
Fluoro-Germanium (IV) Cations with Neutral Co-Ligands—Synthesis, Properties and Comparison with Neutral GeF4 Adducts

 , , and
, , and
Abstract
:
1. Introduction
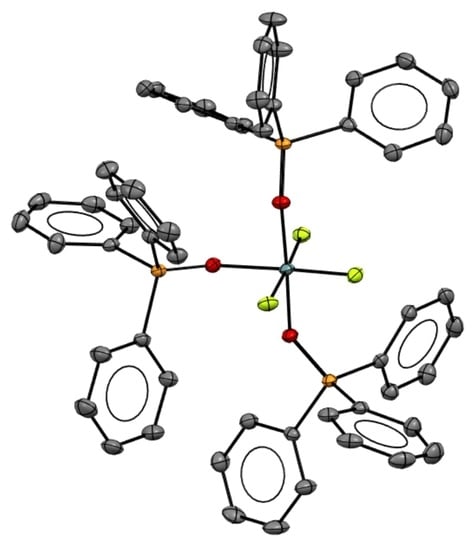
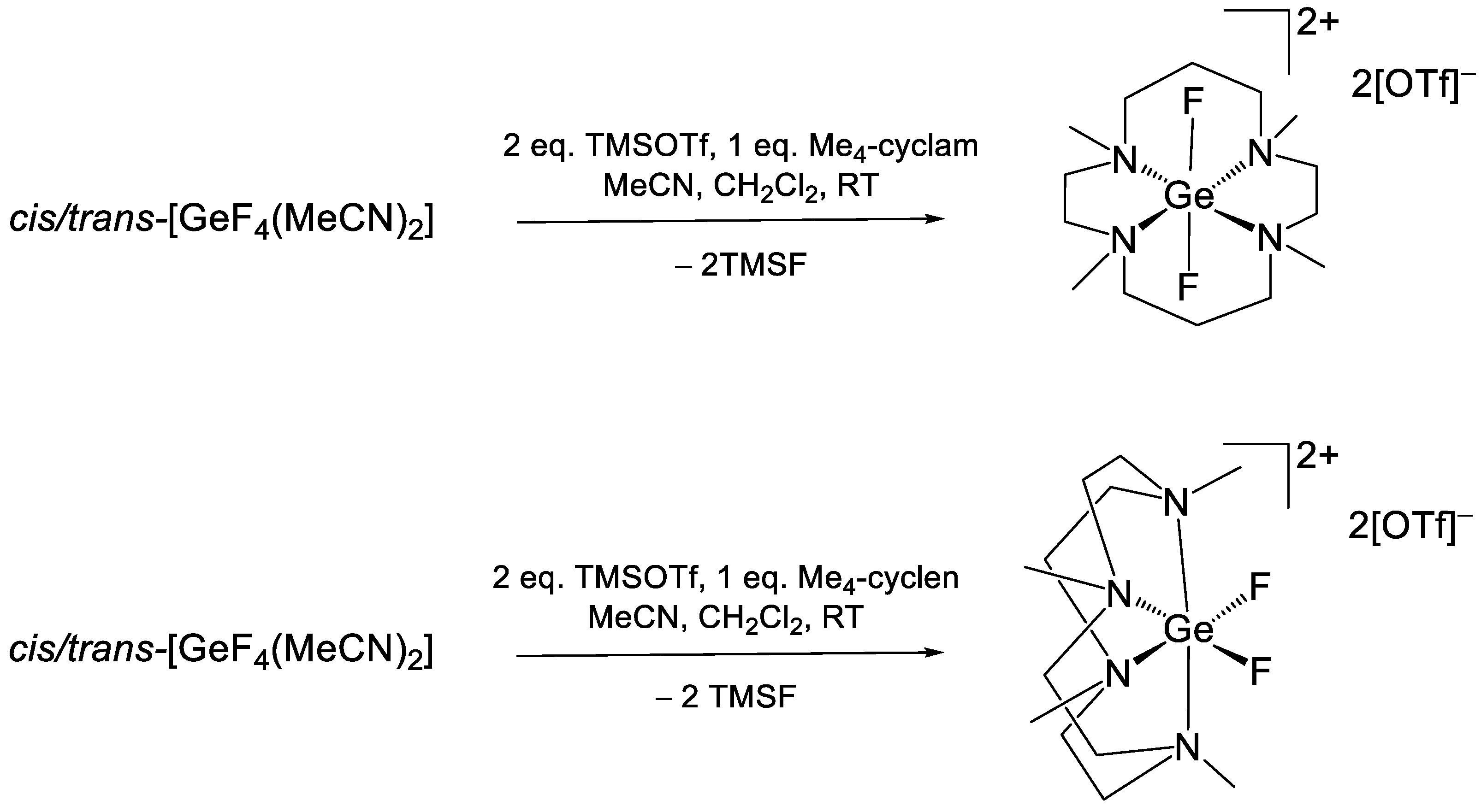
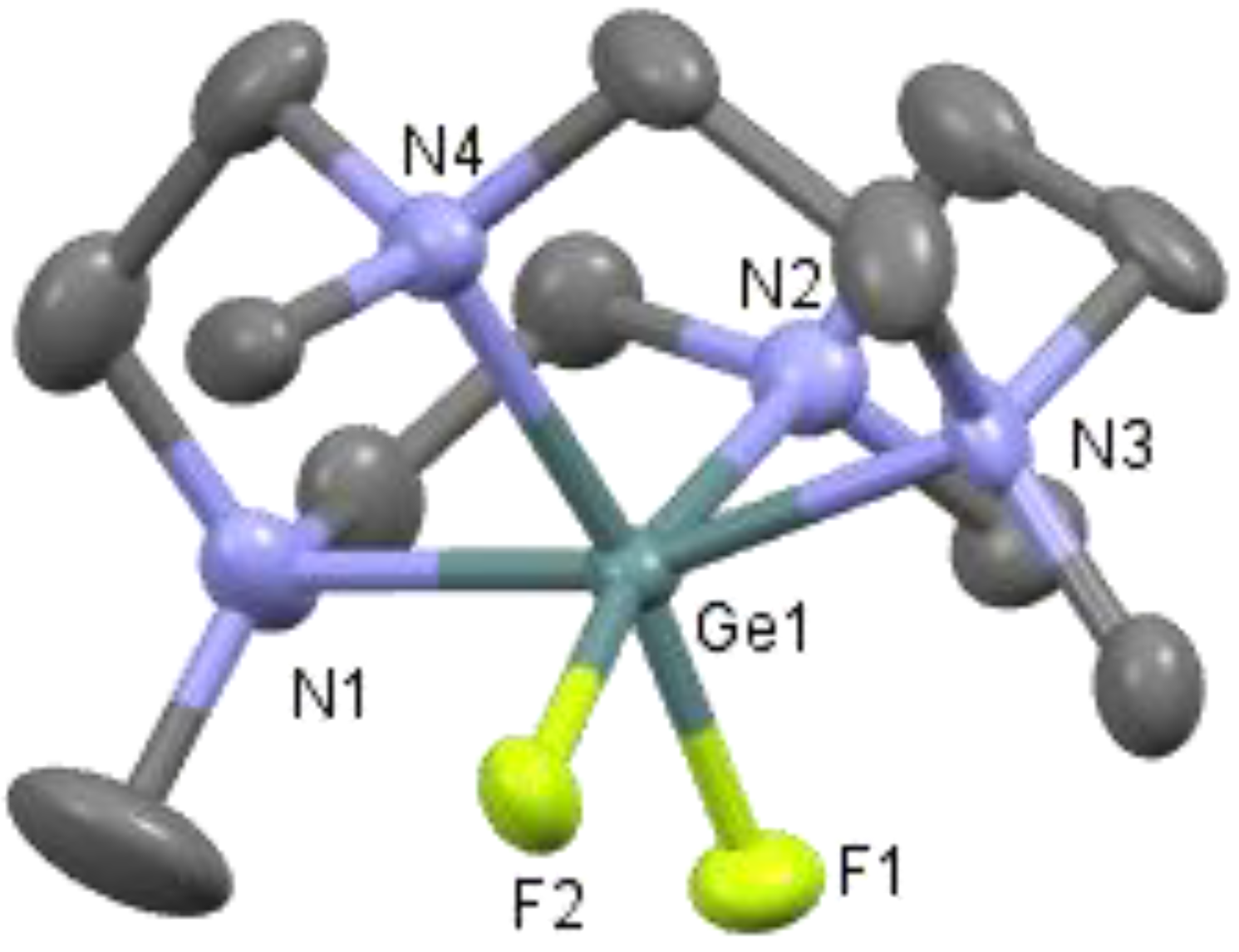
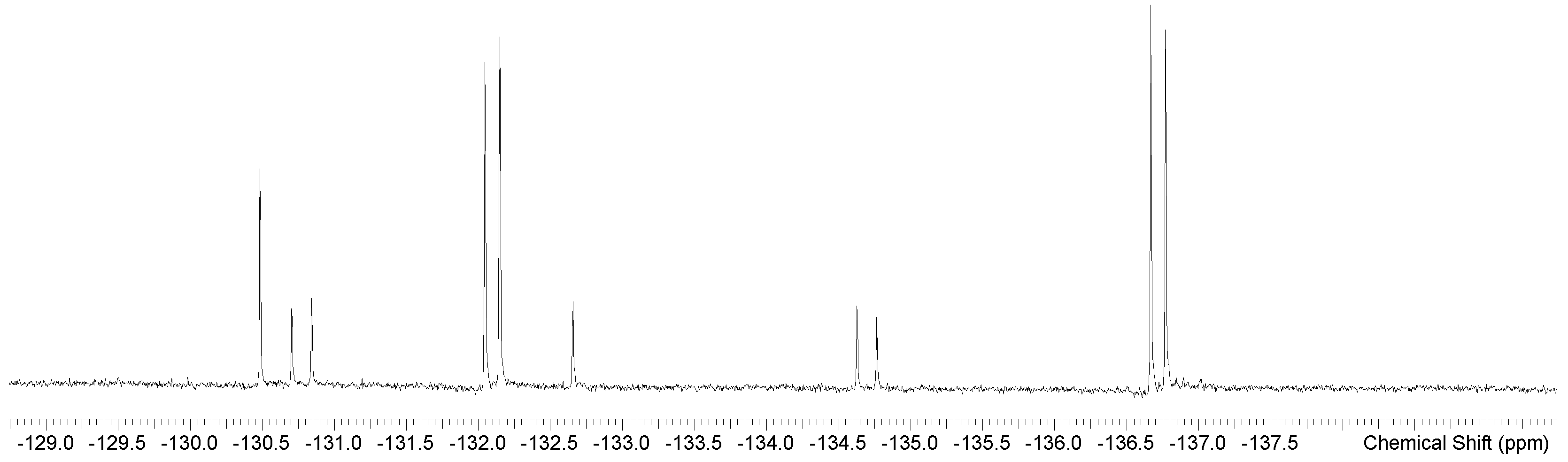
2. Results
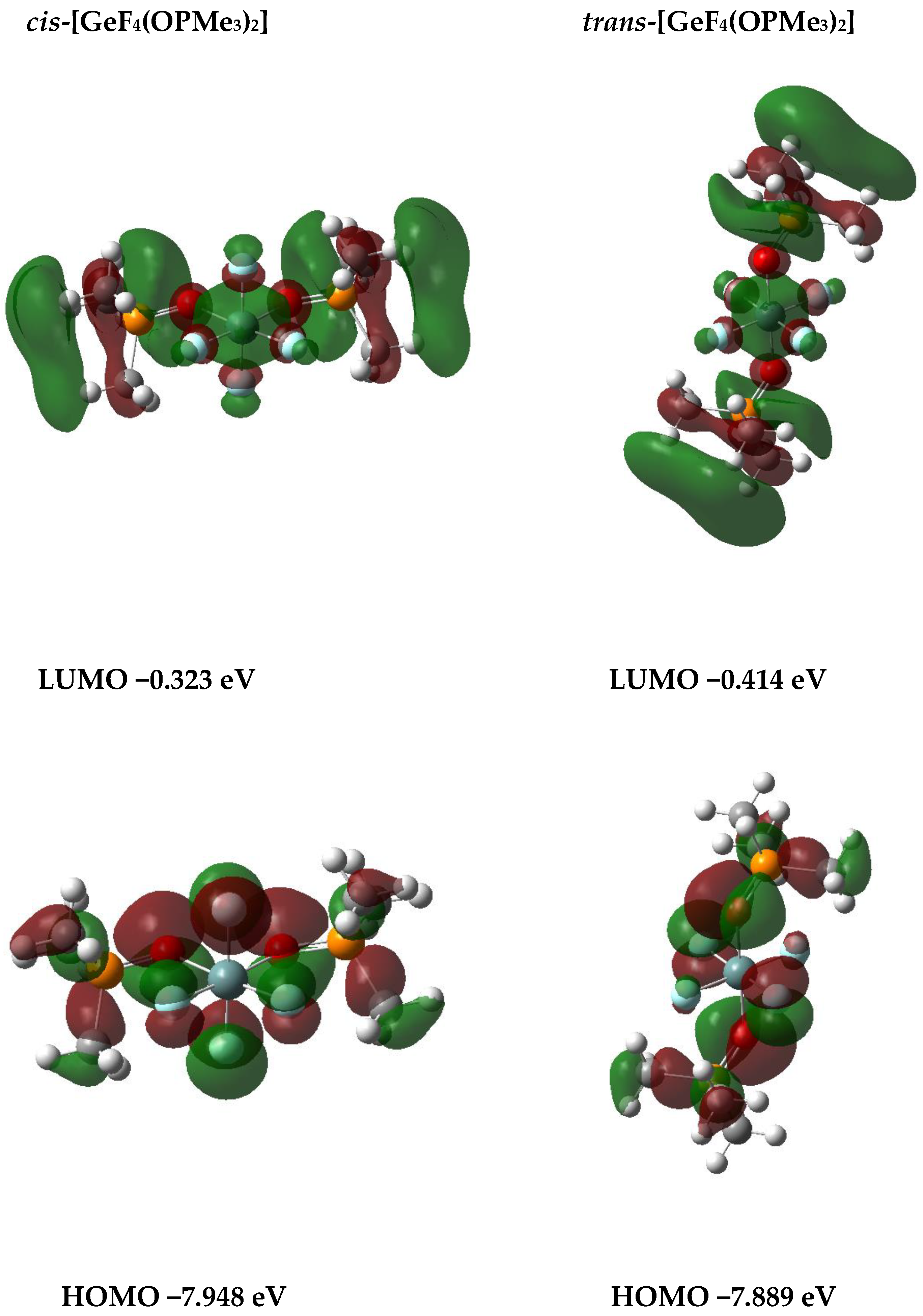
DFT Calculations
3. Materials and Methods
3.1. X-ray Experimental
3.2. DFT Calculations
3.3. Complex Syntheses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCleverty, J.A.; Meyer, T.J. (Eds.) Comprehensive Coordination Chemistry II; Elsevier: Oxford, UK, 2004; Volume 3. [Google Scholar]
- Downs, A.J. Chemistry of Aluminium, Gallium, Indium and Thallium; Blackie & Son Publishing: London, UK, 1993. [Google Scholar]
- Smith, P.J. (Ed.) The Chemistry of Tin; Chapman & Hall: London, UK, 1998. [Google Scholar]
- Levason, W.; Reid, G.; Zhang, W. Coordination complexes of silicon and germanium halides with neutral ligands. Coord. Chem. Rev. 2011, 255, 1319–1341. [Google Scholar] [CrossRef]
- Constable, E.C.; Parkin, G.; Que, L., Jr. (Eds.) Comprehensive Coordination Chemistry III; Elsevier: Oxford, UK, 2021; Volume 3. [Google Scholar]
- Benjamin, S.L.; Levason, W.; Reid, G. Medium and high oxidation state metal/non-metal fluoride and oxide–fluoride complexes with neutral donor ligands. Chem. Soc. Rev. 2013, 42, 1460–1499. [Google Scholar] [CrossRef] [PubMed]
- Levason, W.; Monzittu, F.M.; Reid, G. Coordination chemistry and applications of medium/high oxidation state metal and non-metal fluoride and oxide-fluoride complexes with neutral donor ligands. Coord. Chem. Rev. 2019, 391, 90–130. [Google Scholar] [CrossRef]
- Bhalla, R.; Darby, C.; Levason, W.; Luthra, S.K.; McRobbie, G.; Reid, G.; Sanderson, G.; Zhang, W. Triaza-macrocyclic complexes of aluminium, gallium and indium halides: Fast 18F and 19F incorporation via halide exchange under mild conditions in aqueous solution. Chem. Sci. 2014, 5, 381–391. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, R.; Levason, W.; Luthra, S.K.; McRobbie, G.; Monzittu, F.M.; Palmer, J.; Reid, G.; Sanderson, G.; Zhang, W. Hydrothermal synthesis of Group 13 metal trifluoride complexes with neutral N-donor ligands. Dalton Trans. 2015, 44, 9569–9580. [Google Scholar] [CrossRef] [Green Version]
- Chansaenpak, K.; Vabre, B.; Gabbaï, F.P. [18F]-Group 13 fluoride derivatives as radiotracers for positron emission tomography. Chem. Soc. Rev. 2016, 45, 954–971. [Google Scholar] [CrossRef]
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844. [Google Scholar] [CrossRef]
- Tudela, D.; Rey, F. Reactions of tin (II) fluoride with halogens. Z. Anorg. Allgem. Chem. 1989, 575, 202–208. [Google Scholar] [CrossRef]
- Cheng, F.; Davis, M.F.; Hector, A.L.; Levason, W.; Reid, G.; Webster, M.; Zhang, W. Synthesis, spectroscopic and structural systematics of complexes of germanium(IV) halides (GeX4, X = F, Cl, Br or I) with phosphane oxides and related oxygen donor ligands. Eur. J. Inorg. Chem. 2007, 2007, 2488–2495. [Google Scholar] [CrossRef]
- Cheng, F.; Davis, M.F.; Hector, A.L.; Levason, W.; Reid, G.; Webster, M.; Zhang, W. Synthesis, spectroscopic and structural systematics of complexes of germanium(IV) halides (GeX4, X = F, Cl, Br or I) with mono-, bi- and tri-dentate and macrocyclic nitrogen donor ligands. Eur. J. Inorg. Chem. 2007, 2007, 4897–4905. [Google Scholar] [CrossRef]
- Davis, M.F.; Levason, W.; Reid, G.; Webster, M. Complexes of germanium(IV) fluoride with phosphane ligands: Structural and spectroscopic authentication of germanium(IV) phosphane complexes. Dalton Trans. 2008, 2008, 2261–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.P.; Levason, W.; Reid, G. Neutral and cationic germanium(iv) fluoride complexes with phosphine coordination—Synthesis, spectroscopy and structures. Dalton Trans. 2021, 50, 17751–17765. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.F.; Levason, W.; Reid, G.; Webster, M.; Zhang, W. The first examples of germanium tetrafluoride and tin tetrafluoride complexes with soft thioether coordination—Synthesis, properties and crystal structures. Dalton Trans. 2008, 2008, 533–538. [Google Scholar] [CrossRef]
- Suter, R.; Swidan, A.; Macdonald, C.L.B.; Burford, N. Oxidation of a germanium(II) dication to access cationic germanium(IV) fluorides. Chem. Commun. 2018, 54, 4140–4143. [Google Scholar] [CrossRef]
- MacDonald, E.; Doyle, L.; Chitnis, S.S.; Werner-Zwanziger, U.; Burford, N.; Decken, A. Me3P complexes of p-block Lewis acids SnCl4, SnCl3+ and SnCl22. Chem. Commun. 2012, 48, 7922–7924. [Google Scholar] [CrossRef] [PubMed]
- Greenacre, V.K.; Levason, W.; King, R.P.; Reid, G. Neutral and cationic phosphine and arsine complexes of tin (IV) halides: Synthesis, properties, structures and anion influence. Dalton Trans. 2019, 48, 17097–17105. [Google Scholar] [CrossRef] [Green Version]
- King, R.P.; Woodward, M.S.; Grigg, J.; McRobbie, G.; Levason, W.; Reid, G. Tin (IV) fluoride complexes with neutral phosphine coordination and comparisons with hard N-and O-donor ligands. Dalton Trans. 2021, 50, 14400–14410. [Google Scholar] [CrossRef]
- Everett, M.; Jolleys, A.; Levason, W.; Light, M.E.; Pugh, D.; Reid, G. Cationic aza-macrocyclic complexes of germanium (II) and silicon (IV). Dalton Trans. 2015, 44, 20898–20905. [Google Scholar] [CrossRef] [Green Version]
- Waller, A.W.; Weiss, N.M.; Decato, D.A.; Phillips, J.A. Structural and energetic properties of haloacetonitrile–GeF4 complexes. J. Mol. Struct. 2017, 1130, 984–993. [Google Scholar] [CrossRef] [Green Version]
- Levason, W.; Patel, B.; Popham, M.C.; Reid, G.; Webster, M. Scandium, yttrium and lanthanum nitrate complexes of tertiary arsine oxides: Synthesis and multinuclear spectroscopic studies. X-ray structures of [M(Me3AsO)6](NO3)3 (M = Sc or Y), [Sc(Ph3AsO)3(NO3)2]NO3, [M″(Ph3AsO)4(NO3)2]NO3 (M″ = Y or La) and [La(Ph3AsO)2(EtOH)(NO3)3]. Polyhedron 2001, 20, 2711–2720. [Google Scholar]
- Jura, M.; Levason, W.; Petts, E.; Reid, G.; Webster, M.; Zhang, W. Taking TiF4 complexes to extremes-the first examples with phosphine co-ligands. Dalton Trans. 2010, 39, 10264–10271. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, S.L.; Levason, W.; Pugh, D.; Reid, G.; Zhang, W. Preparation and structures of coordination complexes of the very hard Lewis acids ZrF4 and HfF4. Dalton Trans. 2012, 41, 12548–12557. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, J.E.; Irish, D.E. Vibrational spectrum of the hexafluorogermanate ion. Inorg. Chem. 1964, 3, 1134–1137. [Google Scholar] [CrossRef]
- Wieghardt, K.; Chaudhuri, P.; Nuber, B.; Weiss, J. New triply hydroxo-bridged complexes of chromium(III), cobalt(III), and rhodium(III): Crystal structure of tris(.mu.-hydroxo)bis[(1,4,7-trimethyl-1,4,7-triazacyclononane)chromium(III)] triiodide trihydrate. Inorg. Chem. 1982, 21, 3086–3090. [Google Scholar] [CrossRef]
- Barefield, E.K.; Wagner, F. Metal complexes of 1, 4, 8, 11-tetramethyl-1, 4, 8, 11-tetraazacyclotetradecane, N-tetramethylcyclam. Inorg. Chem. 1973, 12, 2435–2439. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. Synthesis and Structure of Ferrocenol Esters. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16W, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Yu, Y.-R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Solvent Temperature | 19F{1H} NMR/ppm a | 2JFF/Hz | 31P{1H} NMR/ppm | Reference |
|---|---|---|---|---|---|
| [GeF4(dmso)2] cis trans | CD3NO2 253 K | −115.3 (t), −129.8 (t) −115.4 (s) | 61 | This work | |
| [GeF3(dmso)3][OTf] mer fac | CD3NO2 253 K | −109.8 (d), −121.7 (t) −122.2 (s) | 71 | This work | |
| [GeF4(dmf)2] cis trans | CD3NO2 253 K | −125.4 (t), −135.8 (t) −125.4 (s) | 59 | This work | |
| [GeF3(dmf)3][OTf] mer fac | CD3NO2 253 K | −126.1 (d) −135.0 (t) −134.5 (s) | 64 | This work | |
| [GeF4(py)2] trans | CDCl3 253 K | −125.7 (s) | Ref. [14] | ||
| [GeF3(py)3][OTf] mer fac | CD2Cl2 298 K | −122.0 (d), −137.3 (t) −149.2 (s) | 55 | This work | |
| [GeF4(pyNO)2] cis trans | CD3NO2 298 K 253 K | −142.8 (br s) −136.1 (t), −133.1 (t) −129.8 (s) | 58 | This work | |
| [GeF3(pyNO)3][OTf] mer fac | CD3NO2 253 K | −136.8 (d), −143.0 (t) −141.8 (s) | 65 | This work | |
| [GeF4(OPPh3)2] cis trans | CDCl3 253 K | −100.9 (t), −120.6 (t) −105.3 (s) | 64 | 40.8 (s) 40.2 (s) | Ref. [13] |
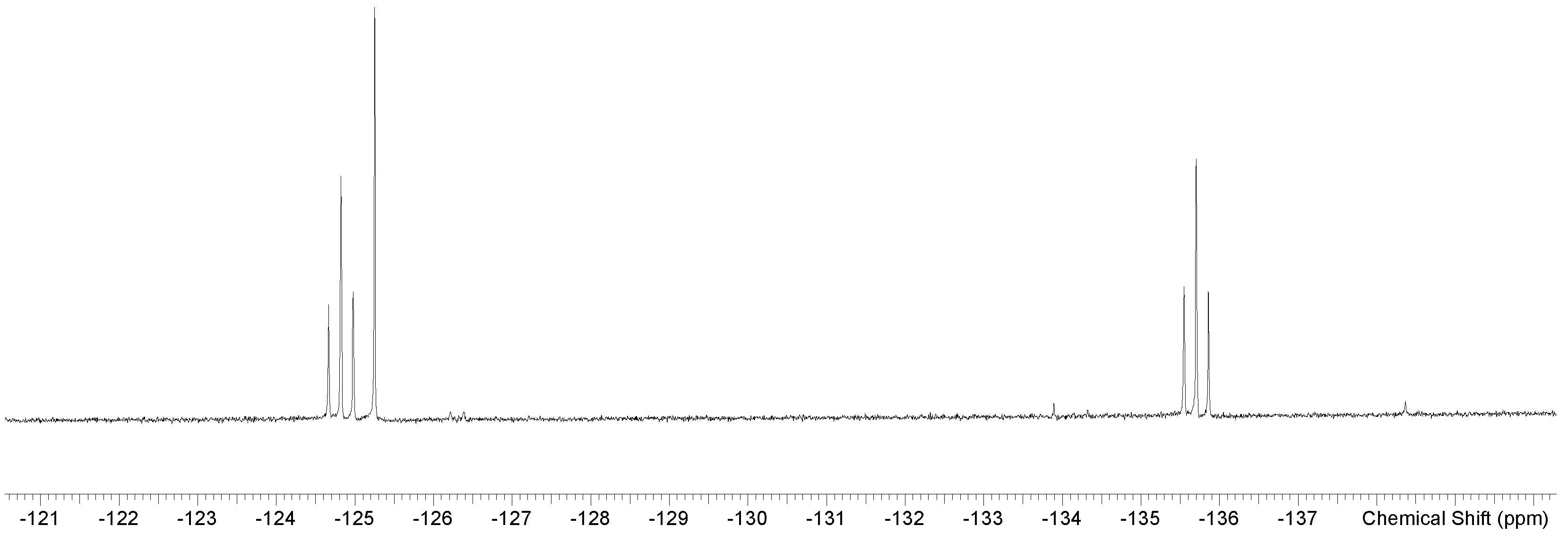
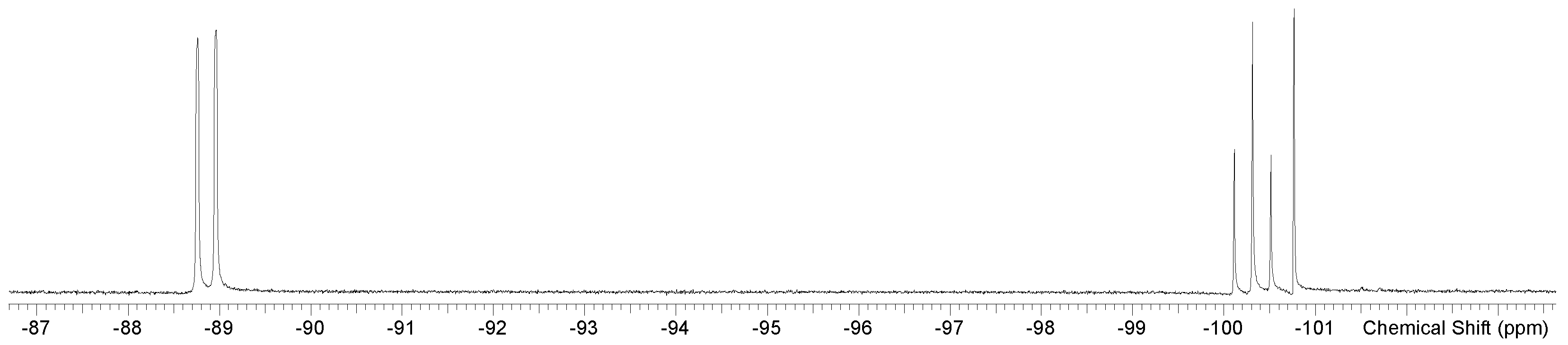
| [GeF3(OPPh3)3][OTf] mer fac | CD2Cl2 298 K | −89.0(d), −100.4(t) −100.9(s) | 76 | 44.1 (s) 41.7 (s) 43.7 (s) | This work |
| [GeF4(OPMe3)2] cis trans | CD2Cl2 298 K | −107.6 (t), −121.6 (t) −109.9 (s) | 58 | 65.1 (s) 65.8 (s) | Ref. [13] |
| [GeF3(OPMe3)3][OTf] mer fac | CD3NO2 298 K | −95.6 (d), −106.6 (t) −93.0 (s) | 64 | 67.4 (m), 66.9 (m) 70.4 (s) | This work |
| [GeF4(OAsPh3)2] cis trans | CD2Cl2 298 K | −94.4 (t), −112.9 (t) −98.2 (s) | 60 | Ref. [13] | |
| [GeF3(OAsPh3)3]OTf b mer fac | CD3NO2 298K | −79.3 (d), −89.5 (t) −89.9 (s) | 68 | This work | |
| [GeF2(OAsPh3)4][OTf]2 b cis trans | CD3NO2 298K | −65.1 (s) −59.1 (s) | This work | ||
| [GeF4(MeCN)2] cis trans | CD2Cl2 180 K | −101.2 (t), −134.2 (t) −108.2 (s) | 55 | Ref. [13] | |
| fac-[GeF3(Me3-tacn)][OTf] | CD3NO2 298 K | −151.7 (s) | This work | ||
| mer-[GeF3(terpy)][OTf] | CD2Cl2 298 K | −115.9 (d), −153.0 (t) | 68 | This work | |
| [GeF2(Me4-cyclen)][OTf]2 cis | CD3NO2 298 K | −132.3 (s) | This work | ||
| [GeF2(Me4-cyclam)][OTf]2 trans | CD3NO2 298 K | −136.8 (d) −134.8 (d) −132.7 (s) −132.2 (d) −130.8 (d) −130.5 (s) | 38 52 38 52 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woodward, M.S.; King, R.P.; Bannister, R.D.; Grigg, J.; McRobbie, G.; Levason, W.; Reid, G. Fluoro-Germanium (IV) Cations with Neutral Co-Ligands—Synthesis, Properties and Comparison with Neutral GeF4 Adducts. Inorganics 2022, 10, 107. https://doi.org/10.3390/inorganics10080107
Woodward MS, King RP, Bannister RD, Grigg J, McRobbie G, Levason W, Reid G. Fluoro-Germanium (IV) Cations with Neutral Co-Ligands—Synthesis, Properties and Comparison with Neutral GeF4 Adducts. Inorganics. 2022; 10(8):107. https://doi.org/10.3390/inorganics10080107
Chicago/Turabian StyleWoodward, Madeleine S., Rhys P. King, Robert D. Bannister, Julian Grigg, Graeme McRobbie, William Levason, and Gillian Reid. 2022. "Fluoro-Germanium (IV) Cations with Neutral Co-Ligands—Synthesis, Properties and Comparison with Neutral GeF4 Adducts" Inorganics 10, no. 8: 107. https://doi.org/10.3390/inorganics10080107
APA StyleWoodward, M. S., King, R. P., Bannister, R. D., Grigg, J., McRobbie, G., Levason, W., & Reid, G. (2022). Fluoro-Germanium (IV) Cations with Neutral Co-Ligands—Synthesis, Properties and Comparison with Neutral GeF4 Adducts. Inorganics, 10(8), 107. https://doi.org/10.3390/inorganics10080107