
Crystallographic and Theoretical Study of Osme Bonds in Nitrido-Osmium(VI) Complexes



Abstract
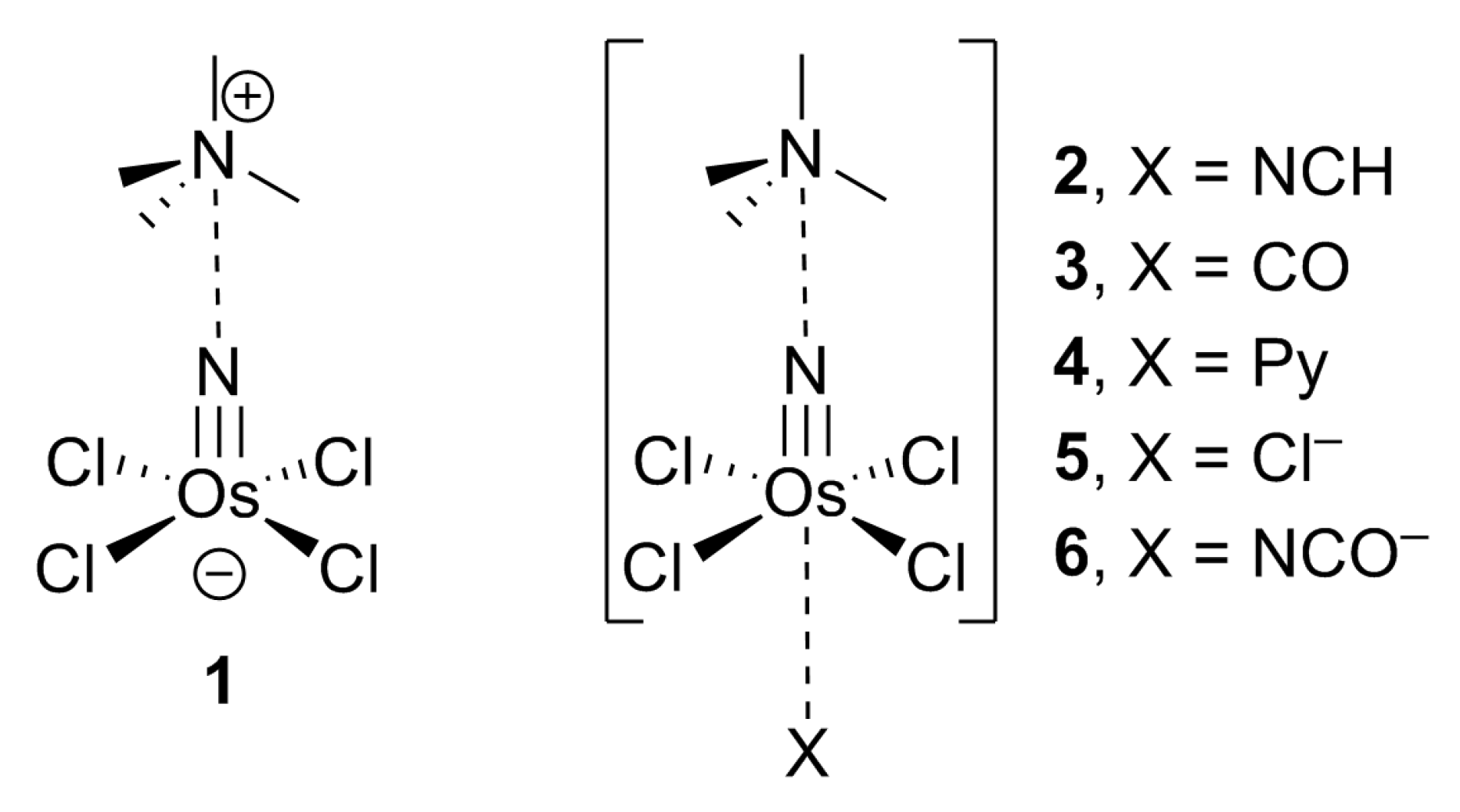
:1. Introduction
2. Results and Discussion
2.1. CSD Search
2.2. MEP Surface Anaysis
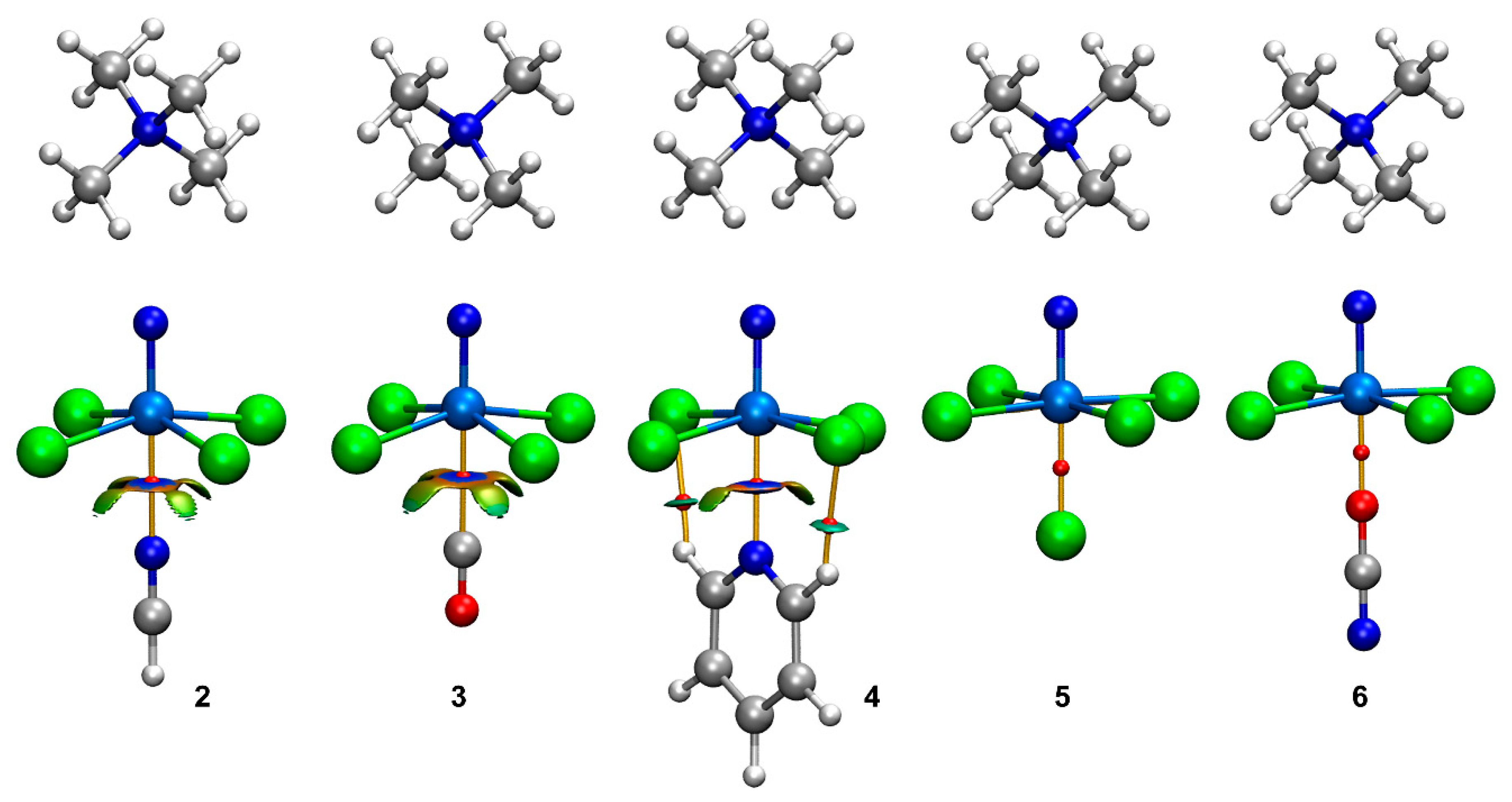
2.3. Energetic and Geometric Analyses
2.4. Combined QTAIM/NCIplot Analysis
2.5. NBO Analysis
2.6. Electron Density (ED) vs. Electrostatic Potential (ESP) Analysis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming interactions from the electrophilic site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Scheiner, S.; Michalczyk, M.; Zierkiewicz, W. Coordination of anions by noncovalently bonded σ-hole ligands. Coord. Chem. Rev. 2020, 405, 213136. [Google Scholar] [CrossRef]
- Bauza, A.; Frontera, A. σ/π-Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Beer, P.D. Sigma-hole interactions in anion recognition. Chem 2018, 4, 731–783. [Google Scholar] [CrossRef]
- Kolar, M.H.; Hobza, P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C(sp3) atoms as tetrel bond donors: A crystallographic survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The chalcogen bond in crystalline solids: A world parallel to halogen bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Strakova, K.; Assies, L.; Goujon, A.; Piazzolla, F.; Humeniuk, H.V.; Matile, S. Dithienothiophenes at work: Access to mechanosensitive fluorescent probes, chalcogen-bonding catalysis, and beyond. Chem. Rev. 2019, 119, 10977–11005. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, J.; Ling, K.B.; Cockroft, S.L. The origin of chalcogen-bonding interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef]
- Riwar, L.J.; Trapp, N.; Root, K.; Zenobi, R.; Diederich, F. Supramolecular capsules: Strong versus weak chalcogen bonding. Angew. Chem. Int. Ed. 2018, 57, 17259–17264. [Google Scholar] [CrossRef]
- Macchione, M.; Goujon, A.; Strakova, K.; Humeniuk, H.V.; Licari, G.; Tajkhorshid, E.; Sakai, N.; Matile, S. A chalcogen-bonding cascade switch for Planarizable push-pull probes. Angew. Chem. Int. Ed. 2019, 58, 15752–15756. [Google Scholar] [CrossRef] [PubMed]
- Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S.M. Chalcogen bonding catalysis of a nitro-Michael reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927. [Google Scholar] [CrossRef]
- Wang, W.; Zhu, H.; Liu, S.; Zhao, Z.; Zhang, L.; Hao, J.; Wang, Y. Chalcogen-chalcogen bonding catalysis enables assembly of discrete molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef]
- Borissov, A.; Marques, I.; Lim, J.Y.C.; Félix, V.; Smith, M.D.; Beer, P.D. Anion recognition in water by charge-neutral halogen and chalcogen bonding foldamer receptors. J. Am. Chem. Soc. 2019, 141, 4119–4129. [Google Scholar] [CrossRef]
- Benz, S.; Macchione, M.; Verolet, Q.; Mareda, J.; Sakai, N.; Matile, S. Anion transport with chalcogen bonds. J. Am. Chem. Soc. 2016, 138, 9093–9096. [Google Scholar] [CrossRef] [Green Version]
- Heywood, V.L.; Alford, T.P.J.; Roeleveld, J.J.; Lekanne Deprez, S.J.; Verhoofstad, A.; van der Vlugt, J.I.; Domingos, S.R.; Schnell, M.; Davis, A.P.; Mooibroek, T.J. Observations of tetrel bonding between sp3-carbon and THF. Chem. Sci. 2020, 11, 5289–5293. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. σ-holes and π-holes: Similarities and differences. J. Comput. Chem. 2018, 39, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.H.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.H.; Brinck, T. Extending the σ-hole concept to metals: An electrostatic interpretation of the effects of nanostructure in gold and platinum catalysis. J. Am. Chem. Soc. 2017, 139, 11012–11015. [Google Scholar] [CrossRef]
- Legon, A.C.; Walker, N.R. What’s in a name? ‘Coinage-metal’ non-covalent bonds and their definition. Phys. Chem. Chem. Phys. 2018, 20, 19332–19338. [Google Scholar] [CrossRef]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium bonds: Noncovalent interactions involving group 12 elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Noncovalent interactions involving group 6 in biological systems: The case of molybdopterin and Tungstopterin cofactors. Chem Eur. J. 2022, 28, e202201660. [Google Scholar] [CrossRef]
- Daolio, A.; Pizzi, A.; Terraneo, G.; Frontera, A.; Resnati, G. Anion···anion interactions involving σ-holes of perrhenate, pertechnetate and permanganate anions. ChemPhysChem 2021, 22, 2281–2285. [Google Scholar] [CrossRef]
- Daolio, A.; Pizzi, A.; Calabrese, M.; Terraneo, G.; Bordignon, S.; Frontera, A.; Resnati, G. Molecular electrostatic potential and noncovalent interactions in derivatives of group 8 elements. Angew. Chem. Int. Ed. 2021, 60, 20723–20727. [Google Scholar] [CrossRef]
- Daolio, A.; Pizzi, A.; Calabrese, M.; Terraneo, G.; Bordignon, S.; Frontera, A.; Resnati, G. Osme bond: Anisotropic distribution of electron density in action. Acta Cryst. 2021, A77, C800. [Google Scholar]
- Chan, P.M.; Yu, W.-Y.; Che, C.-M.; Cheung, K.-K. Nitrido-ruthenium(VI) and -osmium(VI) complexes containing chelating multianionic (N, O) ligands. Synthesis, crystal structures and reactions with triphenylphosphine. J. Chem. Soc. Dalton Trans. 1998, 3183–3190. [Google Scholar] [CrossRef]
- Che, C.-M.; Lau, T.-C.; Lam, H.-W.; Poon, C.-K. Metal-nitrido photo-oxidants: Synthesis, photophysics, and photochemistry of [OsVI(NH3)4(N)](X)3(X = Cl, CF3SO3). J. Chem. Soc. Chem. Commun. 1989, 114–116. [Google Scholar] [CrossRef]
- Lam, H.-W.; Che, C.-M.; Wong, K.-Y. Photoredox properties of [OsN(NH3)4]3+ and mechanism of formation of [{Os(NH3)4(CH3CN)}2N2]5+ through a nitrido-coupling reaction. J. Chem Soc. Dalton Trans. 1992, 1411–1416. [Google Scholar] [CrossRef]
- Chin, K.-F.; Cheung, K.-K.; Yip, H.-K.; Mak, T.C.-W.; Che, C.-M. Luminescent nitridometal complexes. Photophysical and photochemical properties of the 3[(dxy)1(dπ*)1] excited state of nitridoosmium(VI) complexes with polypyridine ligands. J. Chem. Soc. Dalton Trans. 1995, 657–663. [Google Scholar] [CrossRef]
- Che, C.-M.; Wong, K.-Y.; Lam, H.-W.; Chin, K.-F.; Zhou, Z.-Y.; Mak, T.C.-W. Photophysical properties, electrochemistry and crystal structure of nitridoosmium(VI) complexes of 2,3-diamino-2,3-dimethylbutane. J. Chem. Soc. Dalton Trans. 1993, 857–861. [Google Scholar] [CrossRef]
- Pipes, D.W.; Bakir, M.; Vitols, S.E.; Hodgson, D.J.; Meyer, T.J. Reversible interconversion between a nitrido complex of osmium(VI) and an ammine complex of osmium(II). J. Am. Chem. Soc. 1990, 112, 5507–5514. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Avilov, A.S.; Lepeshov, G.G.; Kulygin, A.K.; Stahn, J.; Pietsch, U.; Spence, J.C.H. Zero-flux surfaces of the electrostatic potential: The border of influence zones of nucleophilic and electrophilic sites in crystalline environment. J. Phys. Chem. B 2001, 105, 5068–5074. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. Topological properties of the electrostatic potential in weak and moderate N···H hydrogen bonds. J. Phys. Chem. A 2007, 111, 6425–6433. [Google Scholar] [CrossRef]
- Bartashevich, E.V.; Matveychuk, Y.V.; Troitskaya, E.A.; Tsirelson, V.G. Characterizing the multiple non-covalent interactions in N, S-heterocycles–diiodine complexes with focus on halogen bonding. Comput. Theor. Chem. 2014, 1037, 53–62. [Google Scholar] [CrossRef]
- Bartashevich, E.; Yushina, I.; Kropotina, K.; Muhitdinova, S.; Tsirelson, V. Testing the tools for revealing and characterizing the iodine-iodine halogen bond in crystals. Acta Cryst. 2017, B73, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bartashevich, E.; Mukhitdinova, S.; Yushina, I.; Tsirelson, V. Electronic criterion for categorizing the chalcogen and halogen bonds: Sulfur-iodine interactions in crystals. Acta Cryst. 2019, B7, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory; Theoretical Chemistry Institute, University of Wisconsin-Madison: Madison, WI, USA, 2018. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Codes | ||
|---|---|---|
| BAGRIK | LIFJUF | SEMHEA |
| FEJZUP | MIZWAT | SEMHIE |
| FEKBEC | MOQQUE | VECMUL |
| FOCWOK | NOGZEO | WAKGUK |
| FUTNAK | NOGZIS | WAKHAR |
| HUZYUW | NOGZOY | YACDIP |
| HUZZAD | QADJOV | YUNYUY |
| HUZZEH | QADJUB | |
| JOMKAX | QUFYAU | |
| Complex | ΔE | d 1 | α 2 | ρ |
|---|---|---|---|---|
| 2 | −3.10 | 2.523 (2.15) | 100.488 | 0.038 |
| 3 | −5.04 | 2.604 (2.21) | 101.565 | 0.037 |
| 4 | −13.82 | 2.555 (2.15) | 100.503 | 0.042 |
| 5 | −9.11 | 2.452 (1.46) | 92.227 | 0.067 |
| 6 | −3.33 | 2.095 (2.00) | 94.290 | 0.082 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomila, R.M.; Frontera, A. Crystallographic and Theoretical Study of Osme Bonds in Nitrido-Osmium(VI) Complexes. Inorganics 2022, 10, 133. https://doi.org/10.3390/inorganics10090133
Gomila RM, Frontera A. Crystallographic and Theoretical Study of Osme Bonds in Nitrido-Osmium(VI) Complexes. Inorganics. 2022; 10(9):133. https://doi.org/10.3390/inorganics10090133
Chicago/Turabian StyleGomila, Rosa M., and Antonio Frontera. 2022. "Crystallographic and Theoretical Study of Osme Bonds in Nitrido-Osmium(VI) Complexes" Inorganics 10, no. 9: 133. https://doi.org/10.3390/inorganics10090133
APA StyleGomila, R. M., & Frontera, A. (2022). Crystallographic and Theoretical Study of Osme Bonds in Nitrido-Osmium(VI) Complexes. Inorganics, 10(9), 133. https://doi.org/10.3390/inorganics10090133