cyclooctadiene ligand (COD) could be displaced by the bis(phosphinine) and whether a chelated bis(phosphinine) complex was preferred over two monodentate ligands. The reaction of [PdCl2(COD)] with two equivalents of 4 led to the chelate complex 7 in preference to [PdCl2(4)2]; matching the stoichiometry 1:1 gave 7 in a 51% yield as a poorly soluble red solid. The reaction was selective with only one product observed. Unfortunately, we were unsuccessful in growing single crystals suitable for X-ray diffraction. 31P{1H} NMR spectroscopy showed a singlet at 204 ppm, which was at a lower frequency when compared with 4 (261 ppm). This shift to a lower frequency upon coordination was not seen for [RuCp*(Cl)(4)] or [RuCp*(H)(4)], which gave higher frequency resonances than the free ligand (273 and 286 ppm, respectively) although the [Mo(CO)4(6)] and [W(CO)4(6)] complexes gave lower 31P chemical shifts for the coordinated phosphinine ligand compared with 6. 1H NMR spectroscopy revealed 2 multiplets for the H atoms at the 4- and 5-positions along with 1 resonance for the 3-Me group and 1 SiMe3 environment in agreement with a plane of symmetry passing through the palladium and the SiMe2 group. However, the presence of two resonances for the SiMe2 group indicated that the ligand was not coplanar with the PdCl2 fragment, as has been seen previously in complexes of this ligand with other metal fragments [58]. We attributed this flexibility to bind to metals out of the plane of the ligand to the high s-character of the phosphinine lone pairs, which render them less directional [11]. Mass spectrometry revealed the correct isotope distribution and accurate mass for the [M-Cl]+ ion and an elemental analysis was also in agreement with the empirical formula of 7. Pd-phosphinine complexes are relatively rare. Monodentate complexes of 2,4,6-tri-tert-butylphosphinine with Pd(II), Pt(II) and Au(I) have been reported [59] along with chelating complexes of 2-pyridyl-4,6-diphenylphosphinine to PdCl2, which was poorly soluble and featured a 31P NMR chemical shift of 159 ppm [60], as well as [PdCl2{2-(PPh2O)PC5H4}], which gave a doublet at 165.9 ppm for the phosphinine P atom [61]. Other Pd complexes usually feature P-substituted phosphahexadienyl anions [38,39,40,45,62]. Interestingly, the phosphine-sulfide-substituted phosphinine 1,2-{P(S)Ph2}2-3,5-Ph2PC5H reacted with [PdCl2(COD)] to give a phosphahexadienyl ligand featuring a P-Cl bond driven by the coordination of the sulfur atoms and the stability of the d8 square planar geometry [37].
Reactions with selenium were attempted to investigate the selenation of the diphenylphosphine groups and to establish the inertness of the phosphinine to oxidation under these conditions. Reactions of 5 with excess selenium gave a mixture of products with short reaction times, which was likely to include the monoselenated products of the PPh2 groups as 31P NMR spectroscopy revealed new phosphinine 31P doublets at 239.4 and 231.9 ppm (103.4 Hz and 100.8 Hz, respectively). For comparison, the phosphinine 31P NMR signal for 5 was a doublet (29 Hz) at 220.8 ppm. Continued reaction gave full conversion to the diselenated product (8) with 31P NMR resonances evident at δ = 234.8 (d, 100 Hz, phosphinine), 32.9 (dd, 100 and 6 Hz, ortho-PPh2) and 32.4 ppm (d, 6 Hz, meta-PPh2) with selenium satellites only present on the lower frequency resonances (728 Hz for the ortho-PPh2 and 734 Hz for the meta-PPh2). The 77Se NMR spectrum showed the expected two doublets with the same coupling constants, allowing the assignment of these signals. A comparison with SePPh3, which has a 1J coupling constant of 732 Hz [29], showed that there was very little difference in the donor ability so the phosphinine substituent acted similarly to a phenyl ring. The characterisation of 8 was performed by mass spectrometry (accurate mass determined for [M + H]+) and by single crystal X-ray diffraction. The molecular structure (Figure 2) revealed disorder in the central phosphinine ring over an inversion centre situated in the middle of the molecule, as was seen in the structure of 5 [36], which prevented a detailed assessment of the bond lengths and angles in the ring. The P=Se bond lengths were determined to be 2.1097(5) Å.
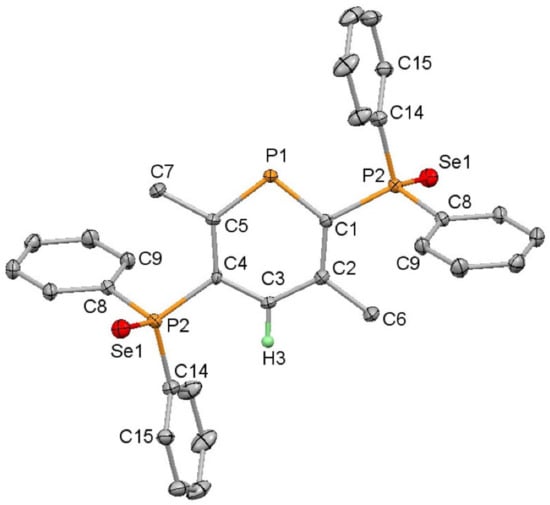
Figure 2.
Molecular structure of 8 (thermal ellipsoids at 50%; all H atoms except for those attached to the heterocyclic ring have been omitted for clarity). Only one position of the disordered phosphinine ring is shown.
Reactions of phosphinophosphinine (6) with grey Se gave the analogous phosphine selenide (9) with no evidence of the formation of a phosphinine-selenide despite extensive heating (100 °C for 1 week) and excess Se (five equivalents). 31P{1H} NMR spectroscopic resonances for 9 were observed as doublets at 249.0 and 34.4 ppm (100.6 Hz) with selenium satellites present on the lower frequency resonance (749 Hz). This indicated a marginally poorer σ-donor ability than for PPh3 (732 Hz) and identical behaviour to diphenyl-2-pyridylphosphine (749 Hz) [30]. Additional characterisation was achieved by 1H NMR spectroscopy as well as accurate mass spectrometry and an elemental analysis. Single crystal X-ray diffraction revealed the anticipated molecular structure (Figure 3). The phosphinine ring was planar with C-C bond lengths similar to those observed in other aromatic phosphinine rings [18,19,36] and benzene. The P-C bond lengths in the ring (1.7516(17) and 1.7282(18) Å) were shorter than the P-C bond length to the PPh2 substituent (1.8235(17) Å). The Se=P bond length was 2.1132(4) Å, indistinguishable within the error to Se=P in 8. The structure of 2-{P(S)Ph2}-3-Me-5,6-Ph2PC5H was similar and featured typical bond lengths and angles for an aromatic phosphinine heterocycle [45].
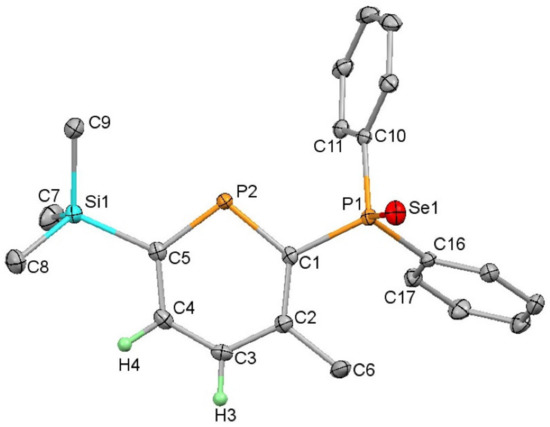
Figure 3.
Molecular structure of 9 (thermal ellipsoids at 50%; all H atoms except for those attached to the heterocyclic ring have been omitted for clarity). Selected bond lengths (Å) and angles (°): Se1-P1 2.1132(4); P2-C1 1.7516(17); C1-C2 1.407(2); C2-C3 1.397(2); C3-C4 1.386(2); C4-C5 1.403(2); P2-C5 1.7282(18); P1-C1 1.8235(17); C5-P2-C1 103.30(8).
We were intrigued by the differences between bis(phosphino)phosphinines and silylphosphinines [63,64,65] with respect to their air and moisture stability. We found the doubly silyl-substituted bis(phosphinine) (4) to be unstable under atmospheric conditions, requiring column chromatography under anaerobic conditions as well as storage and handling under N2. The bis(phosphino)phosphinine (5); on the other hand, was stable to air and moisture and this compound could be extracted into hot hexane by Soxhlet extraction over many days with no precautions required to exclude air or moisture. We found that the stability of silyl-substituted phosphinophosphinine (6) in the solution was intermediate between these two extremes. Monitoring an aerobic CDCl3 solution of 6 by 31P{1H} NMR spectroscopy revealed that after 21 days at room temperature, two additional species were present along with 6. We assigned one product as the phosphine oxide (10) based on: (i) the 31P chemical shifts of the two doublets at 256.2 and 32.1 ppm that were in the correct regions for phosphinine and phosphine oxide; (ii) the 2J coupling of 103 Hz, which was similar to the analogous selenide (6) (101 Hz); and (iii) an accurate mass spectrum identifying a species with m/z = 383.1144 for [10 + H]+. The other product (11) was evident as two doublets in the 31P{1H} NMR spectrum at −3.0 and −14.0 ppm with a 31 Hz coupling. Rough estimations by a 31P NMR integration revealed ca. 10% 10, 70% 6 and 20% 11. Heating to 40 °C for 21 days revealed a continuing reaction but a further 21 days of heating showed that 11 was not stable; only resonances for 10 and decomposition were evident. Due to the lack of a phosphinine resonance, we suspected that 11 resulted from an attack at a P=C double bond, probably by water with further reactivity a possibility including the loss of the SiMe3 group and/or cycloadditions, as has been seen previously [19,36].
3. Conclusions
The structure of the intermediate chlorodiazaphosphacycle (2) in the synthesis of 1,3,2-diazaphosphinine was confirmed. It revealed that its structure was analogous to the parent titanacycle (1) with the same pattern of single and double bonds around the ring, P(Cl){N=C(tBu)CH=C(tBu)-N(H)}. A SiMe2-linked bis(phosphinine) (4) reacted with [PdCl2(COD)] to form the chelating complex and was characterised thoroughly despite its poor solubility. However, single crystals have not been achieved (to date), precluding a structural characterisation by single crystal X-ray diffraction. The reactions of grey selenium with phosphinophosphinines generated the phosphine selenide derivatives whilst leaving the phosphinine ring intact. This was as anticipated as phosphinine selenides are extremely rare. The 1JP-Se coupling constants showed that the phosphinine ring acted similarly to a phenyl ring. This was in line with the favourable binding to transition metals already established as 2-phosphinophosphinines have been shown to act as competent ligands for a variety of transition metal fragments. Chloroform solutions of 2-PPh2-3-Me-6-SiMe3PC5H2 were shown to undergo a slow reaction with oxygen over the course of several weeks to form the phosphine oxide-substituted phosphinine. Side reactions also occurred and 31P NMR spectroscopy showed that the phosphinine ring did not remain intact in these reactions.
4. Materials and Methods
All reactions requiring inert conditions were performed under an oxygen-free nitrogen atmosphere by using standard Schlenk line techniques or by using an MBRUAN UNILab Plus glovebox, unless otherwise noted. Dry toluene was obtained from a solvent purification system (MBRAUN SP-300) and stored over 4 Å molecular sieves prior to use. When required to be dry, C6D6 and CDCl3 were dried over activated 4 Å molecular sieves prior to use. Non-dry solvents were used as received from Fisher Scientific or Goss Scientific (deuterated solvents). Cp2TiMe2 [66], diazaphosphinine [46,47] and the phosphinines 4 [58], 5 [36] and 6 [18] were synthesised as previously described. NMR spectra were obtained using either a Bruker AVIII400 (400 MHz) or an AVIIHD (400 MHz) spectrometer. 1H NMR spectra were recorded at 400 MHz and referenced to the residual protio solvent peak (7.24 for CHCl3 in CDCl3 and 7.16 for C6D5H in C6D6). The 13C{1H} NMR spectra were recorded at 101 MHz and referenced to the solvent peak (77.23 ppm for CDCl3 and 128.39 ppm for C6D6). 31P{1H} NMR spectra were recorded at 162 MHz and referenced to an external standard. Mass spectrometry was conducted at the National Mass Spectrometry Facility at Swansea University using the techniques stated. Elemental analyses were performed by Dr. Brian Hutton (Heriot-Watt University).
Single crystals suitable for X-ray diffraction were covered in inert oil and placed under the cold stream of a Bruker D8 Venture or a Bruker X8 APEXII four circle diffractometer cooled to 100 K. The exposures were collected using Mo Kα radiation (λ = 0.71073). Indexing, data collection and absorption corrections were performed. The structures were then solved using SHELXT [67] and refined by a full-matrix least-squares refinement (SHELXL) [68] interfaced with the programme OLEX2 [69]; 2 was refined as a two component twin (0.4564(18):0.5436(18)) and 8 showed a disorder of the phosphinine ring and a toluene solvate molecule over two positions that was modelled successfully.
4.1. Preparation of Chlorodiazaphosphacycle (2)
Method a: PCl3 and NEt3 were added to titanacycle (1) according to the standard literature method [46,47]. Toluene was removed from the solution under a reduced pressure and the residue was distilled under a reduced pressure into a liquid nitrogen-cooled trap. A few single crystals were observed along with an oil.
Method b: HCl in diethyl ether was added to a solution of diazaphosphinine in toluene. Upon cooling in the freezer, a small amount of precipitate formed, which was isolated by filtration and then analysed.
31P{1H} (162 MHz, CDCl3, 298 K)δ = 109.1 ppm; 1H NMR (400 MHz, CDCl3, 298 K) δ = 6.25 (1 H, d, 4JH-P = 1.5 Hz, CH), 1.26 (18 H, s, tBu).
4.2. Preparation of [PdCl2(κ:η1:η1-4)] (7)
Toluene (50 cm3) was added to a mixture of [PdCl2(COD)] (0.150 g, 0.53 mmol) and bis(phosphinine) (4) (0.526 g, 1.25 mmol) at room temperature. The red-orange suspension was heated for 3 h at 70 °C then allowed to cool to room temperature and settle over 16 h to afford a red solid and a red solution. The solution was separated from the solid and NMR spectroscopic studies of the solid (dissolved in CDCl3) revealed a singlet 31P{1H} resonance at 204 ppm, indicative of chelating η1:η1 coordination. The red solution, which contained bis(phosphinine), was then reacted with a further equivalent of [PdCl2(COD)] (0.150 g, 0.53 mmol) at 70 °C for 3 h, which produced a further red solid. The red solids were then combined (0.266 g, 0.54 mmol, 51%). Unfortunately, crystals suitable for X-ray diffraction studies could not be grown.
31P{1H} (162 MHz, CDCl3, 298 K)δ = 203.9 ppm; 1H NMR (400 MHz, CDCl3, 298 K) δ = 7.94 (m, 2H, meta-Ar-H), 7.18 (m, 2H, para-Ar-H), 2.63 (s, 6H, Ar-Me), 1.54 (s, 3H, SiMe), 1.12 (s, 3H, SiMe), 0.57 ppm (s, 18H, SiMe3). Anal. calcd for C20H34Si3P2Cl2Pd: C 40.17; H 5.73; N 0. Found: C 40.34; H 5.62; N 0. HRMS (ASAP/TOF) m/z: calcd for C20H34Si3P2Cl102Pd: 557.0188[M-Cl]+, found: 557.0192.
4.3. Preparation of 2,5-Bis(diphenylphosphineselenide)-3,6-dimethylphosphinine (8)
Under nitrogen, 5 (198 mg, 0.40 mmol), Se (104 mg, 1.31 mmol) and toluene (2 cm3) were sealed in an ampoule equipped with a J. Young tap and heated at 110 °C for 24 h. The reaction mixture was filtered to remove the excess selenium and all volatiles were removed under a reduced pressure. THF (10 cm3) was added to the solid and heated to 50 °C to dissolve the product and the solution was filtered. The THF was then removed under a reduced pressure, yielding a yellow powder (120 mg, 0.18 mmol, 45%).

1H NMR (400 MHz, CDCl3, 298 K) δ = 7.84 (dd, 13.6 and 7.7 Hz, 8H, meta-Ar), 7.51-7.44 (m, 12H, ortho- and para-Ar), 6.90 (dt, 1H, 3JH-P = 16.1 Hz, 4JH-P = 3.4 Hz, HC), 2.58 (d, 3H, 3JH-P = 17.9 Hz, HF), 2.30 (s, 3H, HG); 31P{1H} NMR (162 MHz, CDCl3, 298 K) δ = 234.8 (d, 2JP1-P2 = 100 Hz, P1), 32.9 (dd, 2JP1-P2 = 100 Hz, 5JP2-P3 = 6 Hz, 1JP-Se = 728 Hz, P2), 32.4 (d, 5JP2-P3 = 6 Hz, 1JP-Se = 734 Hz, P3). 77Se NMR (76 Hz, CDCl3, 298 K) δ = −259.4 (d, 728 Hz, Se=P2), −286.3 (d, 734 Hz, Se=P3); 13C{1H}(100.6 MHz, CDCl3, 298 K) δ = 168.6 (d), 167.9 (dd), 147.2 (m), 139.1 (m), 133.4 (dd), 132.9 (d), 132.2 (dd), 131.4 (d), 130.5 (dd), 130.2 (d), 128.9 (dd), 25.3 (d, CG), 24.1(dd, CF); HRMS (ASAP/TOF): calcd. for C31H28P374Se76Se: 642.9821 [M+H]+, found: 642.9837 m/z; calcd. for C31H28P3Se+: 573.0564 [M + H − Se]+, found: 573.0581.

4.4. 2-Diphenylphosphineselenide-3-methyl-6-trimethylsilylphosphinine (9)
An ampoule was charged with 6 (100 mg, 0.27 mmol), selenium powder (50 mg, 0.63 mmol, 2.3 equiv.) and toluene (2 cm3) then sealed with a J. Young tap. The reaction was heated to 120 °C for 18 h then filtered to remove the excess selenium. The filtrate was concentrated to half its volume and stored at −25 °C for one week. Filtration followed by drying under a high vacuum yielded 9 (80 mg, 0.18 mmol, 67%) as a pale yellow crystalline powder.
1H NMR (400 MHz, CDCl3): δ = 8.09-8.03 (m, 4H, ortho-PPh2), 7.68-7.63 (m, 1H, HD), 7.01-6.94 (m, 7H, PPh2 and HC), 2.69 (s, 3H, HG), 0.15 (s, 9H, HF); 31P{1H} NMR (162 MHz, CDCl3): δ = 249.0 (d, P1, 2JP1-P2 = 97.1 Hz), 34.4 (d, P2, 2JP1-P2 = 97.1 Hz, 77Se satellites (7.63% abundant): dd, 1JSe-P2 ≈ 749.1 Hz); HRMS (ASAP/QTof): m/z: calcd. for C21H24P2SeSi: 447.0367 [M+H]+, found: 447.0369; elemental analysis: anal. calcd. for C21H24P2SeSi: C 56.63, H 5.43, found: C 56.89, H 5.37.

4.5. Preparation of 2-Diphenylphosphineoxide-3-methyl-6-trimethylsilylphosphinine (10)
A solution of 2-diphenylphosphenyl-3-methyl-6-trimethylsilylphosphinine (6, 10 mg) in CDCl3 (0.7 cm3) was left under atmospheric oxygen and moisture in an NMR tube. The reaction progress was monitored using multinuclear NMR spectroscopy and the product by mass spectrometry.
1H NMR (400 MHz, CDCl3, 298 K) δ = 7.74 (m, 1H, HD), 7.65-7.39 (m, 11H, PPh2 + HC), 2.54 (s, 3H, HG), 0.20 (s, 9H, HF); 31P{1H} NMR (162 MHz, CDCl3, 298 K) δ = 256.2 (d, P1, 2JP1-P2 = 103 Hz), 32.1 (d, P2, 2JP2-P1 = 103 Hz); 13C{1H} NMR (100 MHz, CDCl3, 298 K) (a few signals too weak to be identified) δ = 138.3 (dd), 130.5-129.8 (multiple signals, phosphinine + PPh2), 126.4 (m), −2.1 (d, SiMe3); HRMS (ASAP/TOF): calcd. for C21H25OSiP2: 383.1150 [M+H]+, found: 383.1144 m/z.
Author Contributions
Conceptualisation, S.M.M.; methodology, P.A.C., R.J.N. and S.M.M.; validation, P.A.C., B.G. and S.M.M.; formal analysis, all authors.; investigation, all authors.; data curation, S.M.M.; writing—original draft preparation, S.M.M. and R.W.; writing—review and editing, all authors; visualisation, S.M.M.; supervision, S.M.M.; project administration, S.M.M.; funding acquisition, S.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Leverhulme Trust (RPG-2016-338 for supporting P.A.C.) and the EPSRC (DTP studentship to R.J.N.).
Data Availability Statement
The crystallographic data in this study are available in the Cambridge Structural Database, deposition numbers CCDC 2127803 (2), 2127804 (8) and 2127805 (9).
Acknowledgments
We thank the National Mass Spectrometry Facility at Swansea University for help with the mass spectrometry analysis. We thank Brian Hutton (Heriot-Watt University) for help with the elemental analysis and Georgina Rosair (Heriot-Watt University) for collecting the X-ray diffraction data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Märkl, G. 2,4,6-triphenylphosphabenzene. Angew. Chem. Int. Ed. 1966, 5, 846–847. [Google Scholar] [CrossRef]
- Ashe, A.J. Phosphabenzene and arsabenzene. J. Am. Chem. Soc. 1971, 93, 3293–3295. [Google Scholar] [CrossRef]
- Ashe, A.J. The route to phosphabenzene and beyond. Eur. J. Inorg. Chem. 2016, 2016, 572–574. [Google Scholar] [CrossRef]
- Nakajima, K.; Takata, S.; Sakata, K.; Nishibayashi, Y. Synthesis of phosphabenzenes by an iron-catalyzed [2+2+2] cycloaddition reaction of diynes with phosphaalkynes. Angew. Chem. Int. Ed. 2015, 54, 7597–7601. [Google Scholar] [CrossRef]
- Coles, N.T.; Sofie Abels, A.; Leitl, J.; Wolf, R.; Grützmacher, H.; Müller, C. Phosphinine-based ligands: Recent developments in coordination chemistry and applications. Coord. Chem. Rev. 2021, 433, 213729. [Google Scholar] [CrossRef]
- Müller, C.; Sklorz, J.A.W.; de Krom, I.; Loibl, A.; Habicht, M.; Bruce, M.; Pfeifer, G.; Wiecko, J. Recent developments in the chemistry of pyridyl-functionalized, low-coordinate phosphorus heterocycles. Chem. Lett. 2014, 43, 1390–1404. [Google Scholar] [CrossRef]
- Müller, C.; Broeckx, L.E.E.; de Krom, I.; Weemers, J.J.M. Developments in the coordination chemistry of phosphinines. Eur. J. Inorg. Chem. 2013, 2013, 187–202. [Google Scholar] [CrossRef]
- Müller, C. Phosphinine ligands. In Phosphorus(iii) Ligands in Homogeneous Catalysis: Design and Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012; pp. 287–307. [Google Scholar]
- Müller, C.; Vogt, D. Recent developments in the chemistry of donor-functionalized phosphinines. Comptes Rendus Chim. 2010, 13, 1127–1143. [Google Scholar] [CrossRef]
- Le Floch, P. Phosphaalkene, phospholyl and phosphinine ligands: New tools in coordination chemistry and catalysis. Coord. Chem. Rev. 2006, 250, 627–681. [Google Scholar] [CrossRef]
- Müller, C.; Vogt, D. Phosphinine-based ligands in homogeneous catalysis: State of the art and future perspectives. In Phosphorus Compounds: Catalysis by Metal Complexes; Peruzzini, M., Gonsalvi, L., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 37. [Google Scholar]
- Müller, C.; Vogt, D. Phosphinines as ligands in homogeneous catalysis: Recent developments, concepts and perspectives. Dalton Trans. 2007, 47, 5505–5523. [Google Scholar] [CrossRef]
- Kollár, L.; Keglevich, G. P-heterocycles as ligands in homogeneous catalytic reactions. Chem. Rev. 2010, 110, 4257–4302. [Google Scholar] [CrossRef]
- Weber, L. Phosphorus heterocycles: From laboratory curiosities to ligands in highly efficient catalysts. Angew. Chem. Int. Ed. 2002, 41, 563–572. [Google Scholar] [CrossRef]
- DiMauro, E.F.; Kozlowski, M.C. Phosphabenzenes as electron withdrawing phosphine ligands in catalysis. J. Chem. Soc. Perkin Trans. 1 2002, 33, 439–444. [Google Scholar] [CrossRef]
- Mansell, S.M. Catalytic applications of small bite-angle diphosphorus ligands with single-atom linkers. Dalton Trans. 2017, 46, 15157–15174. [Google Scholar] [CrossRef]
- Trodden, E.C.; Delve, M.P.; Luz, C.; Newland, R.J.; Andresen, J.M.; Mansell, S.M. A ruthenium cis-dihydride with 2-phosphinophosphinine ligands catalyses the acceptorless dehydrogenation of benzyl alcohol. Dalton Trans. 2021, 50, 13407–13411. [Google Scholar] [CrossRef]
- Newland, R.J.; Wyatt, M.F.; Wingad, R.L.; Mansell, S.M. A ruthenium(ii) bis(phosphinophosphinine) complex as a precatalyst for transfer-hydrogenation and hydrogen-borrowing reactions. Dalton Trans. 2017, 46, 6172–6176. [Google Scholar] [CrossRef]
- Newland, R.J.; Smith, A.; Smith, D.M.; Fey, N.; Hanton, M.J.; Mansell, S.M. Accessing alkyl- and alkenylcyclopentanes from cr-catalyzed ethylene oligomerization using 2-phosphinophosphinine ligands. Organometallics 2018, 37, 1062–1073. [Google Scholar] [CrossRef]
- Newland, R.J.; Lynam, J.M.; Mansell, S.M. Small bite-angle 2-phosphinophosphinine ligands enable rhodium-catalysed hydroboration of carbonyls. Chem. Commun. 2018, 54, 5482–5485. [Google Scholar] [CrossRef]
- Breit, B.; Winde, R.; Mackewitz, T.; Paciello, R.; Harms, K. Phosphabenzenes as monodentate pi-acceptor ligands for rhodium-catalyzed hydroformylation. Chem. Eur. J. 2001, 7, 3106–3121. [Google Scholar] [CrossRef]
- Breit, B. Highly regioselective hydroformylation under mild conditions with new classes of π-acceptor ligands. Chem. Commun. 1996, 2071–2072. [Google Scholar] [CrossRef]
- Müller, C.; López, L.G.; Kooijman, H.; Spek, A.L.; Vogt, D. Chiral bidentate phosphabenzene-based ligands: Synthesis, coordination chemistry, and application in rh-catalyzed asymmetric hydrogenations. Tetrahedron Lett. 2006, 47, 2017–2020. [Google Scholar] [CrossRef][Green Version]
- Rigo, M.; Hettmanczyk, L.; Heutz, F.J.L.; Hohloch, S.; Lutz, M.; Sarkar, B.; Müller, C. Phosphinines versus mesoionic carbenes: A comparison of structurally related ligands in au(i)-catalysis. Dalton Trans. 2017, 46, 86–95. [Google Scholar] [CrossRef]
- Broeckx, L.E.E.; Bucci, A.; Zuccaccia, C.; Lutz, M.; Macchioni, A.; Müller, C. Cyclometalated phosphinine–iridium(iii) complexes: Synthesis, reactivity, and application as phosphorus-containing water-oxidation catalysts. Organometallics 2015, 34, 2943–2952. [Google Scholar] [CrossRef]
- Hayashi, M. Phosphine sulfides: New aspects of organophosphorus compounds. Chem. Lett. 2020, 50, 1–6. [Google Scholar] [CrossRef]
- Hua, G.; Woollins, J.D. Organic phosphorus-selenium chemistry. In Selenium and Tellurium Chemistry: From Small Molecules to Biomolecules and Materials; Woollins, J.D., Laitinen, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–39. [Google Scholar]
- Benton, A.; Durand, D.J.; Copeland, Z.; Watson, J.D.; Fey, N.; Mansell, S.M.; Rosair, G.M.; Welch, A.J. On the basicity of carboranylphosphines. Inorg. Chem. 2019, 58, 14818–14829. [Google Scholar] [CrossRef]
- Allen, D.W.; Taylor, B.F. The chemistry of heteroarylphosphorus compounds. Part 15. Phosphorus-31 nuclear magnetic resonance studies of the donor properties of heteroarylphosphines towards selenium and platinum(ii). J. Chem. Soc. Dalton Trans. 1982, 13, 51–54. [Google Scholar] [CrossRef]
- Beckmann, U.; Süslüyan, D.; Kunz, P.C. Is the 1 j pse coupling constant a reliable probe for the basicity of phosphines? A 31p nmr study. Phosphorus Sulfur Silicon Relat. Elem. 2011, 186, 2061–2070. [Google Scholar] [CrossRef]
- Holah, D.G.; Hughes, A.N.; Knudsen, K.L. Formation and partial characterisation of a stable phosphinine 1-sulphide. J. Chem. Soc. Chem. Commun. 1988, 19, 493–495. [Google Scholar] [CrossRef]
- Hughes, A.N.; Knudsen, K.L. A 31p nmr study of reactions of 2,3-bis(diphenylphosphino)-6-phenyl-λ3-phosphinine with sulfur: Formation, characterization, and further reaction of a stable phosphinine 1-sulfide. Heterocycles 1990, 30, 543–550. [Google Scholar] [CrossRef]
- Moores, A.; Cantat, T.; Ricard, L.; Mézailles, N.; Le Floch, P. Experimental and theoretical study of phosphinine sulfides. New J. Chem. 2007, 31, 1493–1498. [Google Scholar] [CrossRef]
- Pfeifer, G.; Ribagnac, P.; Le Goff, X.-F.; Wiecko, J.; Mézailles, N.; Müller, C. Reactivity of aromatic phosphorus heterocycles—differences between nonfunctionalized and pyridyl-substituted 2,4,6-triarylphosphinines. Eur. J. Inorg. Chem. 2015, 2015, 240–249. [Google Scholar] [CrossRef]
- Wossidlo, F.; Frost, D.S.; Lin, J.; Coles, N.T.; Klimov, K.; Weber, M.; Böttcher, T.; Müller, C. Making aromatic phosphorus heterocycles more basic and nucleophilic: Synthesis, characterization and reactivity of the first phosphinine selenide. Chem. Eur. J. 2021, 27, 12788–12795. [Google Scholar] [CrossRef]
- Newland, R.J.; Delve, M.P.; Wingad, R.L.; Mansell, S.M. Two isomers of a bis(diphenylphosphino)-phosphinine, and the synthesis and reactivity of ru arene/cp* phosphinophosphinine complexes. New J. Chem. 2018, 42, 19625–19636. [Google Scholar] [CrossRef]
- Doux, M.; Bouet, C.; Mézailles, N.; Ricard, L.; Le Floch, P. Synthesis and molecular structure of a palladium complex containing a λ5-phosphinine-based sps pincer ligand. Organometallics 2002, 21, 2785–2788. [Google Scholar] [CrossRef]
- Doux, M.; Mézailles, N.; Ricard, L.; Le Floch, P. Group 10 metal complexes of sps-based pincer ligands: Syntheses, X-ray structures, and dft calculations. Eur. J. Inorg. Chem. 2003, 2003, 3878–3894. [Google Scholar] [CrossRef]
- Moores, A.; Mézailles, N.; Ricard, L.; Jean, Y.; le Floch, P. H2-palladium and platinum(ii) complexes of a λ4-phosphinine anion: Syntheses, X-ray crystal structures, and dft calculations. Organometallics 2004, 23, 2870–2875. [Google Scholar] [CrossRef]
- Blug, M.; Doux, M.; Le Goff, X.; Maître, P.; Ribot, F.; Le Floch, P.; Mézailles, N. The effect of a fourth binding site on the stabilization of cationic sps pincer palladium complexes: Experimental, dft, and mass spectrometric studies. Organometallics 2009, 28, 2020–2027. [Google Scholar] [CrossRef]
- Doux, M.; Mézailles, N.; Ricard, L.; Le Floch, P. Planar discrimination in an sps-based rhodium(i) complex. Organometallics 2003, 22, 4624–4626. [Google Scholar] [CrossRef]
- Doux, M.; Ricard, L.; Le Floch, P.; Mézailles, N. Group 11 metal complexes of sps-based pincer ligands: Syntheses, X-ray structures and reactivity. Dalton Trans. 2004, 2593–2600. [Google Scholar] [CrossRef]
- Doux, M.; Thuéry, P.; Blug, M.; Ricard, L.; Le Floch, P.; Arliguie, T.; Mézailles, N. Anions of tridentate sps ligands: Syntheses, X-ray structures and dft calculations. Organometallics 2007, 26, 5643–5653. [Google Scholar] [CrossRef]
- Arliguie, T.; Blug, M.; Le Floch, P.; Mézailles, N.; Thuéry, P.; Ephritikhine, M. Organouranium complexes with phosphinine-based sps pincer ligands. Variations with the substituent at the phosphorus atom. Organometallics 2008, 27, 4158–4165. [Google Scholar] [CrossRef]
- Dochnahl, M.; Doux, M.; Faillard, E.; Ricard, L.; Le Floch, P. A new mixed p,s-bidentate ligand featuring a λ4-phosphinine anion and a phosphanyl sulfide group—Synthesis, X-ray crystal structures and catalytic properties of its chloro(cymene)ruthenium and allylpalladium complexes. Eur. J. Inorg. Chem. 2005, 2005, 125–134. [Google Scholar] [CrossRef]
- Avarvari, N.; Le Floch, P.; Ricard, L.; Mathey, F. 1,3,2-diazaphosphinines and -diazaarsinines as precursors for polyfunctional phosphinines and arsinines. Organometallics 1997, 16, 4089–4098. [Google Scholar] [CrossRef]
- Avarvari, N.; Le Floch, P.; Mathey, F. 1,3,2-diazaphosphinines: New, versatile precursors of 1,2-azaphosphinines and polyfunctional phosphinines. J. Am. Chem. Soc. 1996, 118, 11978–11979. [Google Scholar] [CrossRef]
- Doxsee, K.M.; Farahi, J.B. Reductive coupling of nitriles via formal [2 + 2] cycloadditions to the titanium-carbon double bond. J. Am. Chem. Soc. 1988, 110, 7239–7240. [Google Scholar] [CrossRef]
- Petasis, N.A.; Fu, D.K. Reactions of dimethyltitanocene with alkynes and nitriles. Organometallics 1993, 12, 3776–3780. [Google Scholar] [CrossRef]
- Spinney, H.A.; Korobkov, I.; DiLabio, G.A.; Yap, G.P.A.; Richeson, D.S. Diamidonaphthalene-stabilized n-heterocyclic pnictogenium cations and their cation-cation solid-state interactions. Organometallics 2007, 26, 4972–4982. [Google Scholar] [CrossRef]
- Spinney, H.A.; Yap, G.P.A.; Korobkov, I.; DiLabio, G.; Richeson, D.S. Construction of a stable n-heterocyclic phosphenium cation with an electron-rich framework and its complexation to rhodium. Organometallics 2006, 25, 3541–3543. [Google Scholar] [CrossRef]
- Kozma, Á.; Rust, J.; Alcarazo, M. Bis[(dialkylamino)cyclopropenimine]-stabilized piii- and pv-centered dications. Chem.-Eur. J. 2015, 21, 10829–10834. [Google Scholar] [CrossRef]
- Sonnenburg, R.; Borkenhagen, F.; Neda, I.; Holger; Thönnessen; Jones, P.G.; Schmutzler, R. N-phosphorylated nitrogen mustards; preparation of 2-chloroethyl- and bis(2-chloroethyl)-amides with the benzodiazaphosphorinone ring system. Phosphorus Sulfur Silicon Relat. Elem. 1997, 126, 11–26. [Google Scholar] [CrossRef]
- Mallov, I.; Spinney, H.; Jurca, T.; Gorelsky, S.; Burchell, T.; Richeson, D. The 2,2′-diindolylmethane dianion supporting scaffold for group 15 compounds. Inorg. Chim. Acta 2012, 392, 5–9. [Google Scholar] [CrossRef]
- Mecke, E.; Frank, W. Crystal structure of 1,3-di-tert-butyl-2-chloro-1,3,2-diazaphosphorinane—A saturated six-membered phosphorus nitrogen heterocycle with a partially flattened chair conformation and a long piii-cl bond. Acta Crystallogr. Sect. E 2019, 75, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Vidovic, D.; Lu, Z.; Reeske, G.; Moore, J.A.; Cowley, A.H. An n,n′-chelated phosphenium cation supported by a β-diketiminate ligand. Chem. Commun. 2006, 33, 3501–3503. [Google Scholar] [CrossRef] [PubMed]
- Lesikar, L.A.; Woodul, W.D.; Richards, A.F. The reactions of phosphorus(iii) halides with β-diketiminato ligands. Polyhedron 2007, 26, 3242–3246. [Google Scholar] [CrossRef]
- Cleaves, P.A.; Mansell, S.M. Unexpected multiple coordination modes in silyl-bridged bis(phosphinine) complexes. Organometallics 2019, 38, 1595–1605. [Google Scholar] [CrossRef]
- Clendenning, S.B.; Hitchcock, P.B.; Lawless, G.A.; Nixon, J.F.; Tate, C.W. Differences in the η1-ligating properties of 2,4,6-tritertiarybutyl-phosphabenzene, pc5h2bu3t and 2,4,6-tritertiarybutyl-1,3,5-triphosphabenzene, p3c3bu3t. J. Organomet. Chem. 2010, 695, 717–720. [Google Scholar] [CrossRef]
- Campos-Carrasco, A.; Broeckx, L.E.E.; Weemers, J.J.M.; Pidko, E.A.; Lutz, M.; Masdeu-Bultó, A.M.; Vogt, D.; Müller, C. Pdii and ptii complexes of 2-(2′-pyridyl)-4,6-diphenylphosphinine: Synthesis, structure, and reactivity. Chem.-Eur. J. 2011, 17, 2510–2517. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Grützmacher, H. Multidentate phosphanyl phosphinines: Synthesis and properties. Chem.-Eur. J. 2018, 24, 8432–8437. [Google Scholar] [CrossRef]
- Doux, M.; Mézailles, N.; Melaimi, M.; Ricard, L.; Le Floch, P. A σ4, λ5-phosphinine palladium complex: A new type of phosphorus ligand and catalyst. Application to the pd-catalyzed formation of arylboronic esters. Chem. Commun. 2002, 1566–1567. [Google Scholar] [CrossRef]
- Habicht, M.H.; Wossidlo, F.; Bens, T.; Pidko, E.A.; Müller, C. 2-(trimethylsilyl)-λ3-phosphinine: Synthesis, coordination chemistry, and reactivity. Chem.-Eur. J. 2018, 24, 944–952. [Google Scholar] [CrossRef]
- Ferro, V.R.; Omar, S.; González-Jonte, R.H.; García de la Vega, J.M. The σ-donating and π-accepting properties of ortho-si(ch3)3 phosphinine macrocycles. Heteroat. Chem. 2003, 14, 160–169. [Google Scholar] [CrossRef]
- Avarvari, N.; Mézailles, N.; Ricard, L.; Floch, P.L.; Mathey, F. Silacalix-[n]-phosphaarenes: Macrocyclic ligands based on dicoordinate phosphorus centers. Science 1998, 280, 1587–1589. [Google Scholar] [CrossRef] [PubMed]
- Payack, J.F.; Hughes, D.L.; Cai, D.; Cottrell, I.F.; Verhoeven, T.R. Dimethyltitanocene. Org. Synth. 2002, 79, 19. [Google Scholar]
- Sheldrick, G. Shelxt-integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of shelx. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).