
Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate

 , , , , and
, , , , and
Abstract
:1. Introduction
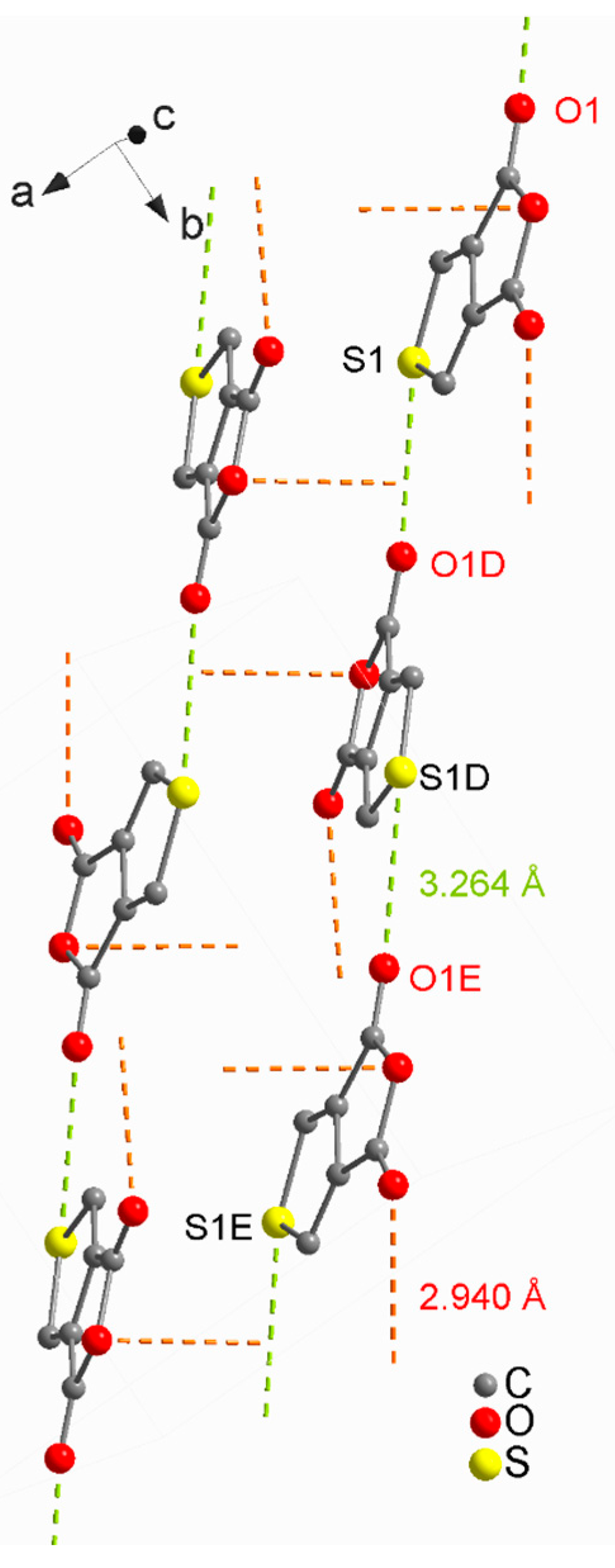
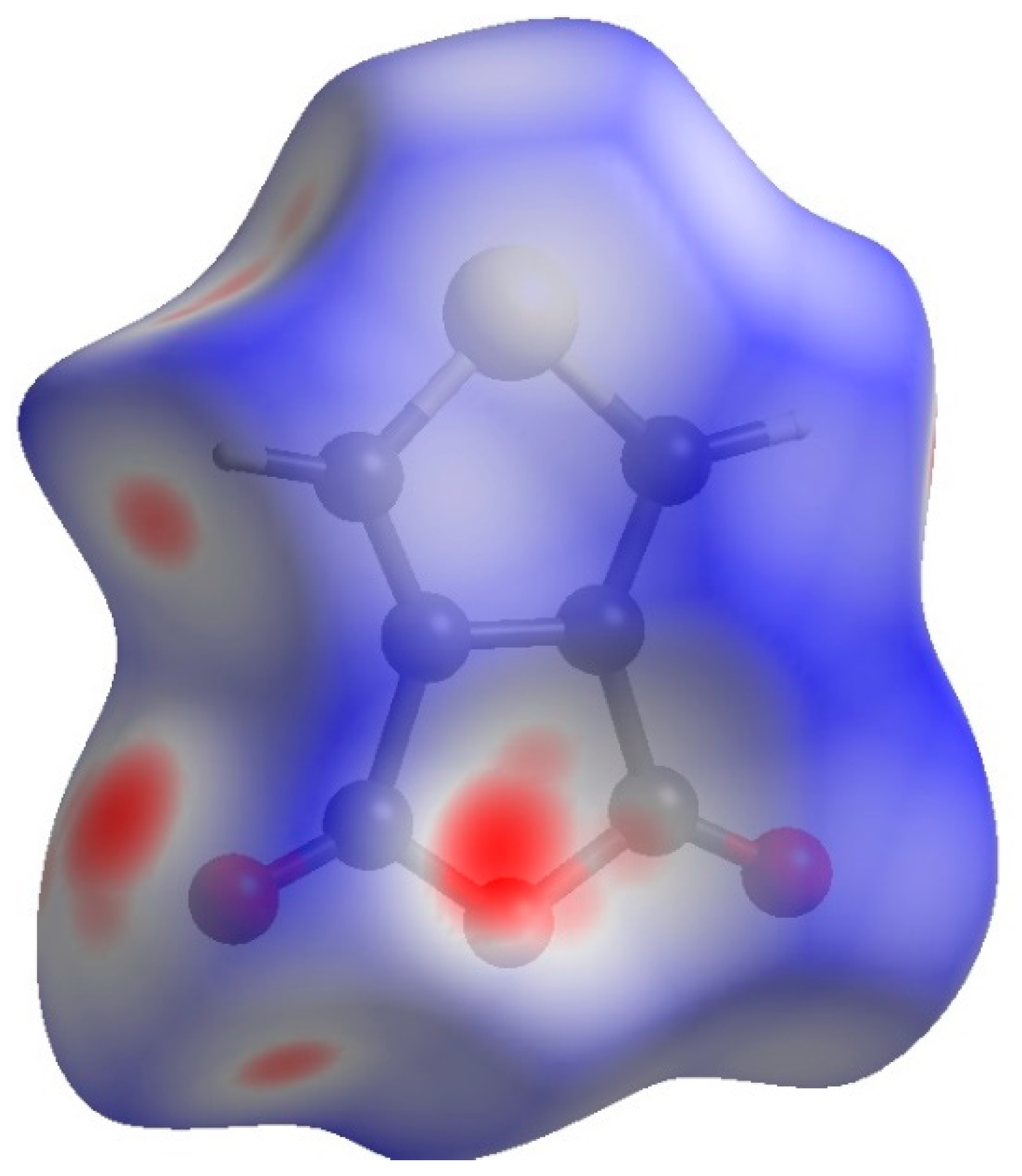
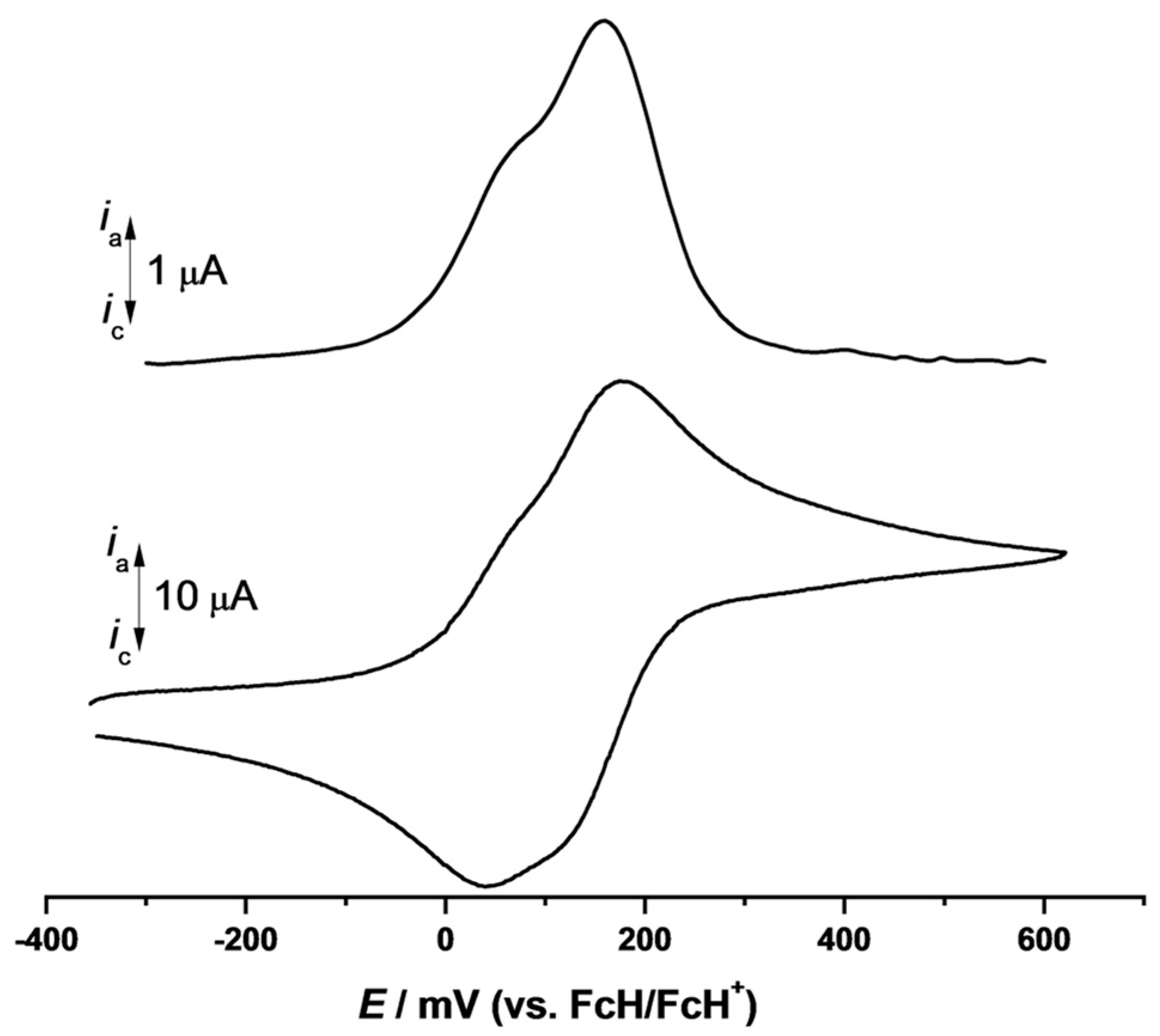
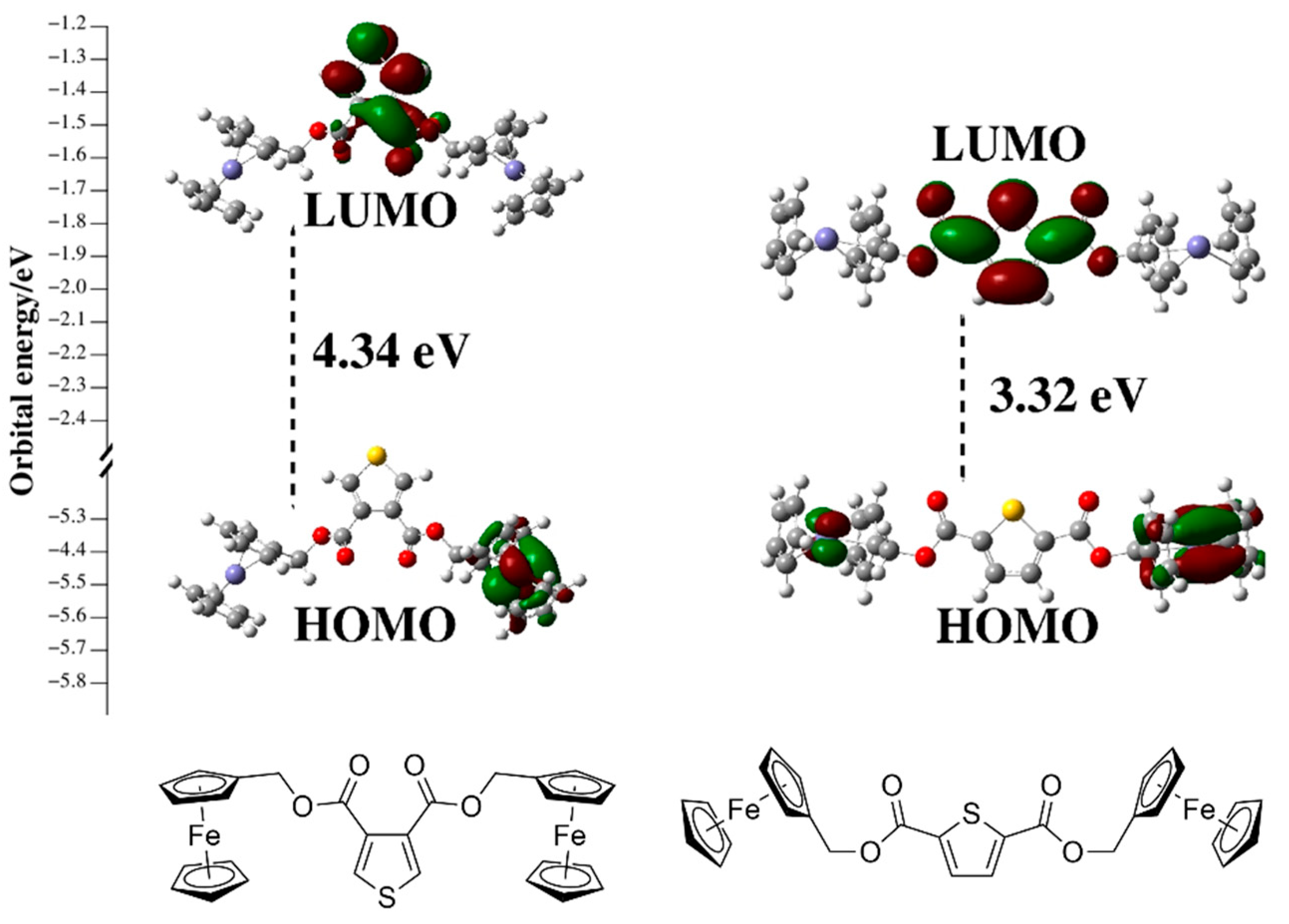
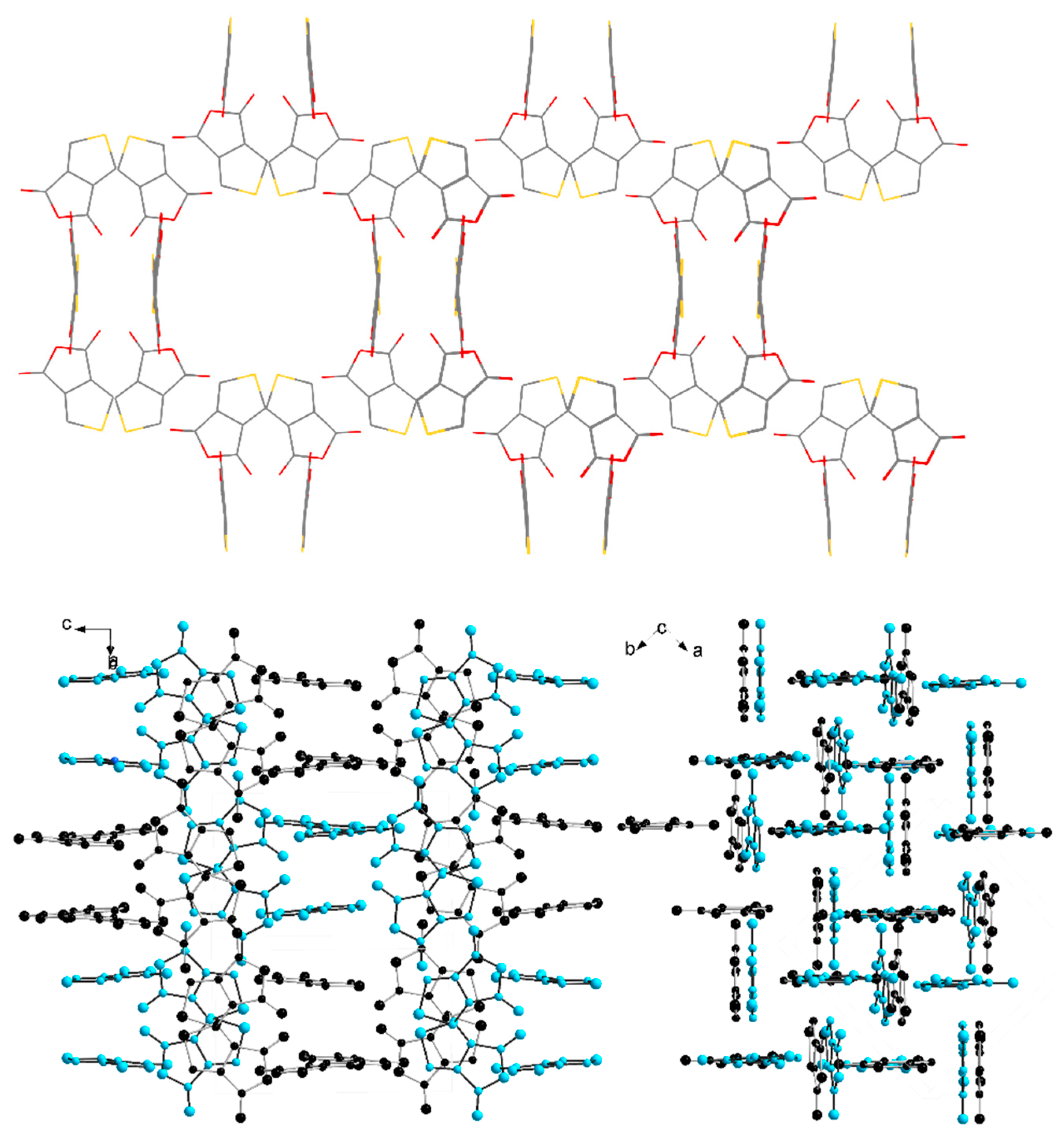
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Supporting Data
References
- Kealy, T.J.; Pauson, P.L. A new type of organo-iron compound. Nature 1951, 168, 1039. [Google Scholar] [CrossRef]
- Miller, S.A.; Tebboth, J.A.; Tremaine, J.F. Dicyclopentadienyliron. J. Chem. Soc. 1952, 114, 632. [Google Scholar] [CrossRef]
- Van Staveren, D.R.; Metzler-Nolte, N. Bioorganometallic chemistry of ferrocene. Chem. Rev. 2004, 104, 5931. [Google Scholar] [CrossRef] [PubMed]
- Štěpnička, P. (Ed.) Ferrocenes: Ligands, Materials and Biomolecules; Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Tong, R.; Zhao, Y.; Wang, L.; Yu, H.; Ren, F.; Saleem, M.; Amer, W.A. Recent research progress in the synthesis and properties of burning rate catalysts based on ferrocene-containing polymers and derivatives. J. Organomet. Chem. 2014, 755, 16. [Google Scholar] [CrossRef]
- Dai, L.-X.; Hou, X.-L. (Eds.) Chiral Ferrocenes in Asymmetric Catalysis; Synthesis and Applications; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Yu, Y.; Bond, A.D.; Leonard, P.W.; Lorenz, U.J.; Timofeeva, T.V.; Vollhardt, K.P.C.; Whitener, G.D.; Yakovenko, A.A. Hexaferrocenylbenzene. Chem. Commun. 2006, 24, 2572. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Bond, A.D.; Leonard, P.W.; Vollhardt, K.P.C.; Whitener, G.D. Syntheses, structures, and reactivity of radial oligocyclopentadienyl metal complexes: Penta (ferrocenyl) cyclopentadienyl and congeners. Angew. Chem. Int. Ed. 2006, 45, 1794. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Rüffer, T.; Erasmus, E.; Swarts, J.C.; Lang, H. A star-shaped supercrowded 2,3,4,5-tetraferrocenylthiophene: Synthesis, solid-state structure, and electrochemistry. Organometallics 2010, 29, 4900–4905. [Google Scholar] [CrossRef]
- Frenzel, P.; Lehrich, S.W.; Korb, M.; Hildebrandt, A.; Lang, H. Ferrocenyloxysilanes: Synthesis, characterization and electrochemical investigations. J. Organomet. Chem. 2017, 845, 98. [Google Scholar] [CrossRef]
- Packheiser, R.; Lang, H. Mixed transition–metal complexes based on multitopic 1,3,5-triethynyl-and 1-diphenylphosphino-3,5-diethynyl-benzene linking units. Inorg. Chim. Acta 2011, 366, 177–183. [Google Scholar] [CrossRef]
- Packheiser, R.; Rüffer, T.; Ecorchard, P.; Lang, H.Z. Transition-Metal Complexes Based on the 1,3,5-Triethynyl Benzene Linking Unit. Anorg. Allg. Chem. 2010, 636, 2607. [Google Scholar] [CrossRef] [Green Version]
- Packheiser, R.; Lang, H. A first mixed heteropentametallic transition metal complex: Synthesis and characterisation. Chem. Commun. 2007, 10, 580. [Google Scholar] [CrossRef]
- Barlow, S.; O’Hare, D. Metal−metal interactions in linked metallocenes. Chem. Rev. 1997, 97, 637. [Google Scholar] [CrossRef] [PubMed]
- Paul, F.; Lapinte, C. Organometallic molecular wires and other nanoscale-sized devices: An approach using the organoiron (dppe) Cp* Fe building block. Coord. Chem. Rev. 1998, 178–180, 431–509. [Google Scholar] [CrossRef]
- Lal, B.; Badshah, A.; Altaf, A.A.; Khan, N.; Ullah, S. Miscellaneous applications of ferrocene-based peptides/amides. Appl. Organomet. Chem. 2011, 25, 843. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A new age for iron: Antitumoral ferrocenes. Organometallics 2013, 32, 5626. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. (Multi) ferrocenyl five-membered heterocycles: Excellent connecting units for electron transfer studies. Organometallics 2013, 32, 5640. [Google Scholar] [CrossRef]
- Ceccon, A.; Santi, S.; Orian, L.; Bisello, A. Electronic communication in heterobinuclear organometallic complexes through unsaturated hydrocarbon bridges. Coord. Chem. Rev. 2004, 248, 683. [Google Scholar] [CrossRef]
- Ward, M.D. Metal-metal interactions in binuclear complexes exhibiting mixed valency; molecular wires and switches. Chem. Soc. Rev. 1995, 24, 121. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. Influencing the electronic interaction in diferrocenyl-1-phenyl-1 H-pyrroles. Dalt. Trans. 2011, 40, 11831. [Google Scholar] [CrossRef]
- Li, Y.; Josowicz, M.; Tolbert, L.M. Diferrocenyl molecular wires. The role of heteroatom linkers. J. Am. Chem. Soc. 2010, 132, 10374. [Google Scholar] [CrossRef]
- Moriuchi, T.; Hirao, T. Imide-bridged diferrocene for protonation-controlled regulation of electronic communication. Tetrahedron Lett. 2007, 48, 5099. [Google Scholar] [CrossRef]
- Guldi, D.M.; Maggini, M.; Scorrano, G.; Prato, M. Intramolecular electron transfer in fullerene/ferrocene based donor−bridge−acceptor dyads. J. Am. Chem. Soc. 1997, 119, 974. [Google Scholar] [CrossRef]
- Sixt, T.; Sieger, M.; Krafft, M.J.; Bubrin, D.; Fiedler, J.; Kaim, W. Ambi-valence taken literally: Ruthenium vs iron oxidation in (1,1′-diphosphinoferrocene) ruthenium (II) hydride and chloride complexes as deduced from spectroelectrochemistry of the heterodimetallic “mixed-valent” intermediates. Organometallics 2010, 29, 5511. [Google Scholar] [CrossRef]
- Nguyen, P.; Elipe, P.G.; Manners, I. Organometallic polymers with transition metals in the main chain. Chem. Rev. 1999, 99, 1515. [Google Scholar] [CrossRef] [PubMed]
- MacDowell, D.W.H.; Jourdenais, R.A.; Naylor, R.W.; Wisowaty, J.C. The Reaction of Thiophene-3,4-dicarbonyl Chloride with Aluminum Chloride and Benzene. J. Org. Chem. 1972, 37, 4406. [Google Scholar] [CrossRef]
- El-Gaby, M.S.A.; Zahran, M.A.; Ismail, M.M.F.; Ammar, Y.A. A novel synthesis of dibenzo [c,f] chromenes, dibenzo [c,h] chromenes and benzo [7,8] chromeno [3,4-f] isoindoles as antimicrobial agents. II Farmaco. 2000, 55, 227. [Google Scholar]
- Meyer, C.; Neue, B.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Würthwein, E.-U.; Itami, K.; Wünsch, B. Exploitation of an additional hydrophobic pocket of σ 1 receptors: Late-stage diverse modifications of spirocyclic thiophenes by C–H bond functionalization. Org. Biomol. Chem. 2011, 9, 8016. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Itami, K.; Wünsch, B. Late-Stage C–H Bond Arylation of Spirocyclic σ1 Ligands for Analysis of Complementary σ1 Receptor Surface. Eur. J. Org. Chem. 2012, 2012, 5972. [Google Scholar] [CrossRef]
- Meyer, C.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Col, V.D.; Laurini, E.; Itami, K.; Pricl, S.; Wünsch, B.J. Pd-catalyzed direct C–H bond functionalization of spirocyclic σ1 ligands: Generation of a pharmacophore model and analysis of the reverse binding mode by docking into a 3D homology model of the σ1 receptor. Med. Chem. 2012, 55, 8047. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Neue, B.; Schepmann, D.; Yanagisawa, S.; Yamaguchi, J.; Würthwein, E.-U.; Itami, K.; Wünsch, B. Improvement of σ1 receptor affinity by late-stage C–H-bond arylation of spirocyclic lactones. Bioorg. Med. Chem. 2013, 21, 1844. [Google Scholar] [CrossRef]
- Al-Mousawi, S.M.; El-Apasery, M.A. Synthesis of some monoazo disperse dyes derived from aminothienochromene. Molecules 2013, 18, 8837. [Google Scholar] [CrossRef] [Green Version]
- Fuse, S.; Takahashi, R.; Takahashi, T. Facile, One-Step Synthesis of 5-Substituted Thieno [3,4-c] pyrrole-4,6-dione by Palladium-Catalyzed Carbonylative Amidation. Eur. J. Org. Chem. 2015, 16, 3430. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, J.; Yun, H.; Baek, Y.; Lee, P.H. Rhodium-catalyzed intramolecular transannulation reaction of alkynyl thiadiazole enabled 5, n-fused thiophenes. J. Org. Chem. 2017, 82, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Benachenhou, F.; Mesli, M.A.; El Borai, M.; Hanquet, B.; Guilard, R.J. Synthesis and structure elucidation of the condensation products between thiophene dicarbaldehydes and aromatic amines. Potential analgesic and anti-inflammatory agents. Heterocycl. Chem. 1988, 25, 1531. [Google Scholar] [CrossRef]
- Nielsen, C.B.; Bjørnholm, T. New regiosymmetrical dioxopyrrolo-and dihydropyrrolo-functionalized polythiophenes. Org. Lett. 2004, 6, 3381. [Google Scholar] [CrossRef] [PubMed]
- Crayston, J.A.; Iraqi, A.; Mallon, P.; Walton, J.C. Preparation and characterisation of thienonaphthoquinones and their radical ions. J. Chem. Soc. Perkin Trans. 1993, 2, 1589. [Google Scholar] [CrossRef]
- Vishnumurthy, K.; Makriyannis, A.J. Novel and Efficient One-Step Parallel Synthesis of Dibenzopyranones via Suzuki− Miyaura Cross Coupling. Comb. Chem. 2010, 12, 664. [Google Scholar] [CrossRef] [Green Version]
- Taher, D.; Awwadi, F.F.; Pfaff, U.; Speck, J.M.; Rüffer, T.; Lang, H. A series of Se-ferrocenyl thiophene carboselenoates–Synthesis, solid-state structure and electrochemistry. J. Organomet. Chem. 2013, 736, 9. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.F.; Speck, J.M.; Korb, M.; Schaarschmidt, D.; Weheabby, S.; Habashneh, A.Y.; Al-Noaimi, M.; El-Khateeb, M.; Abu-Orabi, S.T.; et al. Heterocyclic-based ferrocenyl carboselenolates: Synthesis, solid-state structure and electrochemical investigations. J. Organomet. Chem. 2017, 845, 55. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.F.; Speck, J.M.; Korb, M.; Wagner, C.; Hamed, E.M.; Al-Noaimi, M.; Habashneh, A.Y.; El-khateeb, M.; Abu-Orabi, S.T.; et al. Ferrocenyl thiocarboxylates: Synthesis, solid-state structure and electrochemical investigations. J. Organomet. Chem. 2017, 847, 59. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.F.; Speck, J.M.; Korb, M.; Schaarschmidt, D.; Wagner, C.; Amarne, H.; Merzweiler, K.; van Koten, G.; Lang, H.J. From ferrocenyl selenoesters to diferrocenyl methanols. Organomet. Chem. 2018, 863, 1. [Google Scholar] [CrossRef]
- Taher, D.; Corrigan, J.F. Aryl (trimethylsilyl) selenides as Reagents for the Synthesis of Mono-and Diselenoesters. Organometallics 2011, 30, 5943. [Google Scholar] [CrossRef]
- Ghazzy, A.; Taher, D.; Helal, W.; Korbb, M.; Al-Shewikib, R.K.; Weheabby, S.; Al-Saidd, N.; Abu-Orabi, S.T.; Lang, H. Aryl ferrocenylmethylesters: Synthesis, solid-state structure and electrochemical investigations. Arab. J. Chem. 2020, 13, 3546. [Google Scholar] [CrossRef]
- Taher, D. Synthesis and reactivity of cyclopentadienyl ruthenium complexes containing ferrocenylselenolates. Transit. Met. Chem. 2009, 34, 641. [Google Scholar] [CrossRef]
- Taher, D.; Wallbank, A.I.; Turner, E.A.; Cuthbert, H.L.; Corrigan, J.F. Alk-2-ynyl Trimethylsilyl Chalcogenoethers by Nucleophilic Substitution of Propargyl Bromides. Eur. J. Inor. Chem. 2006, 22, 4616. [Google Scholar] [CrossRef]
- Taher, D.; Awwadi, F.; El-khateeb, M.; Lang, H. Synthesis and reactivity of cyclopentadienyl iron complexes containing ferrocenyl selenolates. Transit. Met. Chem. 2012, 37, 601. [Google Scholar] [CrossRef]
- Taher, D.; Ghazzy, A.; Awwadi, F.F.; Helal, W.; al Khalyfeh, K.; Korb, M.; Hildebrandt, A.; Kovalski, E.; Lang, H. Ferrocenylmethyl-functionalized 5-membered heterocycles: Synthesis, solid-state structure and electrochemical investigations. Polyhedron 2018, 152, 188. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Miesel, D.; Lang, H. Electrostatic interactions within mixed-valent compounds. Coord. Chem. Rev. 2018, 371, 56. [Google Scholar] [CrossRef]
- Lang, H. Organometallic π-tweezers and 1,1′-bis (diphenylphosphanyl) ferrocene as bidentate chelating ligands for the synthesis of heteromultimetallic compounds. Polyhedron 2018, 139, 50. [Google Scholar] [CrossRef]
- Heinze, K.; Lang, H. Ferrocene-Beauty and Function. Organometallics 2013, 20, 5623. [Google Scholar] [CrossRef]
- Naik, V.S.; Pragasam, A.; Jayanna, H.S.; Vinitha, G. Structural, linear optical, second and third-order nonlinear optical properties of two halogenated chalcone derivatives containing thiophene moiety. Chem. Phys. Lett. 2020, 761, 138051. [Google Scholar] [CrossRef]
- Terpstra, J.W.; van Leusen, A.M. A new synthesis of benzo [b] thiophenes and benzo [c] thiophenes by annulation of disubstituted thiophenes. J. Org. Chem. 1986, 51, 230–238. [Google Scholar] [CrossRef]
- Swarts, J.C.; Nafady, A.; Roudebush, J.H.; Trupia, S.; Geiger, W.E. One-electron oxidation of ruthenocene: Reactions of the ruthenocenium ion in gentle electrolyte media. Inorg. Chem. 2009, 48, 2156. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Jin, B.; Liu, P.; Cheng, L.; Cheng, W.; Zhang, S. Synthesis, electrochemistry and IR spectroelectrochemistry of bisferrocenyl bridged benzene derivates. J. Organomet. Chem. 2012, 697, 57. [Google Scholar] [CrossRef]
- Hildebrandt, A.; al Khalyfeh, K.; Nawroth, J.F.; Jordan, R. Electron transfer studies on conjugated ferrocenyl-containing oligomers. Organometallics 2016, 35, 3713. [Google Scholar] [CrossRef]
- Kalita, D.; Morisue, M.; Kobuke, Y. Synthesis and electrochemical properties of slipped-cofacial porphyrin dimers of ferrocene-functionalized Zn-imidazolyl-porphyrins as potential terminal electron donors in photosynthetic models. New J. Chem. 2006, 30, 77. [Google Scholar] [CrossRef]
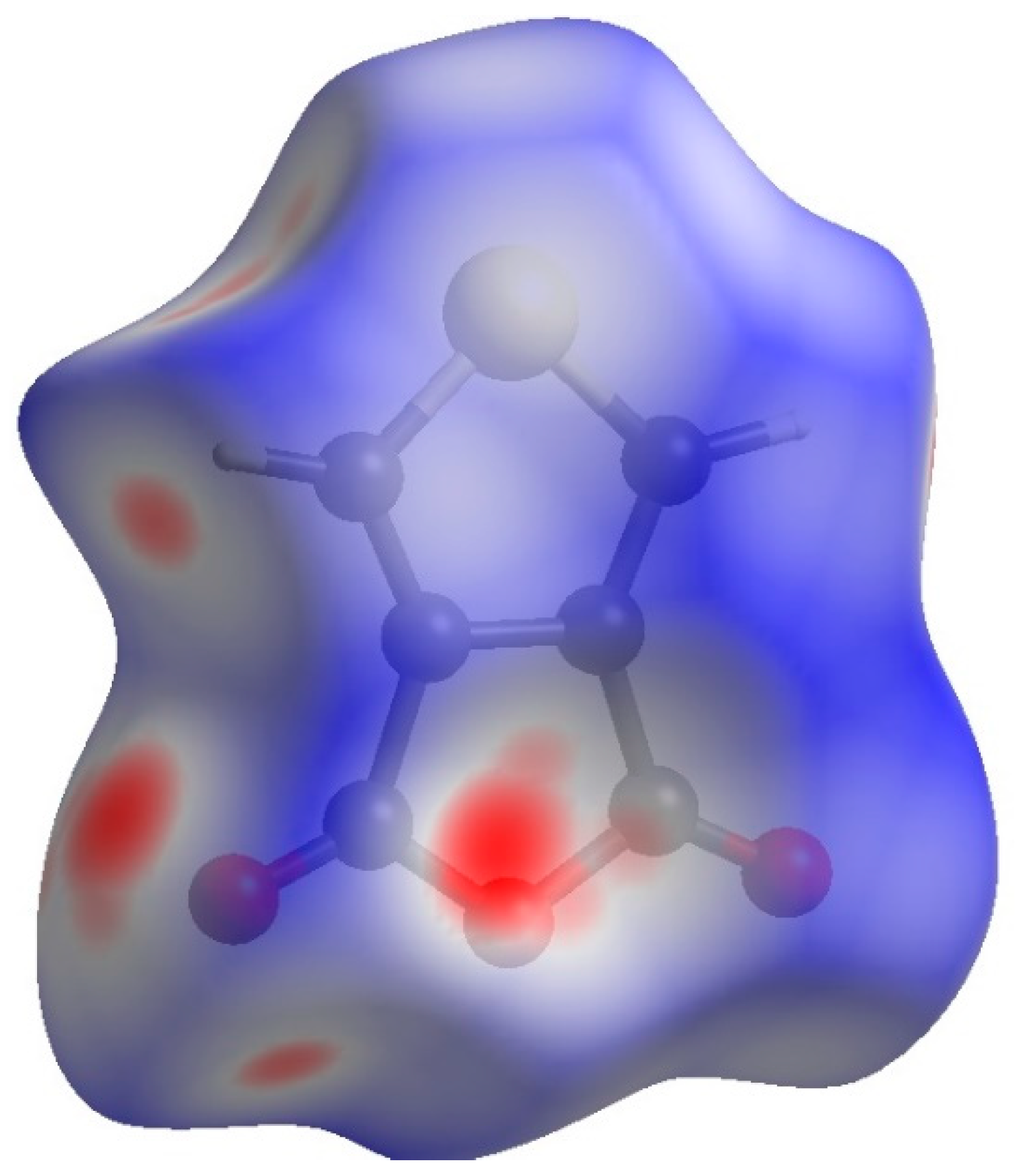
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
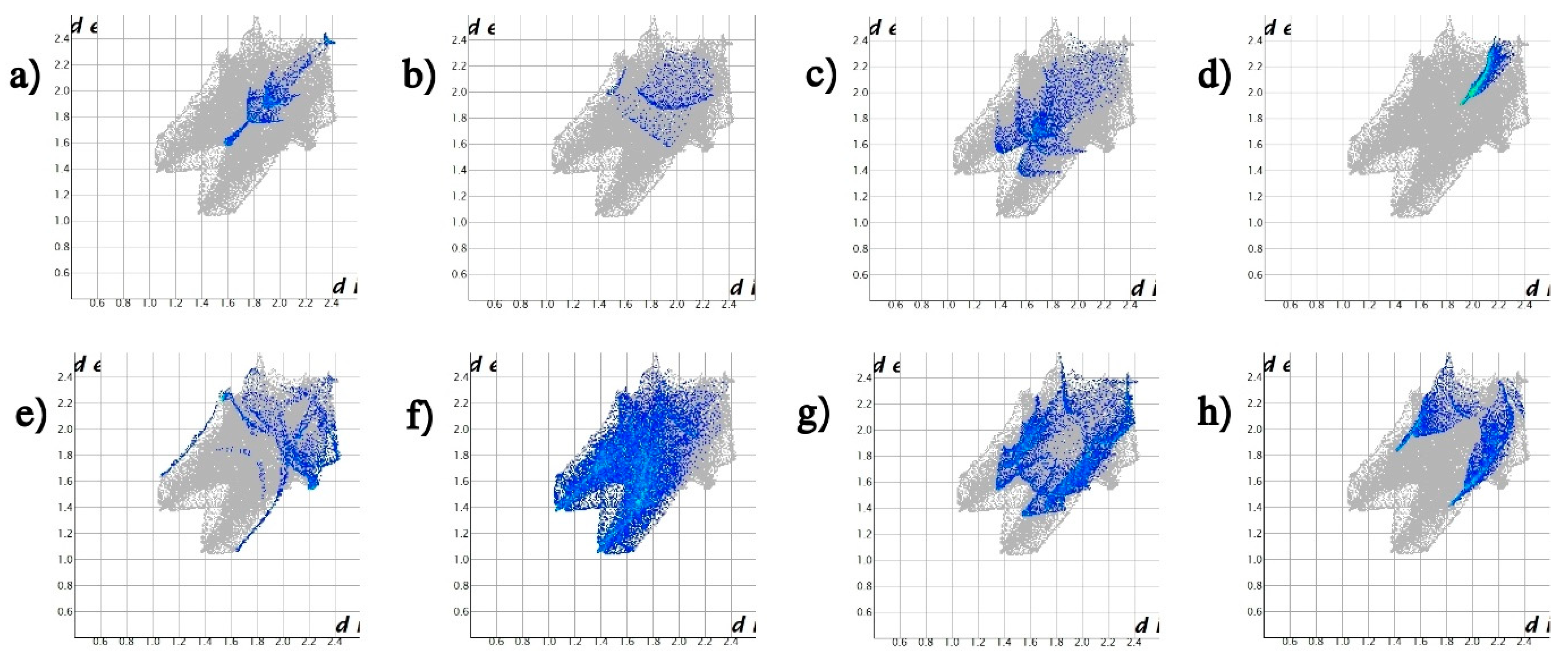
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Comm. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Liu, X.; Du, L.; Zhang, W.; Li, R.; Feng, F.; Feng, X.J. Syntheses, structures and hirshfeld surface analyses of two 3D supramolecules based on nitrogen-heterocyclic tricarboxylate ligand. Mol. Struct. 2019, 1194, 138–143. [Google Scholar] [CrossRef]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents. Pure Appl. Chem. 1984, 56, 461. [Google Scholar] [CrossRef]
- Nafady, A.; Geiger, W.E. Characterization of the successive one-electron oxidation products of the dicobalt fulvalenediyl (Fv) compound Co2Fv (CO) 4 and its phosphine-substituted product. Organometallics 2008, 27, 5624. [Google Scholar] [CrossRef]
- Guillot, B.; Enrique, E.; Huder, L.; Jelsch, C. MS19. O01. Acta Cryst. A 2014, 70, C279. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E 2019, 75, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claus, R.; Lewtak, J.P.; Müller, T.J.; Swarts, J.C. Structural influences on the electrochemistry of 1, 1′-di (hydroxyalkyl) ferrocenes. Structure of [Fe {η5-C5H4–CH (OH)–(CH2) 3OH} 2]. J. Organomet. Chem. 2013, 740, 61. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Paganelli, F.; Tagliavini, E.; Casolari, S.; Grepioni, F.J. Organometallic building blocks for crystal engineering. Synthesis, structure and hydrogen bonding interactions in [Fe (η5-C5H4-CH2 (CH3) OH) 2],[Fe (η5-C5H3 (CH3) COOH) 2], [Fe (η5-C5H4CH (CH3) NH (η5-C5H4CH (CH3))] and in the diaminecyclohexane salt [Fe (η5-C5H4COO) 2] 2−[(1S, 2S)-(NH3) 2C6H10] 2+·2 [H2O]. Organomet. Chem. 2001, 609, 637–639. [Google Scholar]
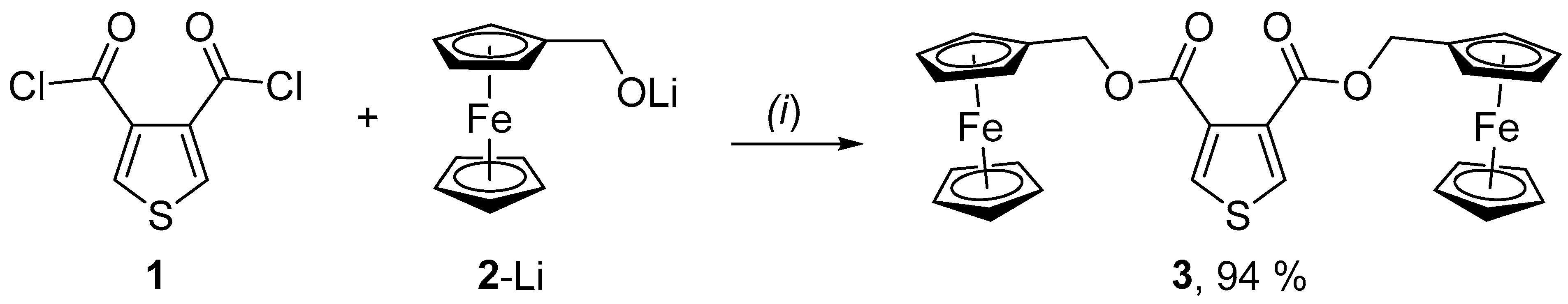


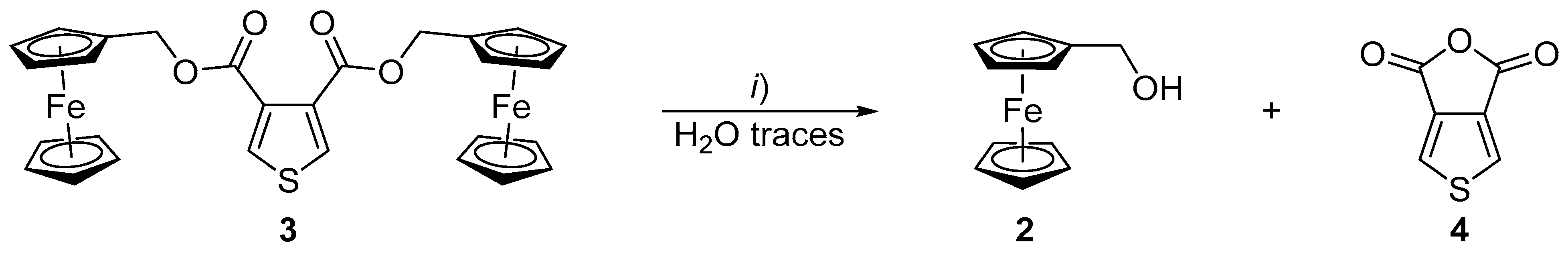




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
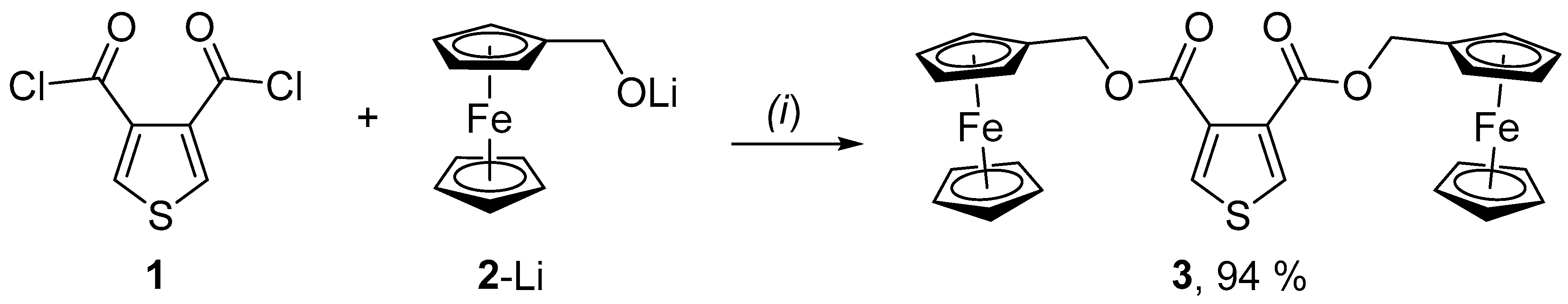{kind=link}
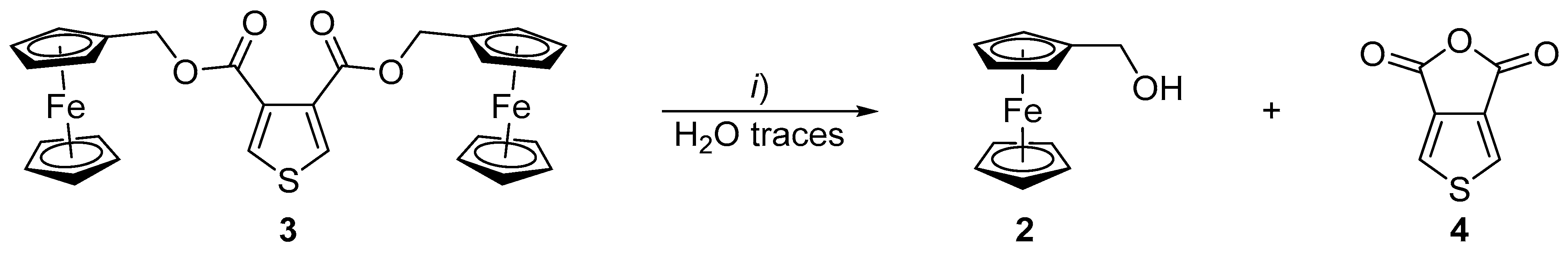{kind=link}
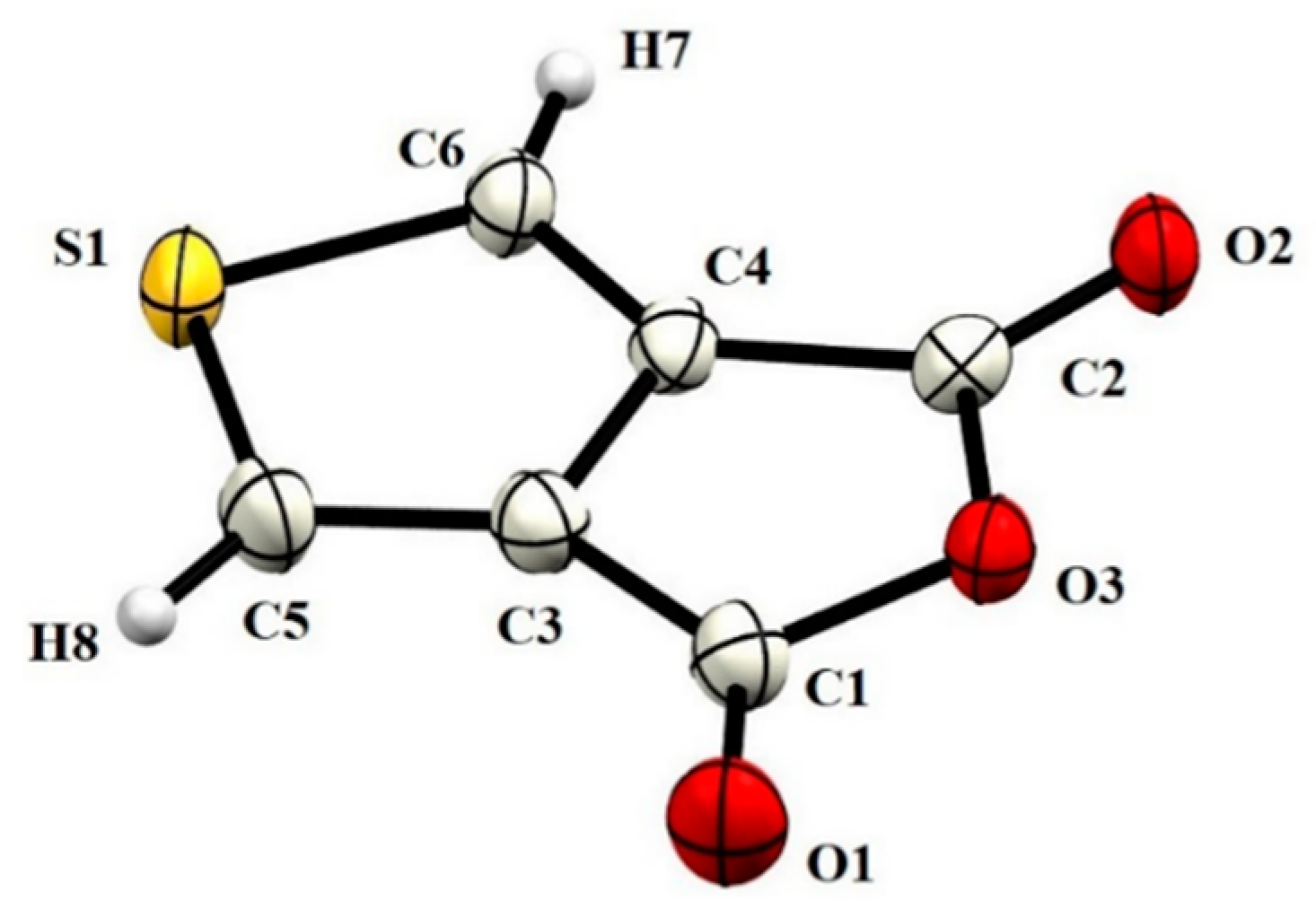
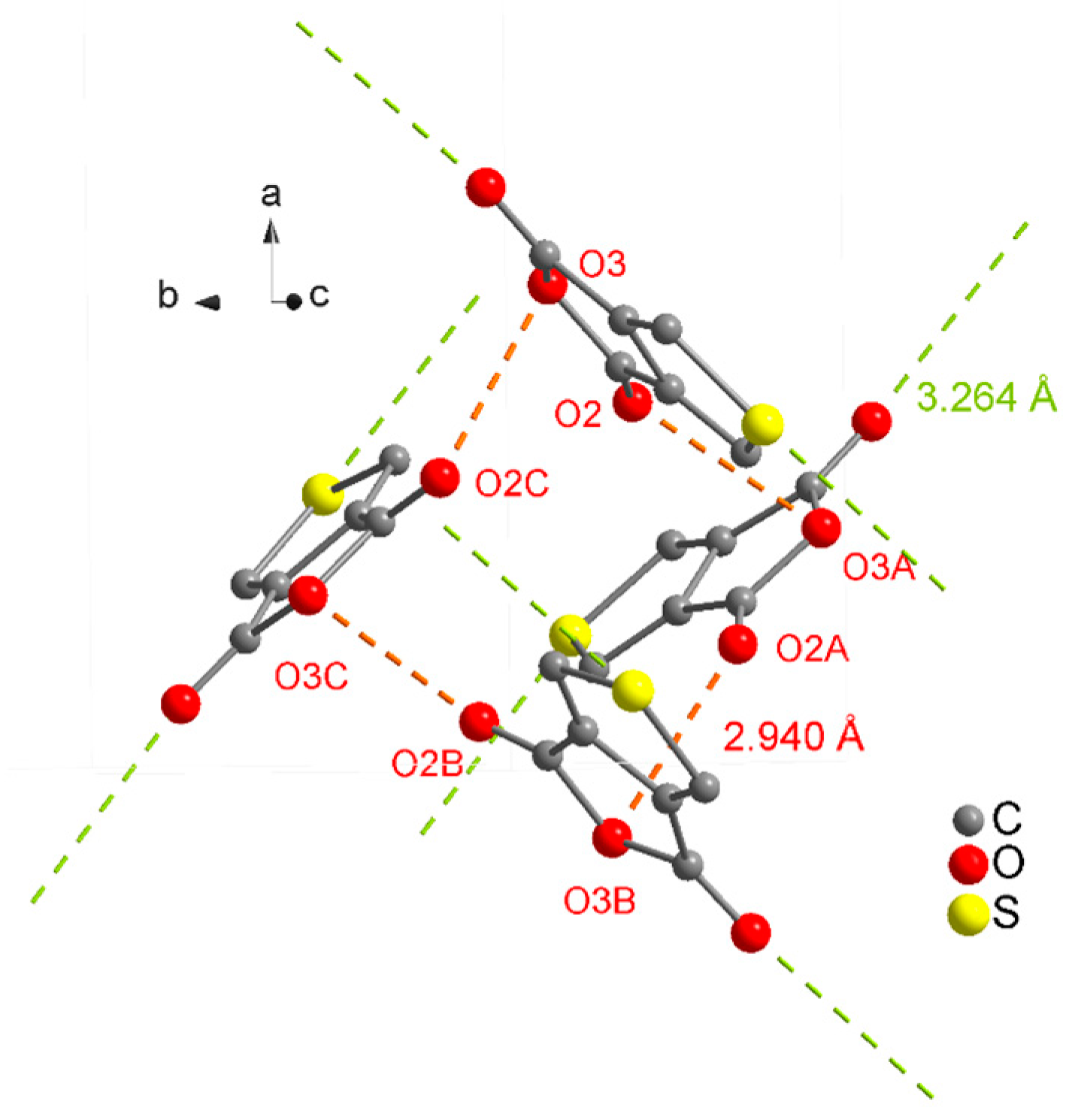
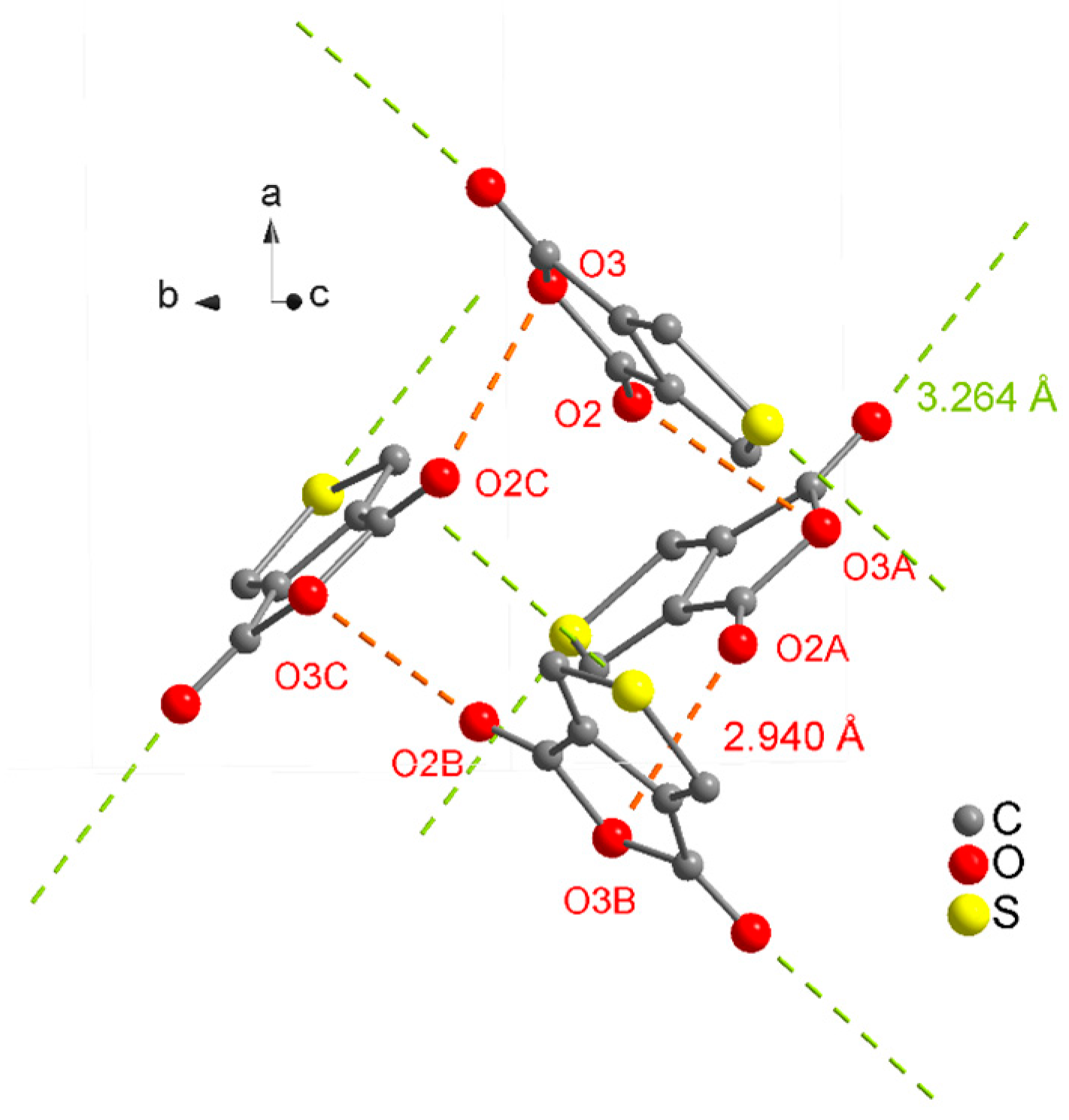
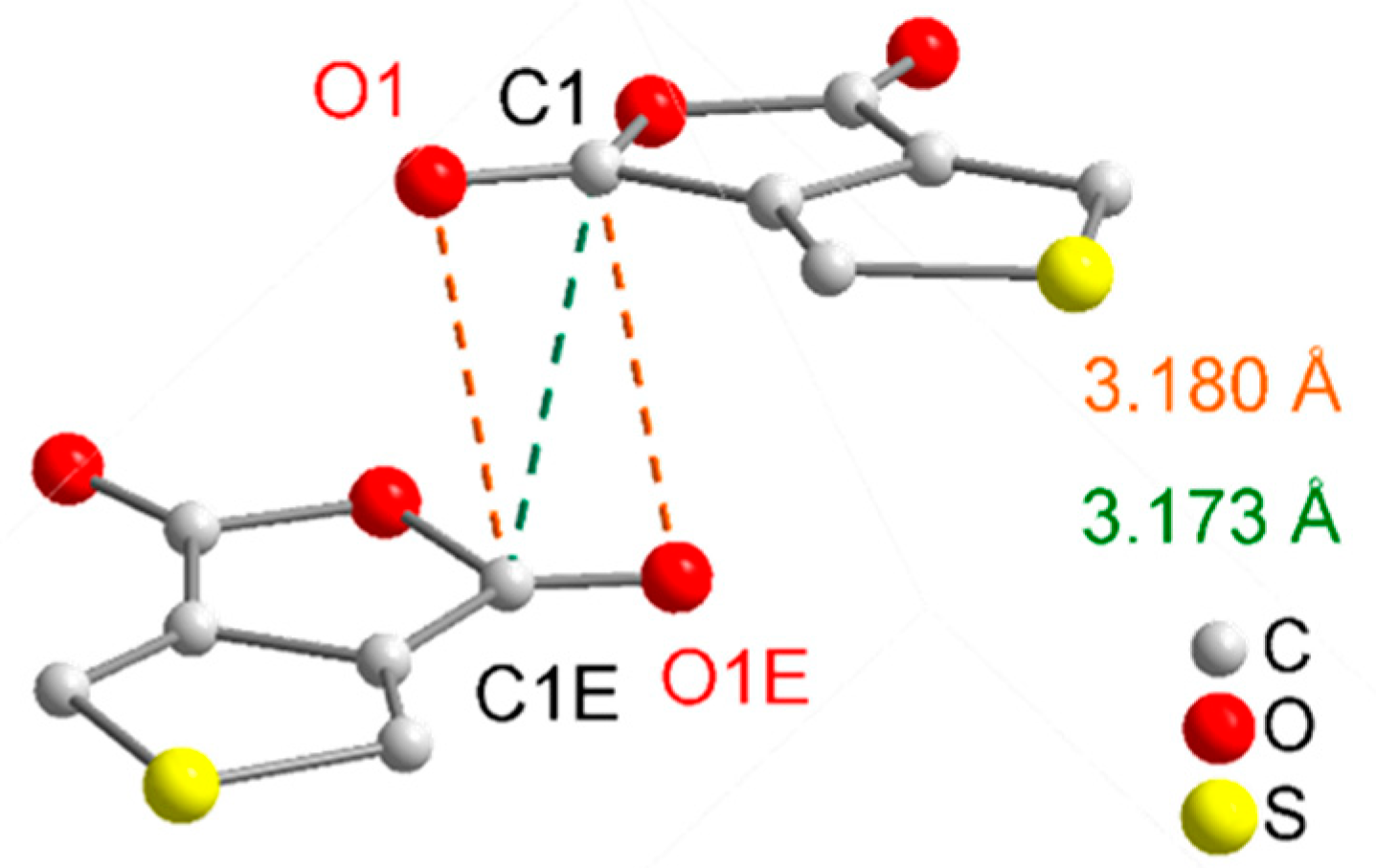
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| C6–H7···O2i | 0.93 | 2.54 | 3.2708(4) | 136 |
| C6–H7···O1ii | 0.93 | 2.57 | 3.1211(4) | 118 |
| Atoms | H | C | O | S |
|---|---|---|---|---|
| Surface interior (%) | 17.53 | 30.67 | 31.41 | 20.39 |
| Surface exterior (%) | 17.86 | 28.16 | 33.22 | 20.76 |
| Actual contacts merged (%) | ||||
| H | 0.68 | - | - | - |
| C | 6.37 | 10.00 | - | - |
| O | 24.98 | 22.97 | 2.50 | - |
| S | 2.68 | 9.48 | 11.69 | 8.65 |
| Equiprobable contacts merged (%) | ||||
| H | 3.13 | - | - | - |
| C | 10.41 | 8.64 | - | - |
| O | 11.43 | 19.03 | 10.43 | - |
| S | 7.28 | 12.11 | 13.30 | 4.23 |
| Enrichment reciprocal contacts merged (%) | ||||
| H | 0.22 | - | - | - |
| C | 0.61 | 1.16 | - | - |
| O | 2.18 | 1.21 | 0.24 | - |
| S | 0.37 | 0.78 | 0.88 | 2.04 |
| Contacts | Symmetry Operation | E_ele | E_pol | E_dis | E_rep | E_tot |
|---|---|---|---|---|---|---|
| O···O, O···H, and O···C | y, −x + 1/2, −z + 1/2 | −25.7 | −5.0 | −17.6 | 22.2 | −32.5 |
| S···O, O···H, and O···C | x + 1/2, y + 1/2, −z | −13.4 | −2.8 | −6.6 | 9.5 | −16.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghazzy, A.; Taher, D.; Korb, M.; Al Khalyfeh, K.; Helal, W.; Amarne, H.; Rüffer, T.; Ishtaiwi, Z.; Lang, H. Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate. Inorganics 2022, 10, 96. https://doi.org/10.3390/inorganics10070096
Ghazzy A, Taher D, Korb M, Al Khalyfeh K, Helal W, Amarne H, Rüffer T, Ishtaiwi Z, Lang H. Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate. Inorganics. 2022; 10(7):96. https://doi.org/10.3390/inorganics10070096
Chicago/Turabian StyleGhazzy, Asma, Deeb Taher, Marcus Korb, Khaled Al Khalyfeh, Wissam Helal, Hazem Amarne, Tobias Rüffer, Zakariyya Ishtaiwi, and Heinrich Lang. 2022. "Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate" Inorganics 10, no. 7: 96. https://doi.org/10.3390/inorganics10070096
APA StyleGhazzy, A., Taher, D., Korb, M., Al Khalyfeh, K., Helal, W., Amarne, H., Rüffer, T., Ishtaiwi, Z., & Lang, H. (2022). Rearrangement of Diferrocenyl 3,4-Thiophene Dicarboxylate. Inorganics, 10(7), 96. https://doi.org/10.3390/inorganics10070096