
A Computational Chemistry Investigation of the Influence of Steric Bulk of Dithiocarbamato-Bound Organic Substituents upon Spodium Bonding in Three Homoleptic Mercury(II) Bis(N,N-dialkyldithiocarbamato) Compounds for Alkyl = Ethyl, Isobutyl, and Cyclohexyl




Abstract
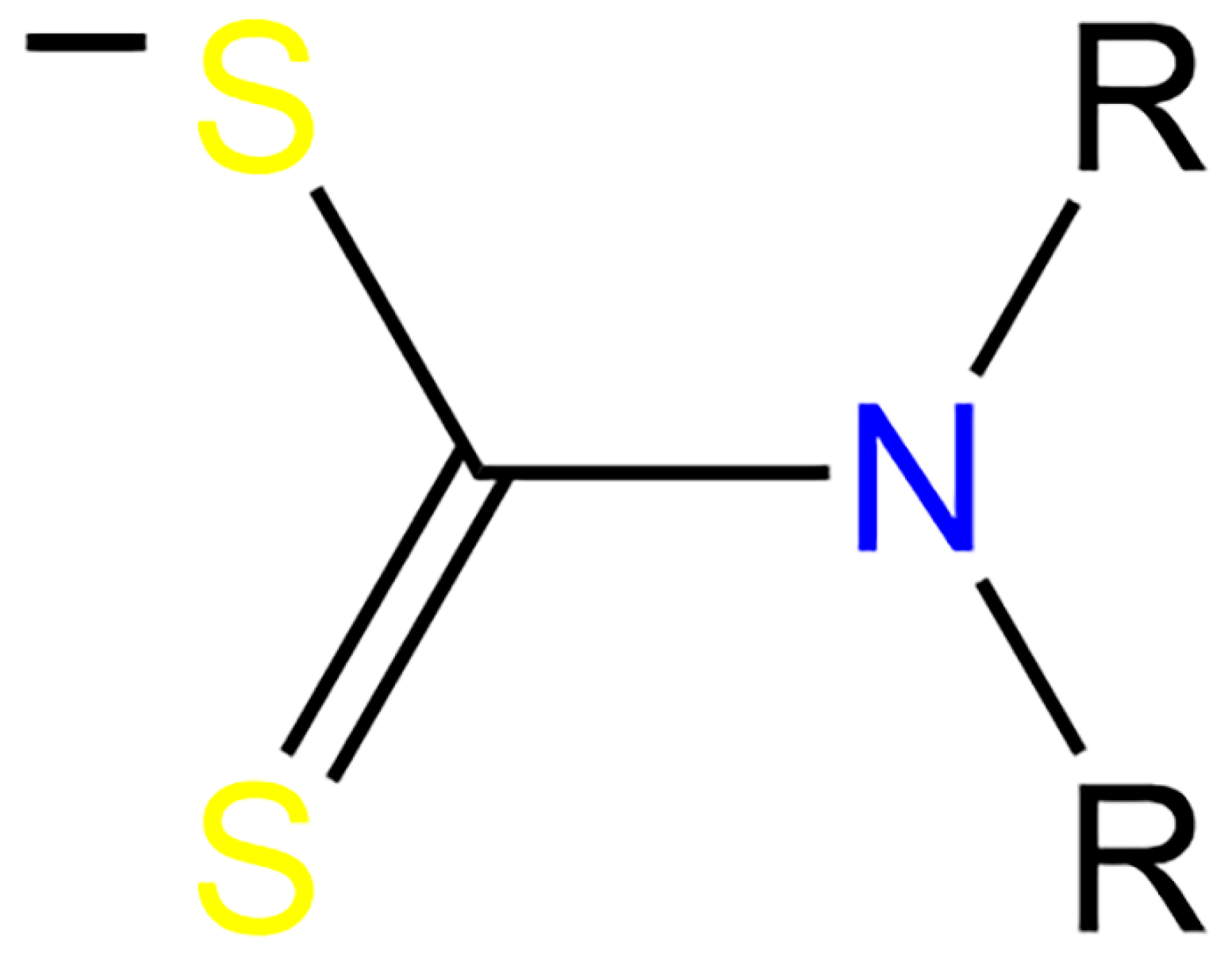
:1. Introduction
2. Methods
3. Results and Discussion
3.1. Literature Survey
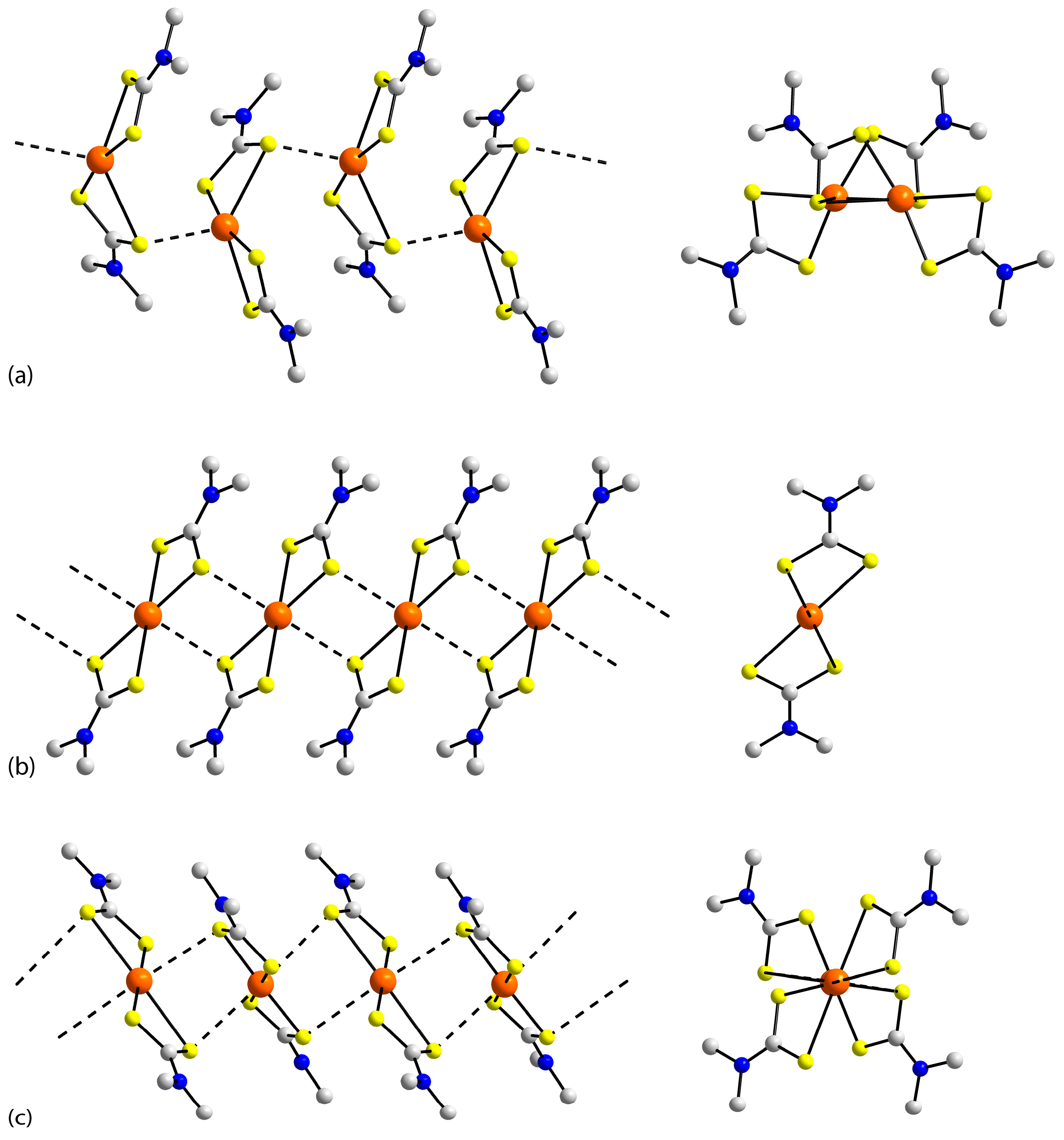
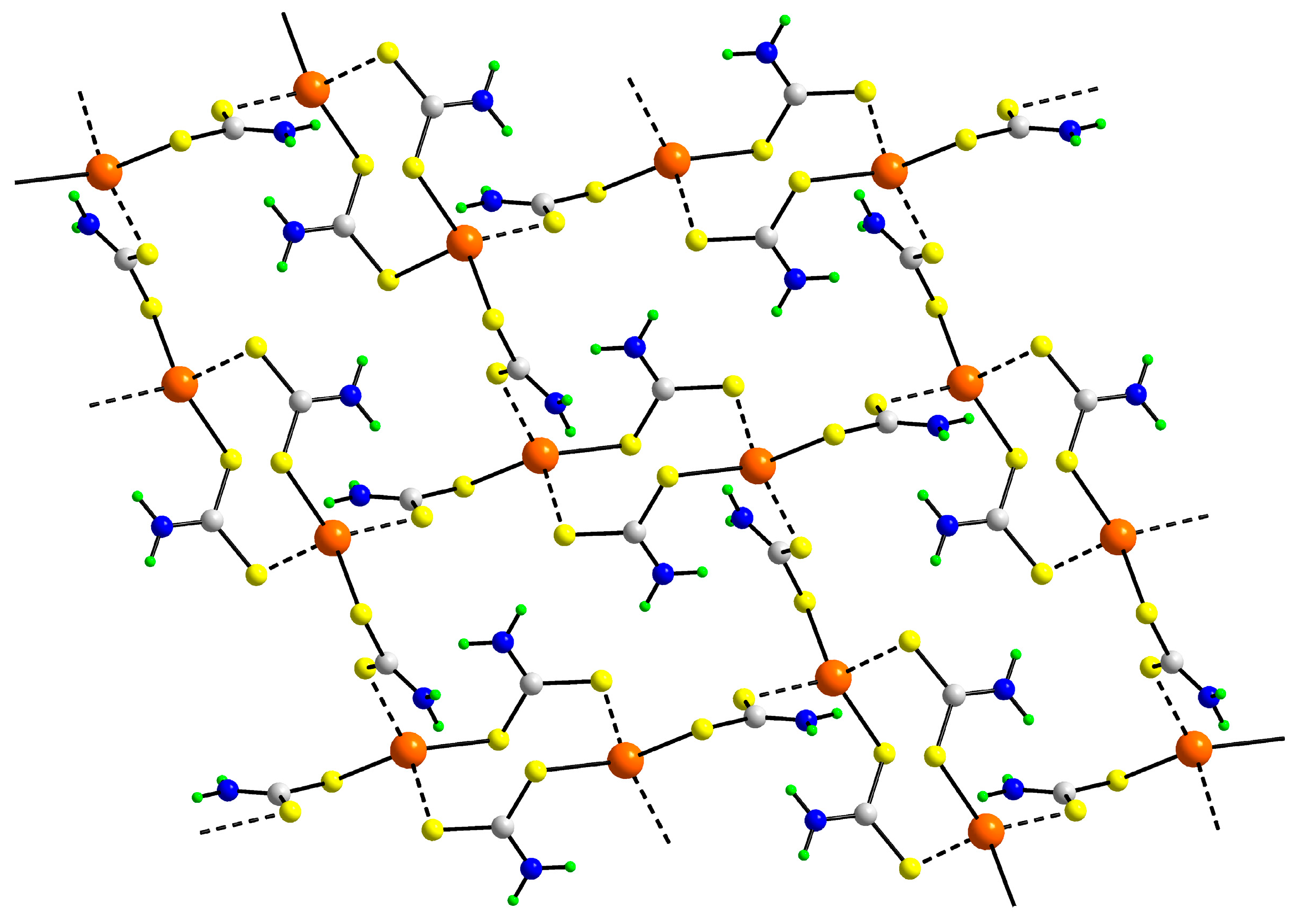
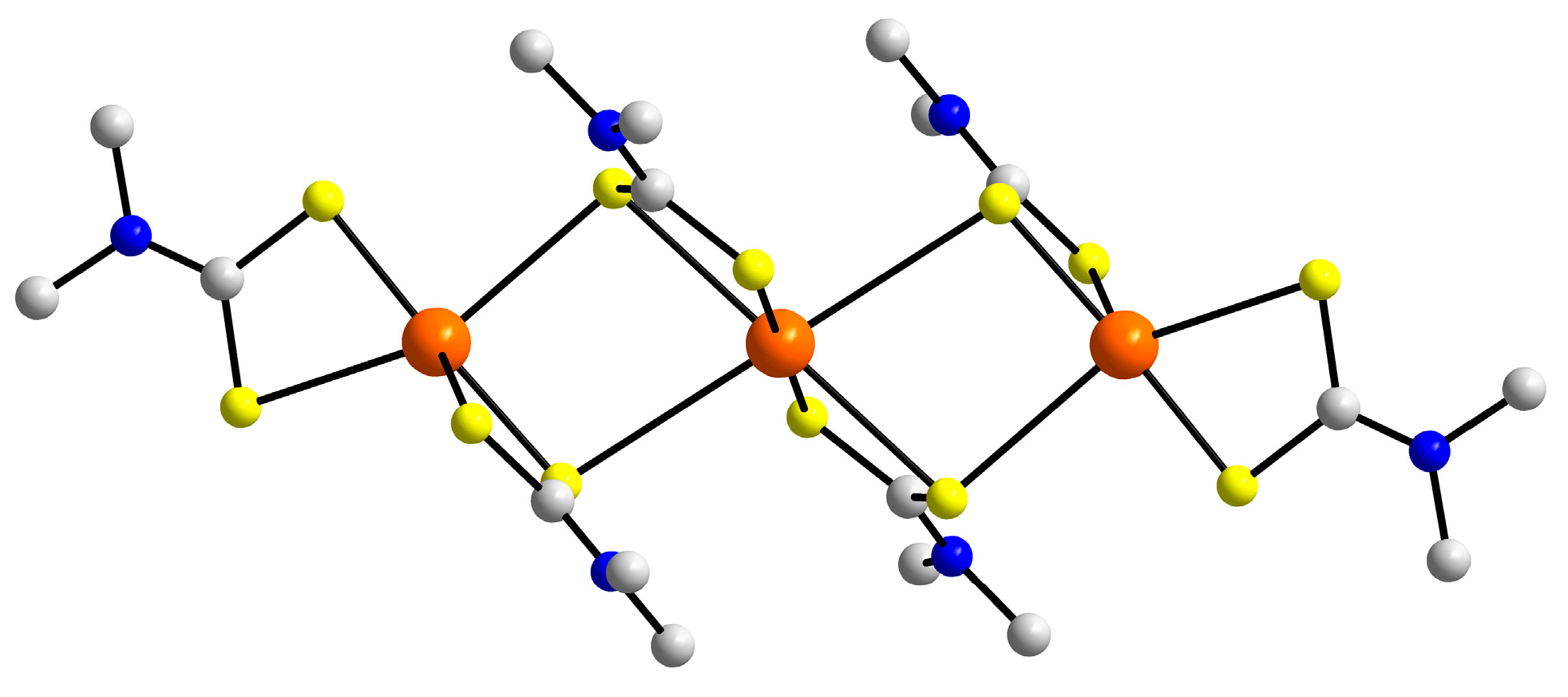
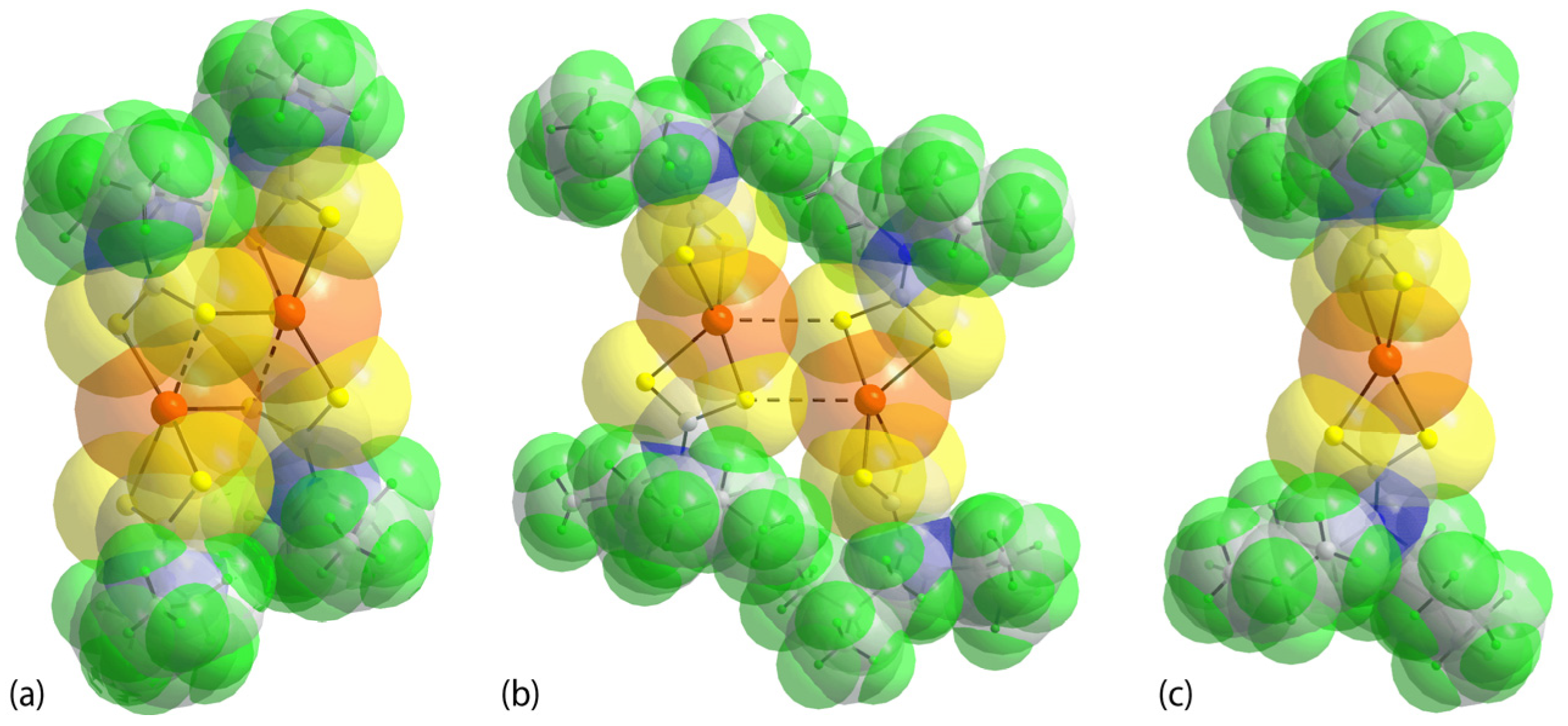
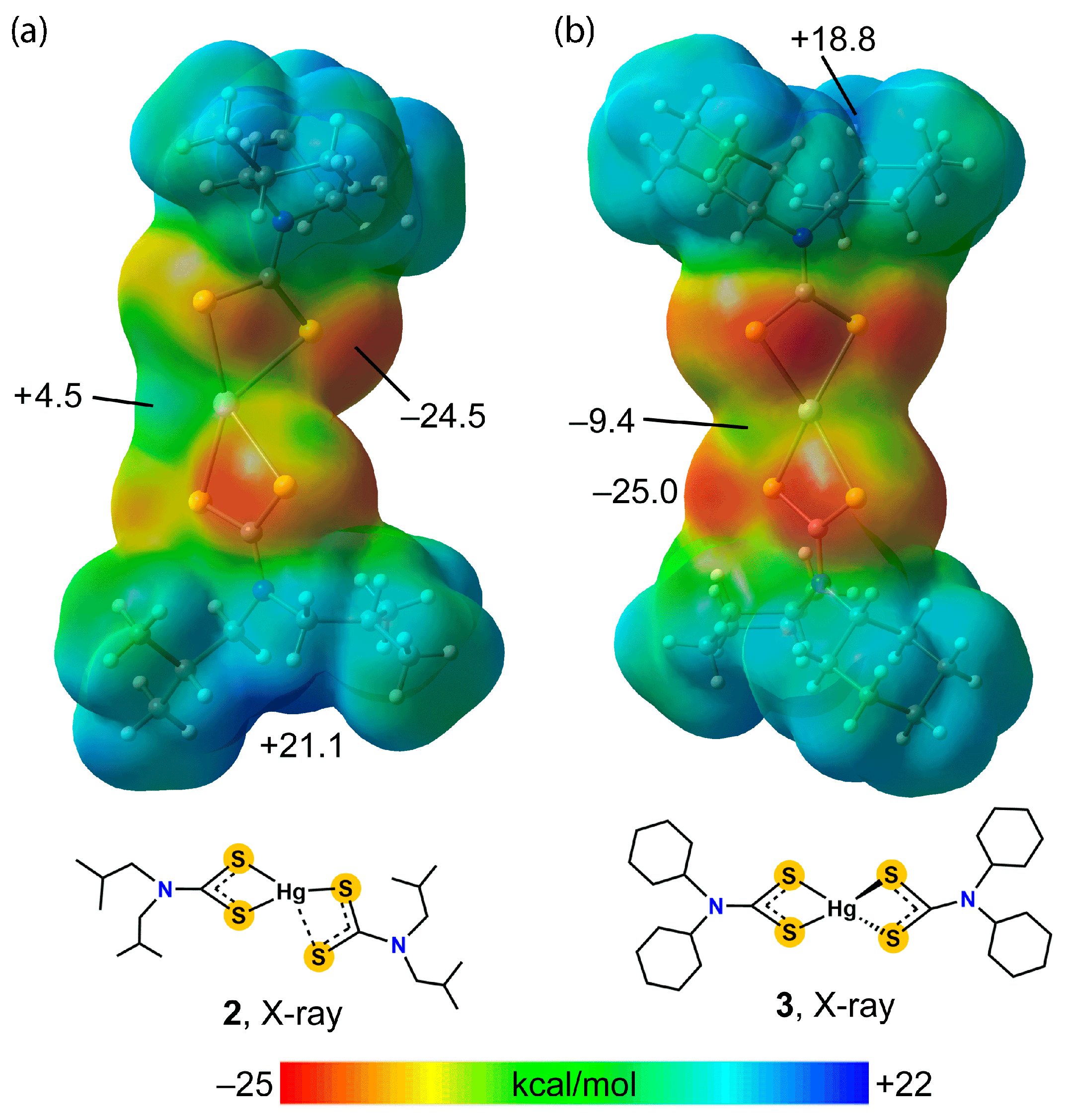
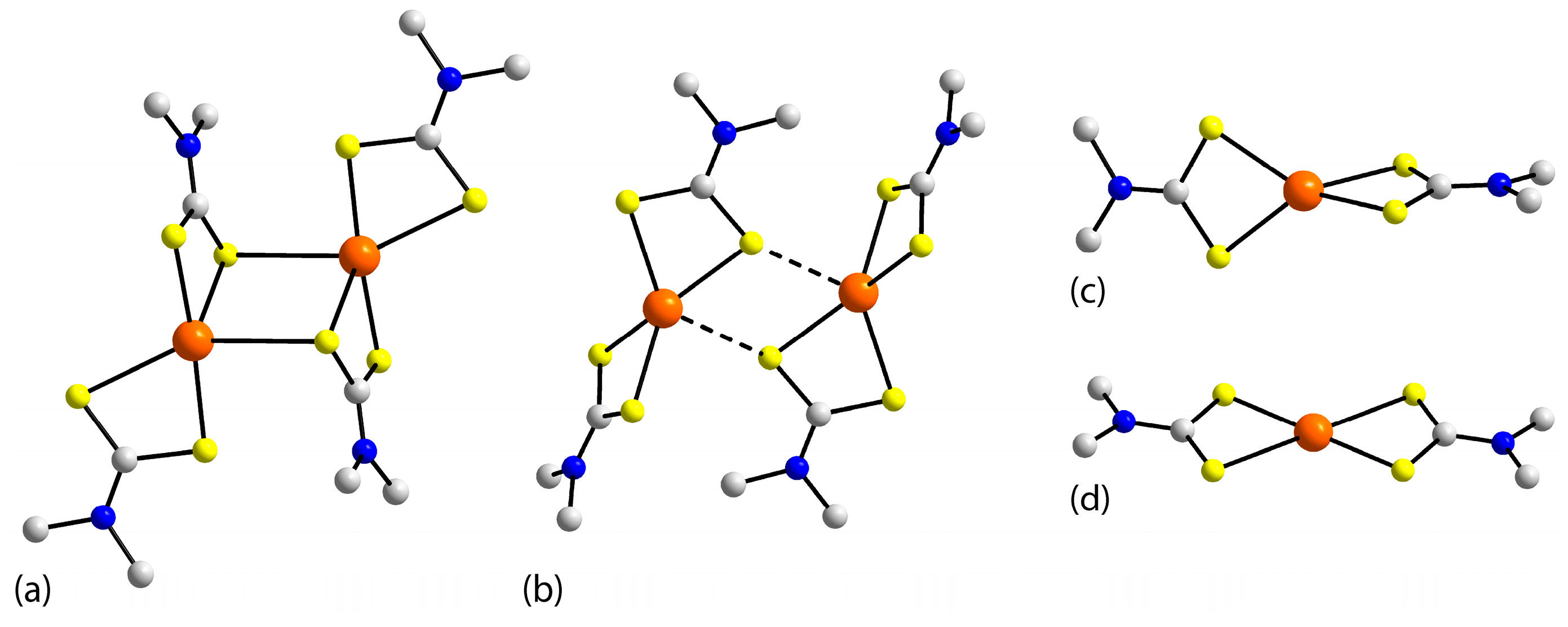
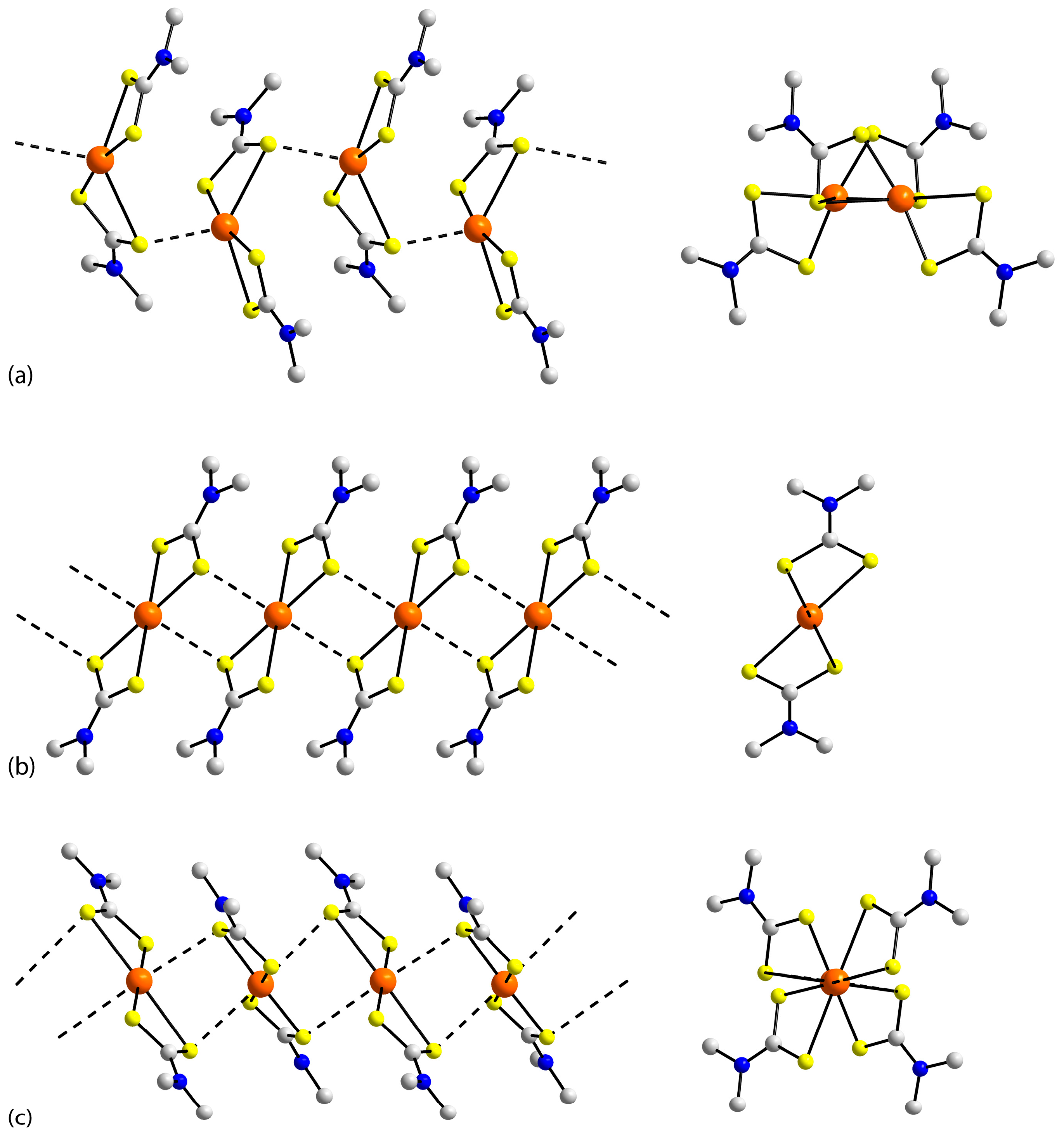
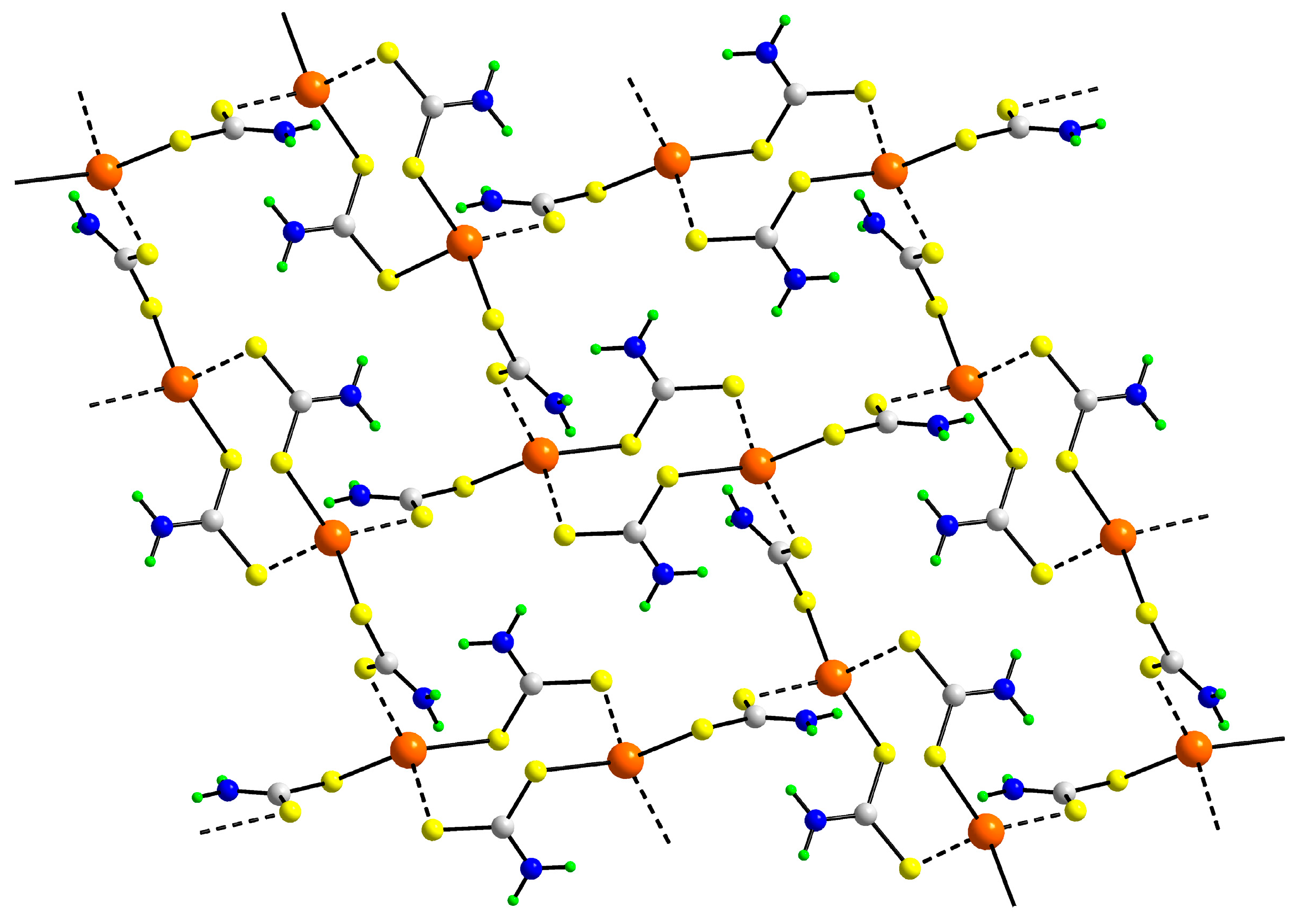
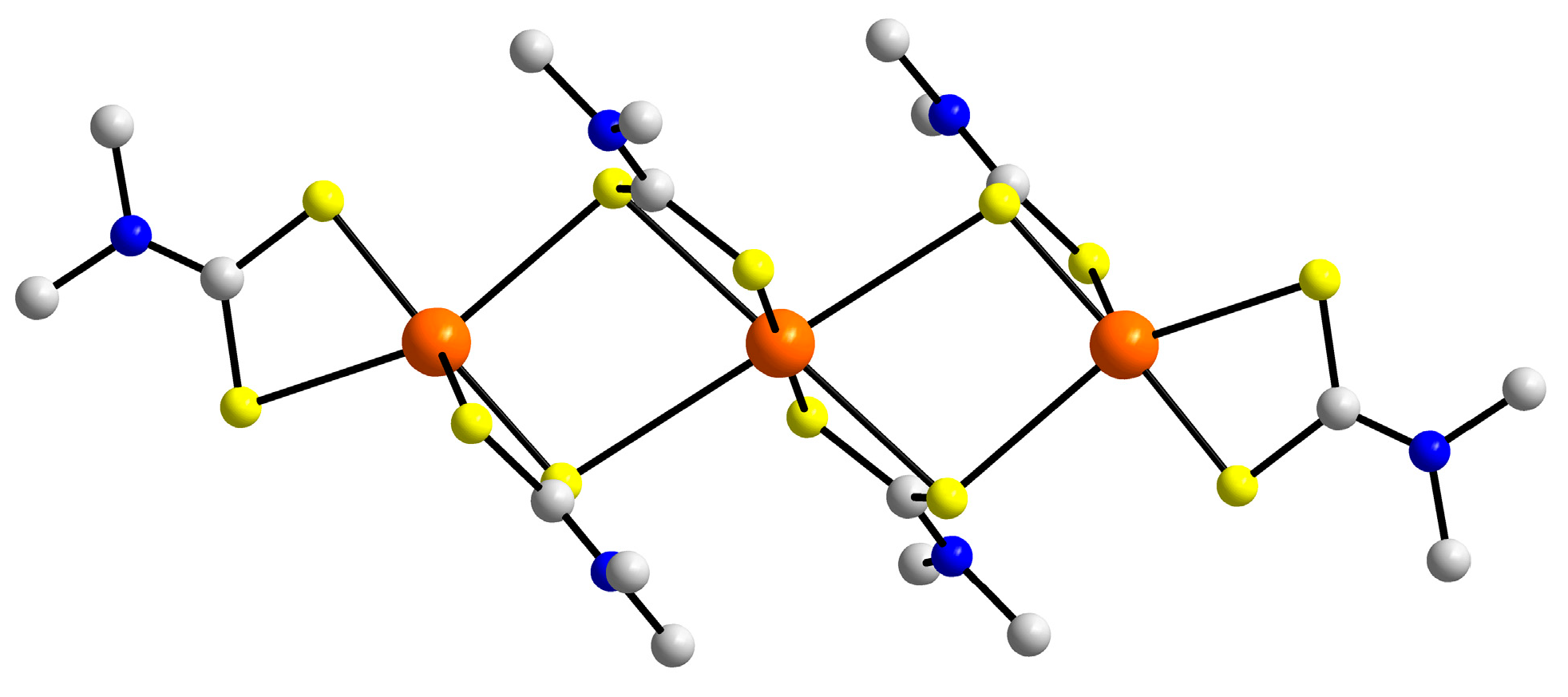
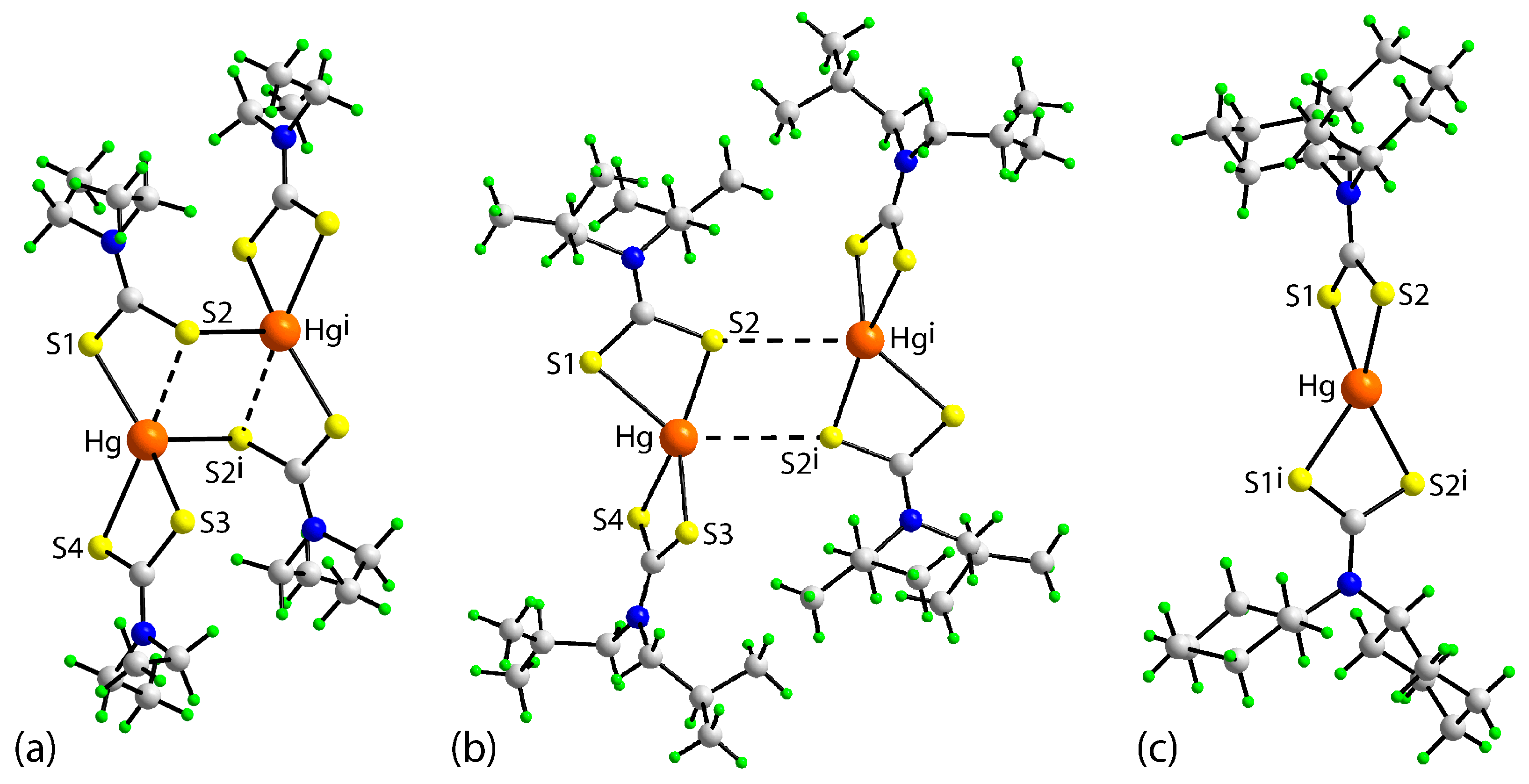
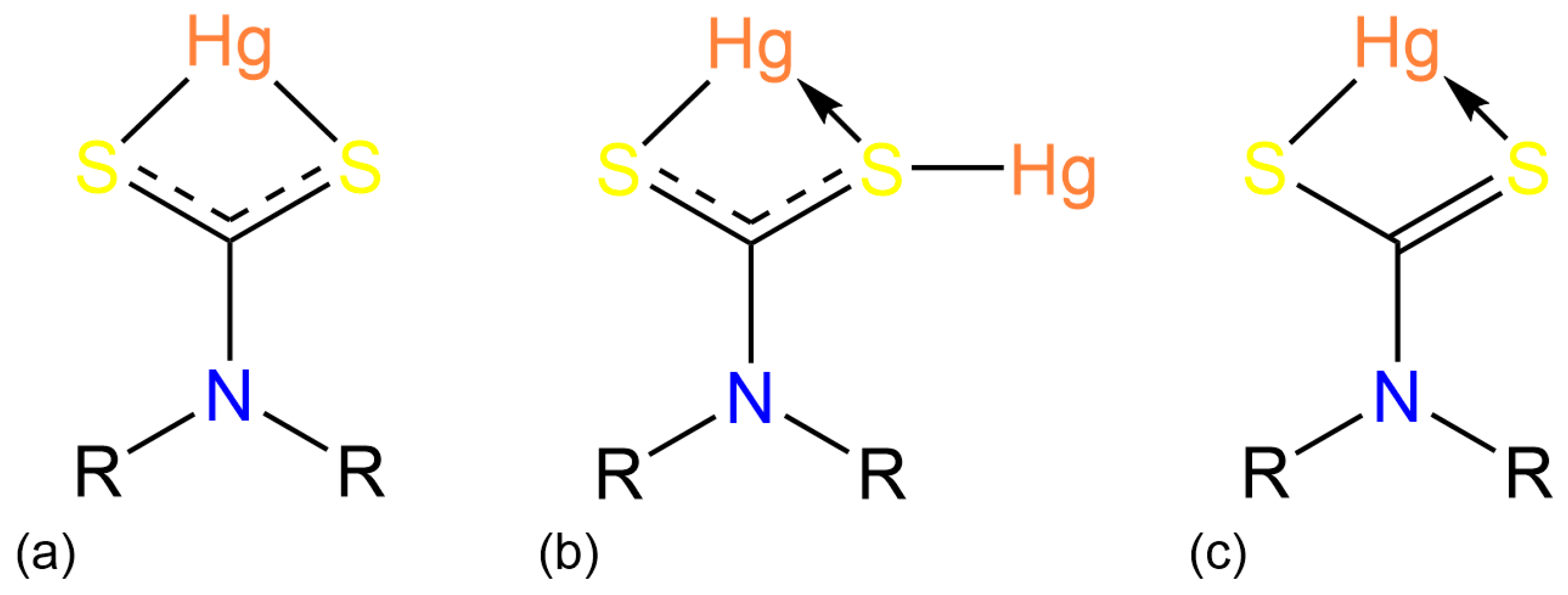
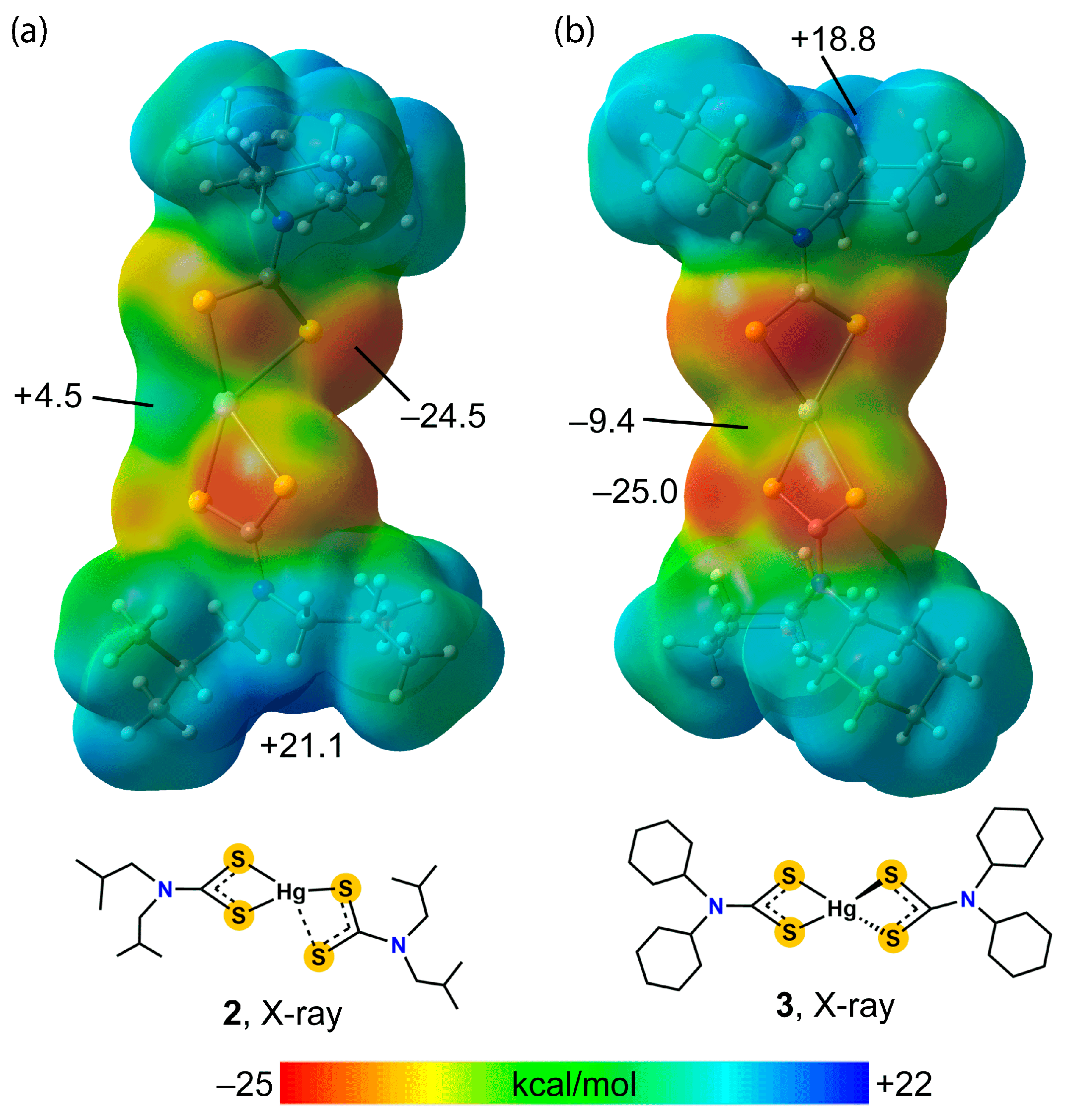
3.2. Experimental Structures
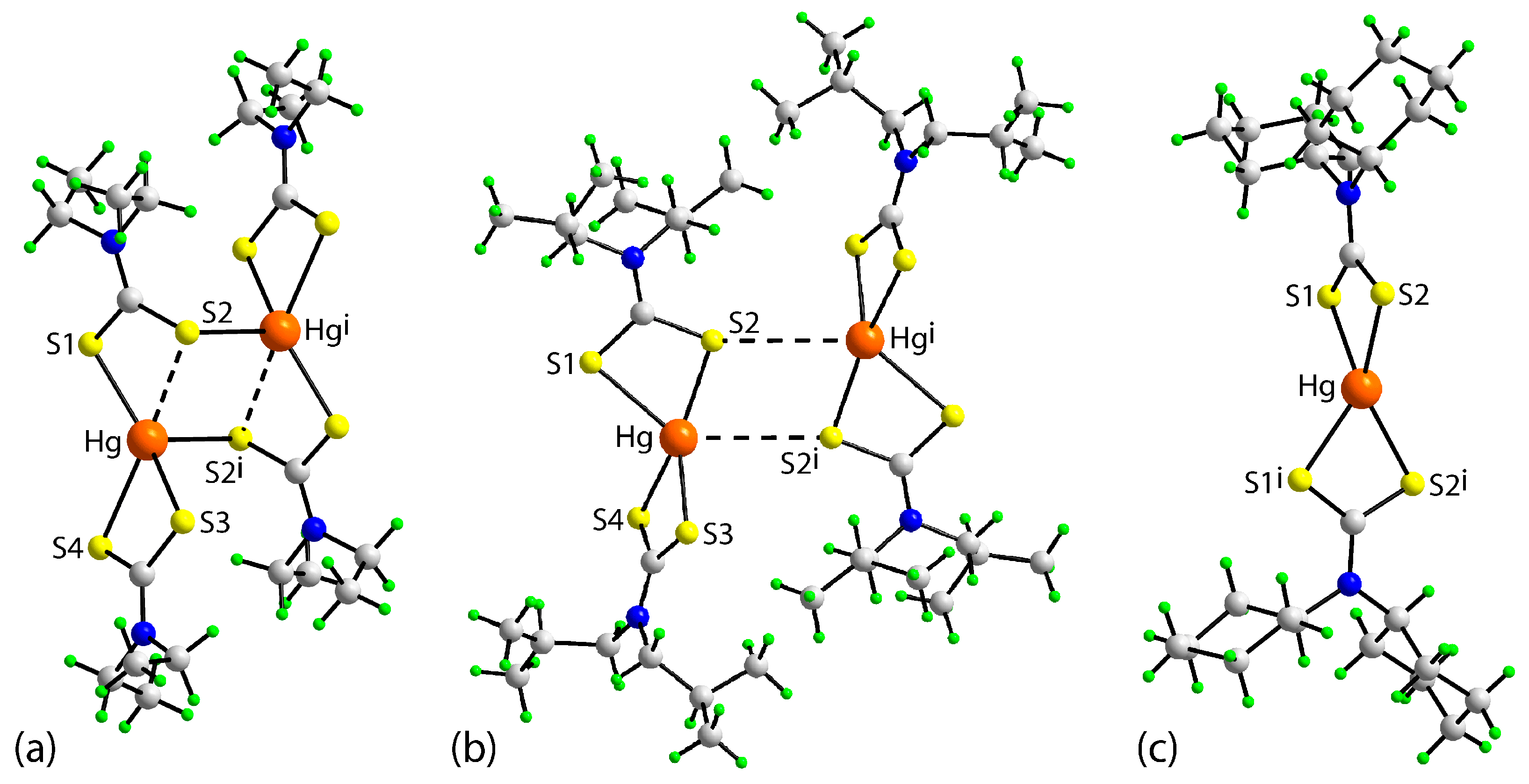
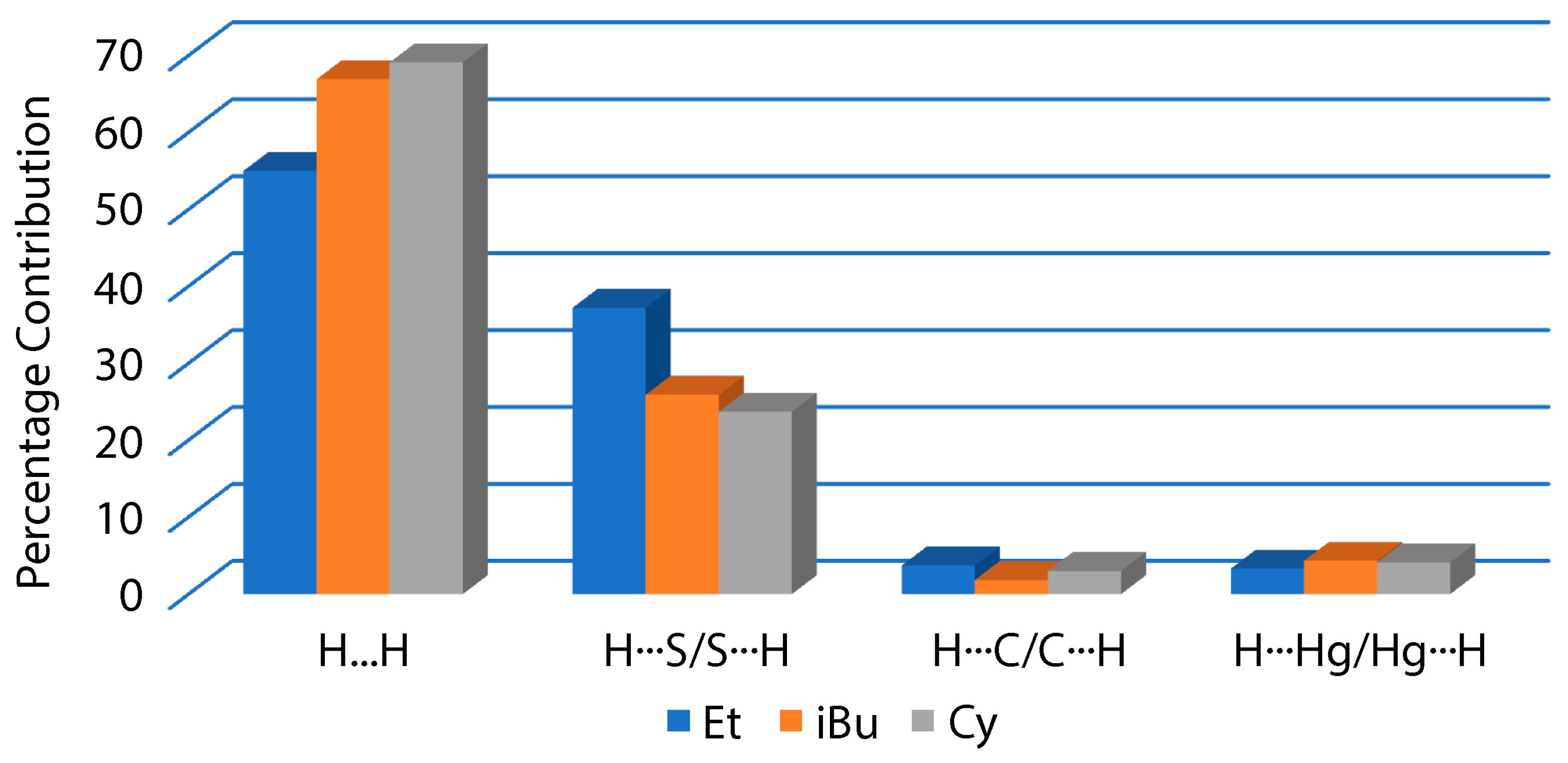
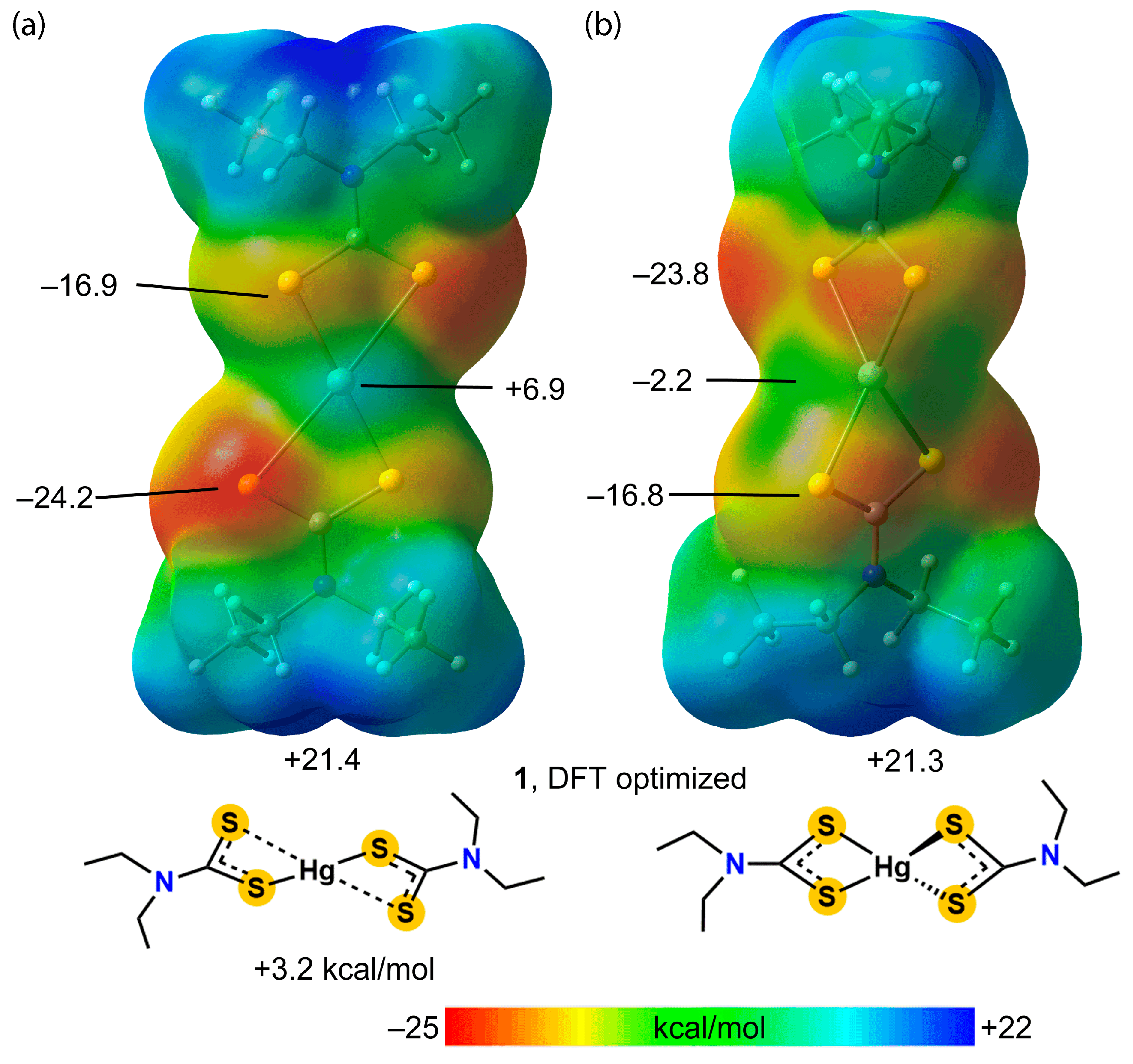
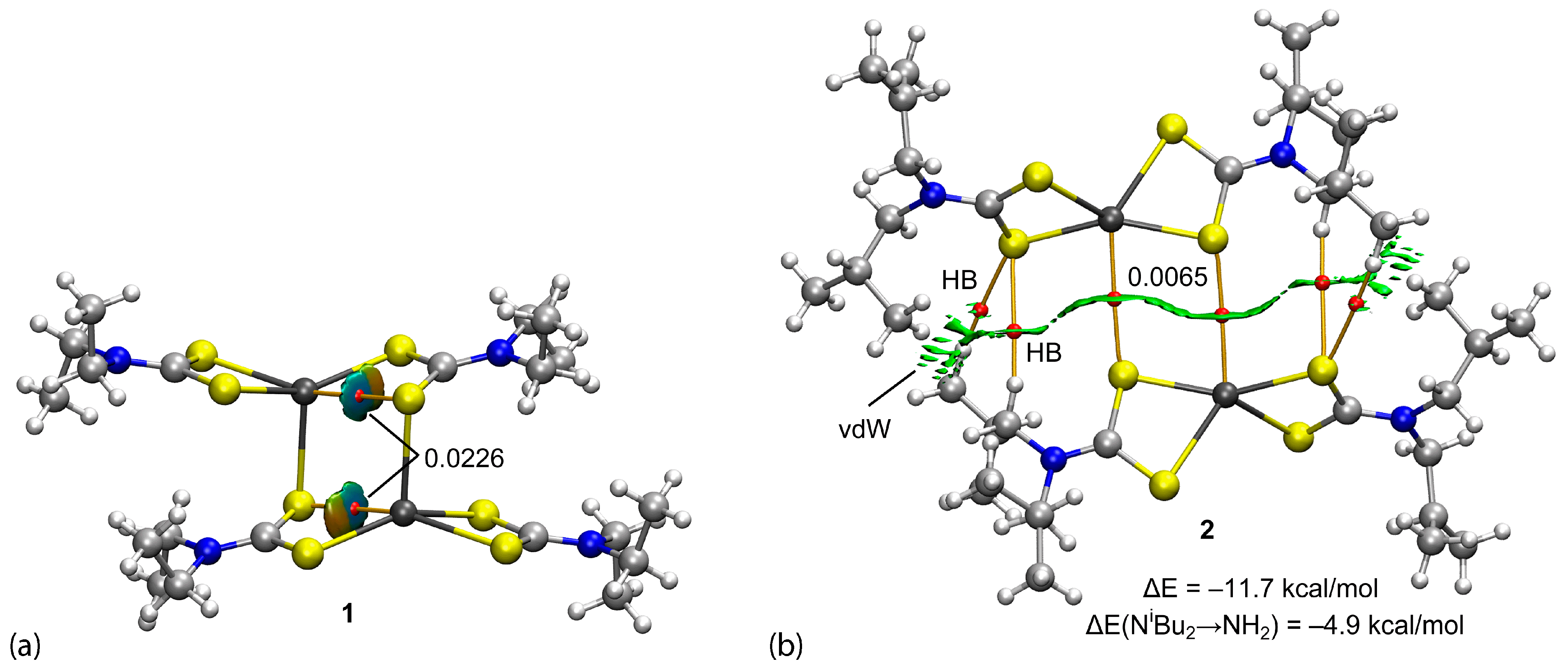
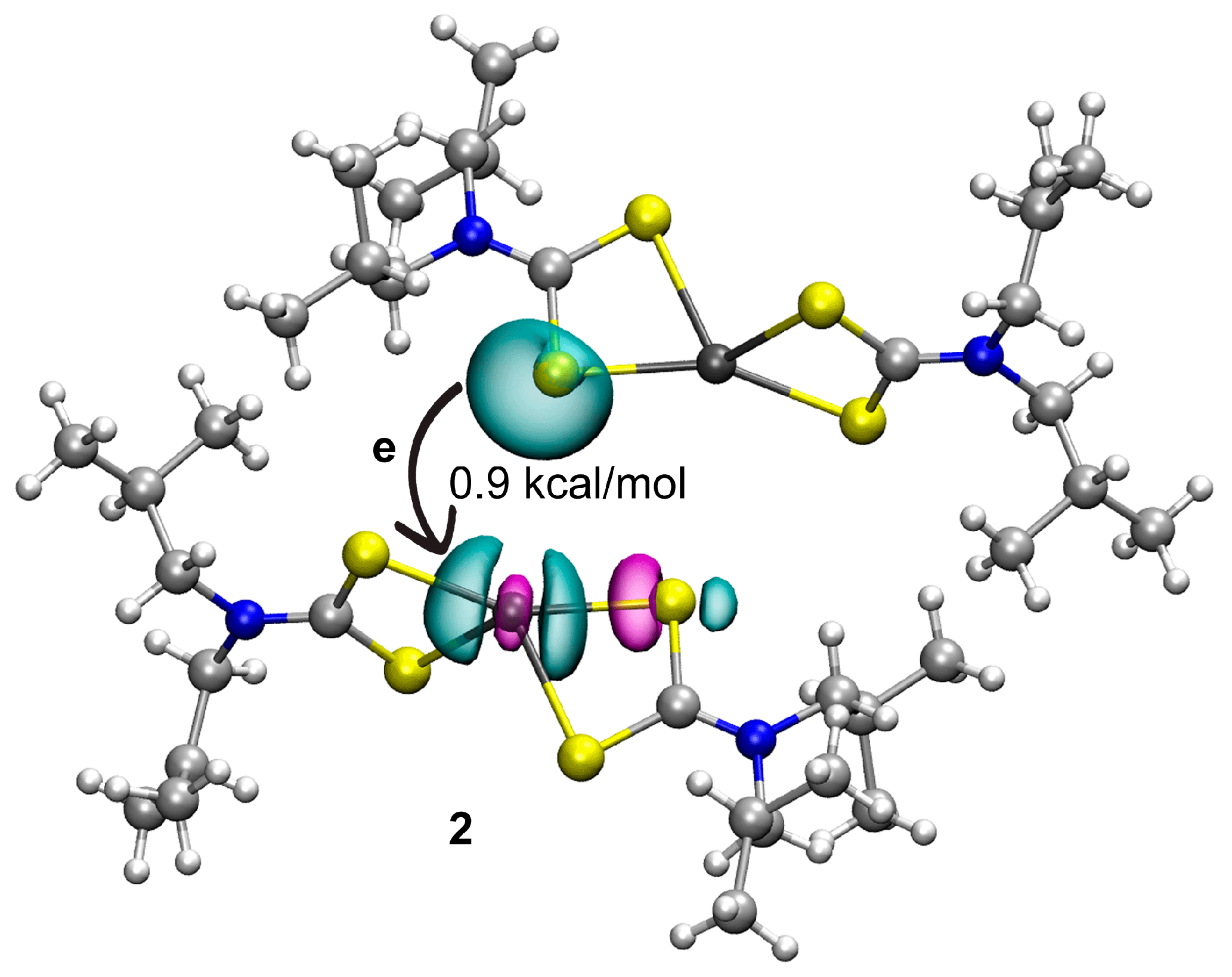
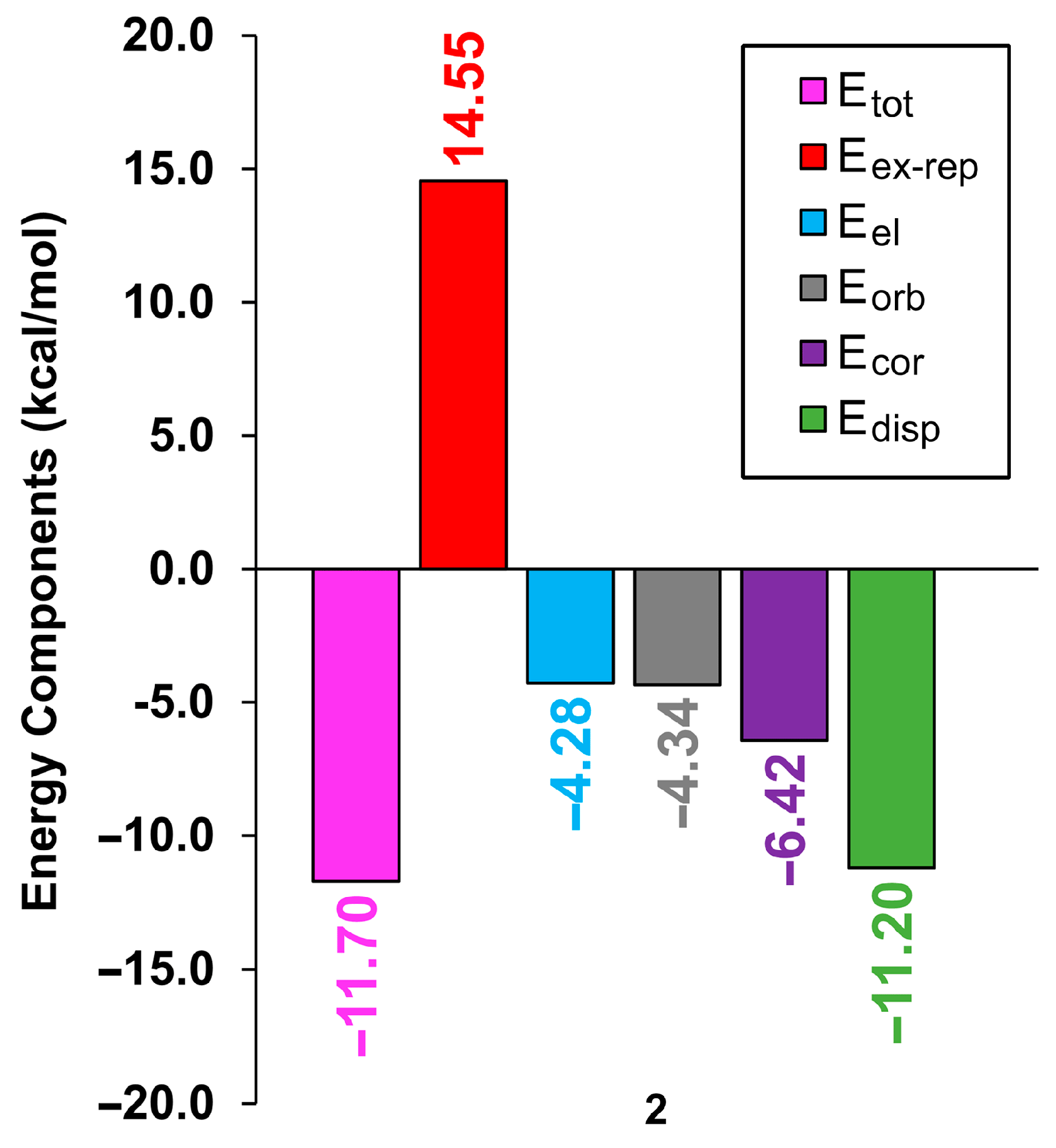
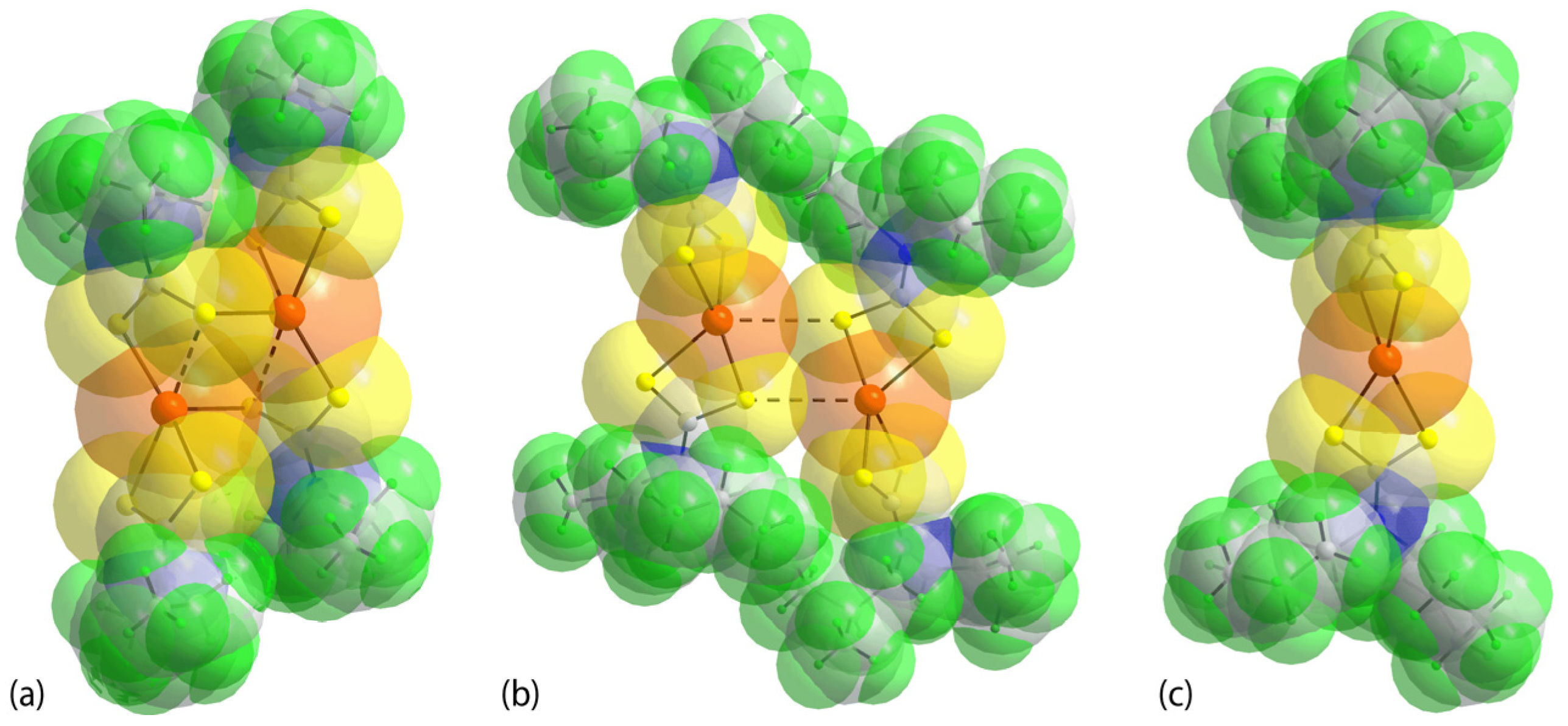
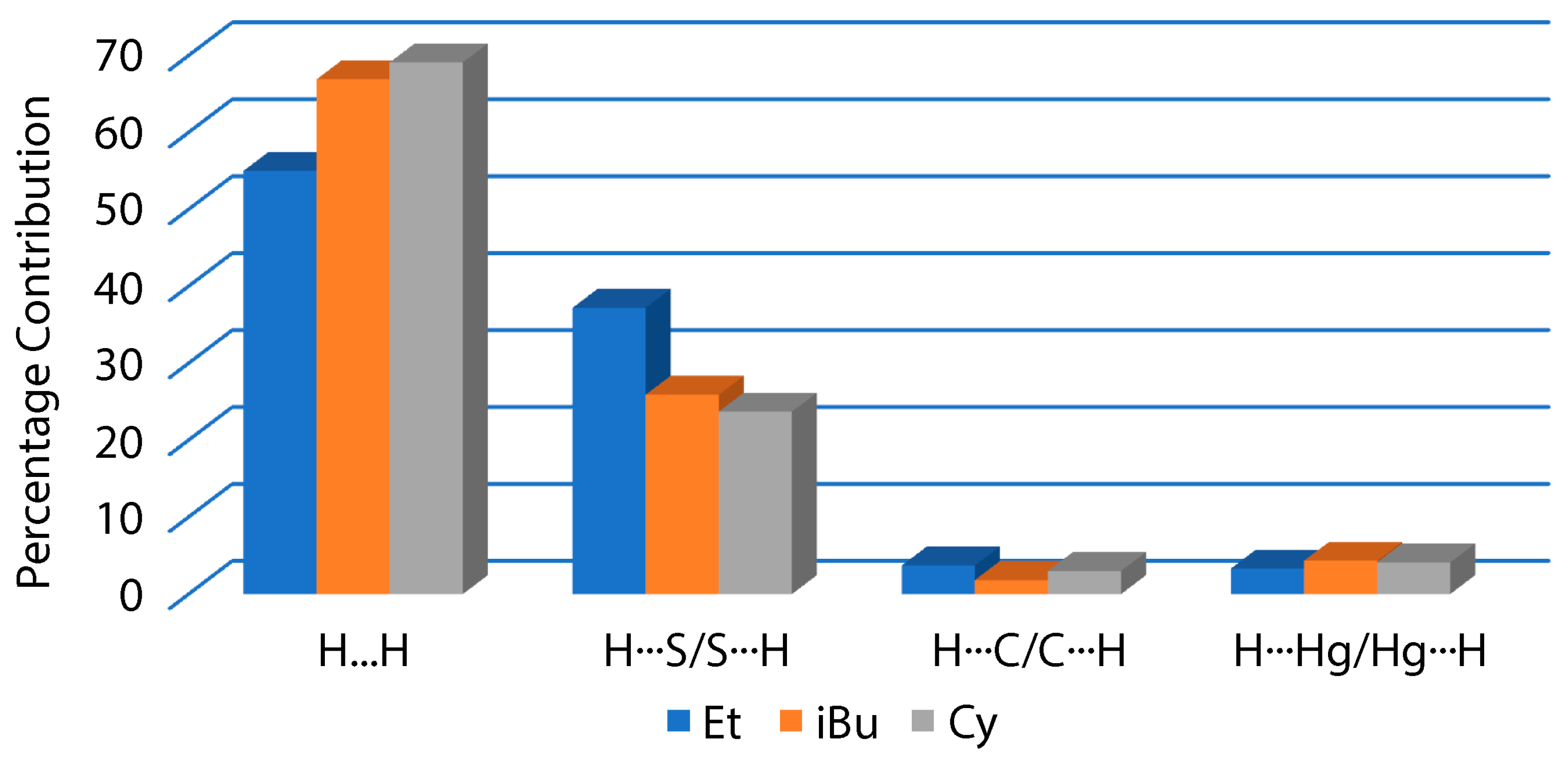
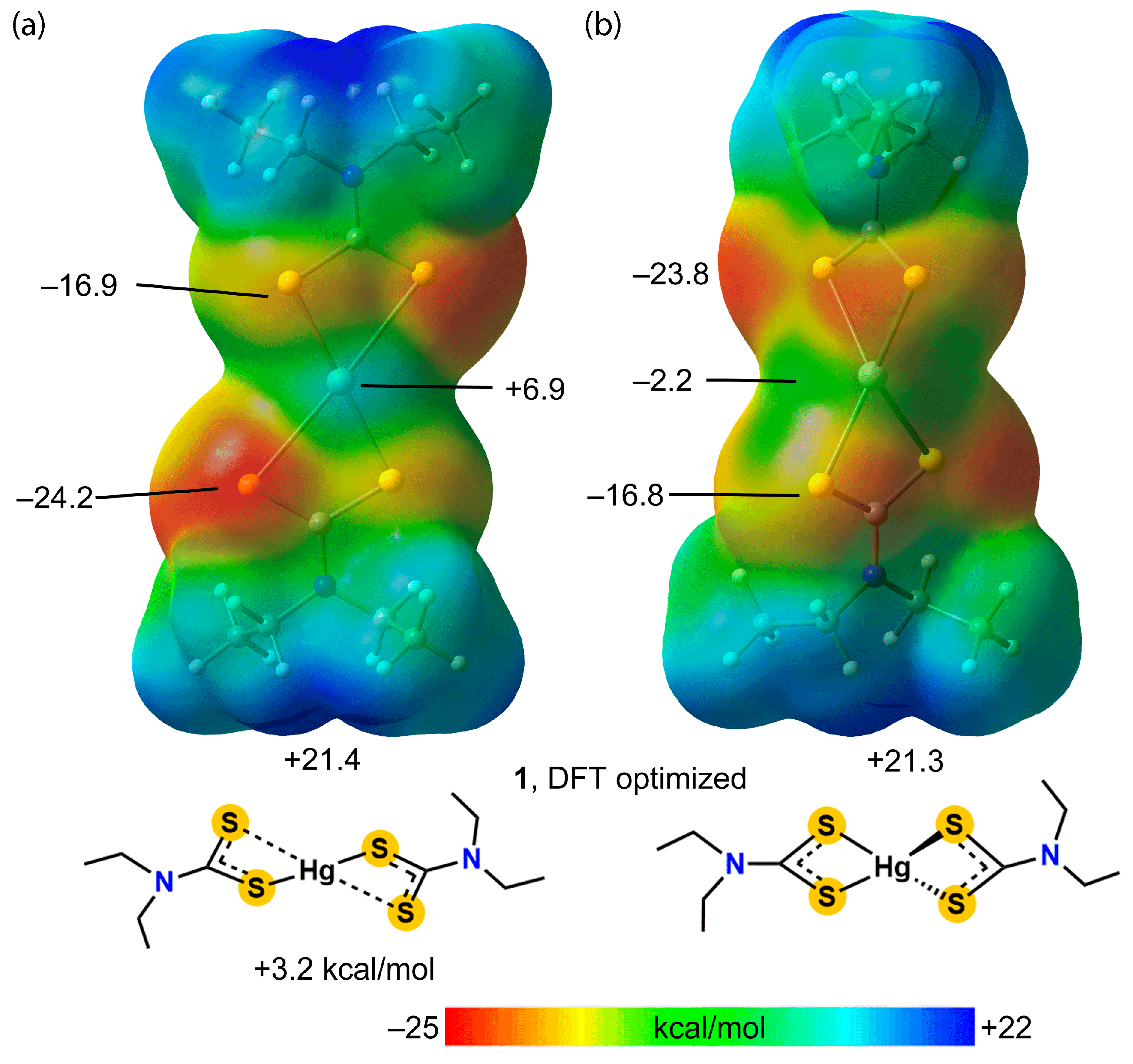
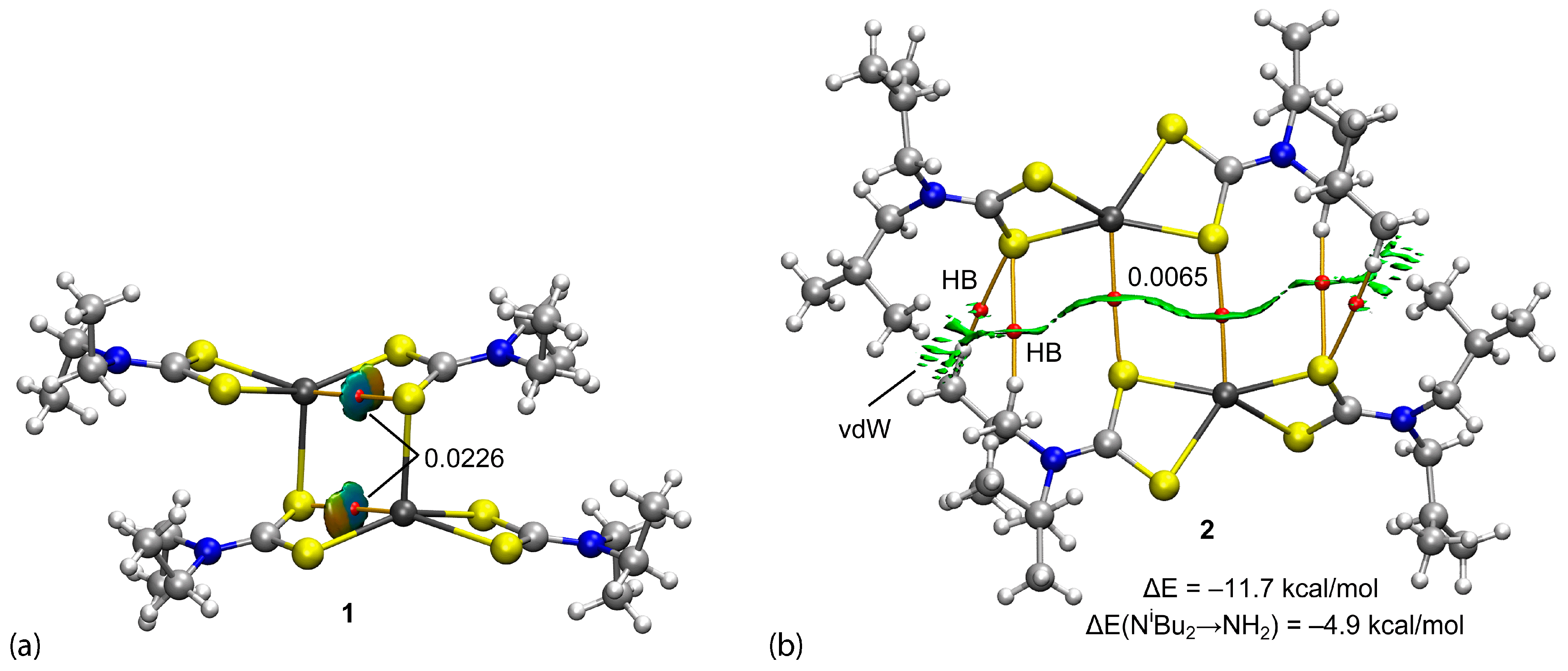
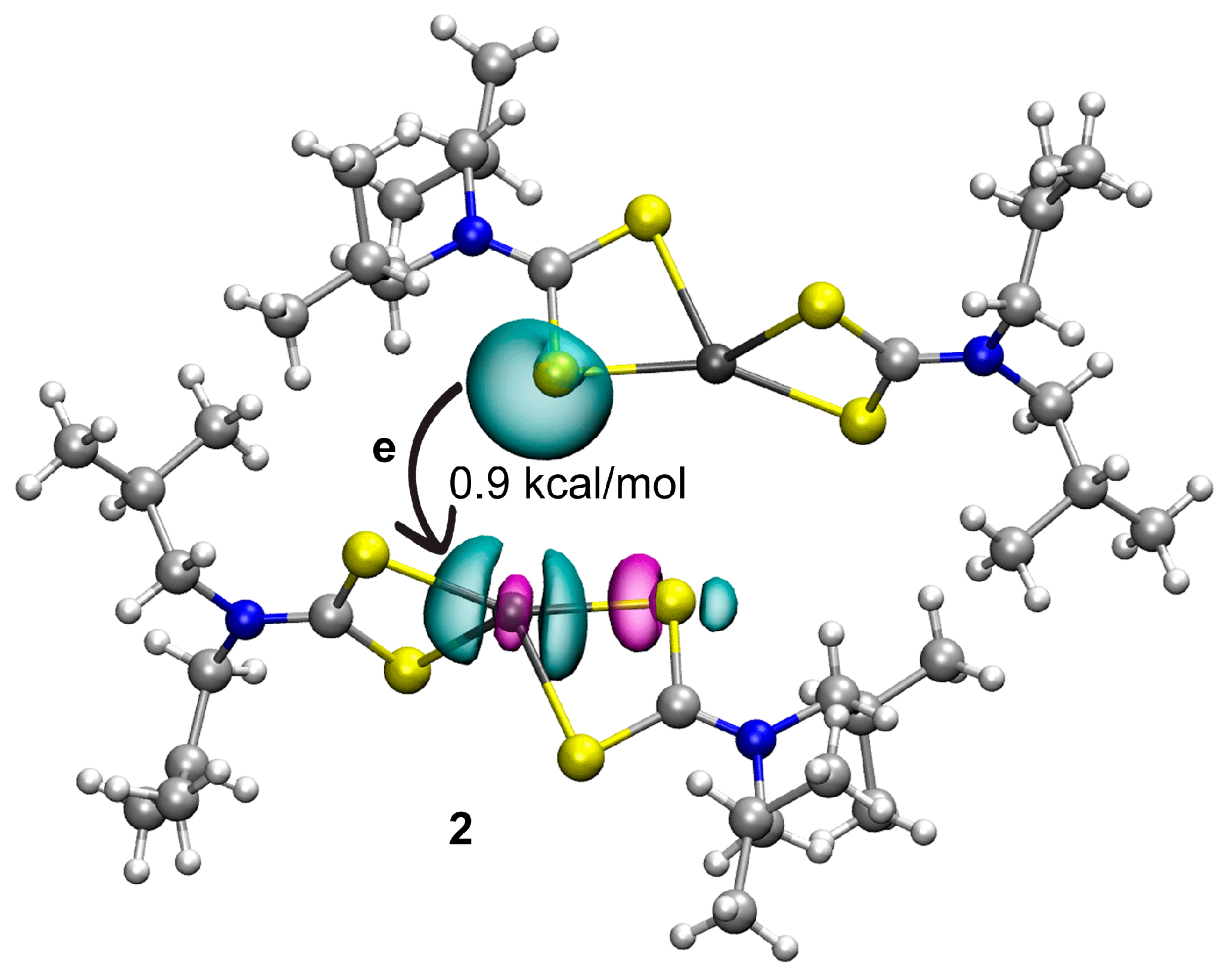
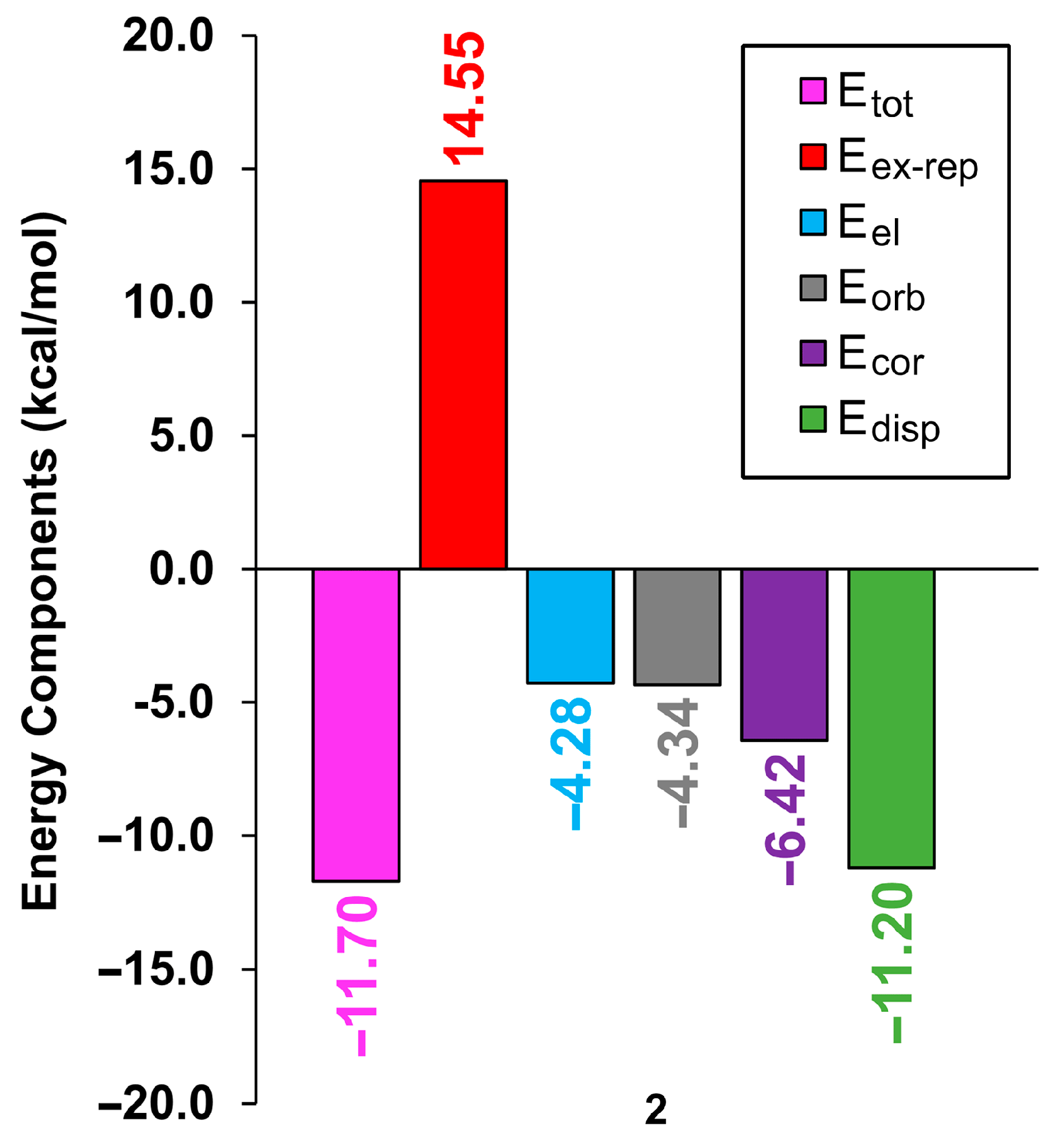
3.3. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coucouvanis, D. The chemistry of the dithioacid and 1,1-dithiolate complexes. Prog. Inorg. Chem. 1970, 11, 234–371. [Google Scholar] [CrossRef]
- Eisenberg, R. Structural systematics of 1,1- and 1,2-dithiolato chelates. Prog. Inorg. Chem. 1970, 12, 295–369. [Google Scholar] [CrossRef]
- Coucouvanis, D. The chemistry of the dithioacid and 1,1-dithiolate complexes, 1968–1977. Prog. Inorg. Chem. 1979, 26, 301–469. [Google Scholar] [CrossRef]
- Hogarth, G. Transition metal dithiocarbamates: 1978–2003. Prog. Inorg. Chem. 2005, 53, 71–561. [Google Scholar] [CrossRef]
- Heard, P.J. Main group dithiocarbamate complexes. Prog. Inorg. Chem. 2005, 53, 1–69. [Google Scholar] [CrossRef]
- Haiduc, I. 1,1-Dithiolato ligands and related selenium and tellurium compounds. In Handbook of Chalcogen Chemistry; Devillanova, F.A., Ed.; Royal Society of Chemistry: Cambridge, UK, 2007; pp. 593–643. ISBN 978-0-85404-366-8. [Google Scholar]
- Tiekink, E.R.T. Molecular architecture and supramolecular association in the zinc-triad 1,1-dithiolates. Steric control as a design element in crystal engineering? CrystEngComm 2003, 5, 101–113. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Exploring the topological landscape exhibited by binary zinc-triad 1,1-dithiolates. Crystals 2018, 8, 292. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. On the coordination role of pyridyl-nitrogen in the structural chemistry of pyridyl-substituted dithiocarbamate ligands. Crystals 2021, 11, 286. [Google Scholar] [CrossRef]
- Lee, S.M.; Tiekink, E.R.T. A structural survey of poly-functional dithiocarbamate ligands and the aggregation patterns they sustain. Inorganics 2021, 9, 7. [Google Scholar] [CrossRef]
- Mulliken, R.S. Structures of complexes formed by halogen molecules with aromatic and with oxygenated solvents. J. Am. Chem. Soc. 1950, 72, 600–608. [Google Scholar] [CrossRef]
- Bent, H.A. Structural chemistry of donor-acceptor interactions. Chem. Rev. 1968, 68, 587–648. [Google Scholar] [CrossRef]
- Hassel, O. Structural Aspects of interatomic charge-transfer bonding. Science 1970, 170, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Alcock, N.W. Secondary bonding to nonmetallic elements. Adv. Inorg. Chem. Radiochem. 1972, 15, 1–58. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.J.; Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Intermolecular interactions in molecular crystals: What’s in a name? Faraday Discuss. 2017, 203, 93–112. [Google Scholar] [CrossRef]
- Scheiner, S.; Michalczyk, M.; Zierkiewicz, W. Coordination of anions by noncovalently bonded σ-hole ligands. Coord. Chem. Rev. 2020, 405, 213136. [Google Scholar] [CrossRef]
- Brammer, L.; Peuronen, A.; Roseveare, T.M. Halogen bonds, chalcogen bonds, pnictogen bonds, tetrel bonds and other σ-hole interactions: A snapshot of current progress, Acta Crystallogr. Sect. C Struct. Chem. 2023, 79, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Alkorta, I.; Elguero, J.; Mooibroek, T.J.; Frontera, A. Spodium bonds: Noncovalent interactions involving Group 12 elements. Angew. Chem. Int. Ed. 2020, 59, 17482–17487. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Rocchigiani, L. Assessing the orbital contribution in the “spodium bond” by natural orbital for chemical valence−charge displacement analysis. Inorg. Chem. 2021, 60, 4683–4692. [Google Scholar] [CrossRef]
- Liu, N.; Xie, X.; Li, Q. Chalcogen bond involving zinc(II)/cadmium(II) carbonate and its enhancement by spodium bond. Molecules 2021, 26, 6443. [Google Scholar] [CrossRef]
- Wysokiński, R.; Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Anion⋯anion (MX3−)2 dimers (M = Zn, Cd, Hg; X = Cl, Br, I) in different environments. Phys. Chem. Chem. Phys. 2021, 23, 13853–13861. [Google Scholar] [CrossRef]
- Singh, A.; Kociok-Köhn, G.; Dutta, A.; Kumar, A.; Muddassir, M. Diaminopyridine Hg(II)-based 1D supramolecular polymer: Crystallographic and computational insights into spodium bonding. J. Solid State Chem. 2022, 315, 123517. [Google Scholar] [CrossRef]
- Kumar, P.; Banerjee, S.; Radha, A.; Firdoos, T.; Sahoo, S.C.; Pandey, S.K. Role of non-covalent interactions in the supramolecular architectures of mercury(II) diphenyldithiophosphates: An experimental and theoretical investigation. New J. Chem. 2021, 45, 2249–2263. [Google Scholar] [CrossRef]
- Liu, N.; Li, Q.; Scheiner, S. Spodium and tetrel bonds involving Zn(II)/Cd(II) and their interplay. Chem. Phys. 2022, 556, 111470. [Google Scholar] [CrossRef]
- Gao, M.; Zhao, Q.; Yu, H.; Fu, M.; Li, Q. Insight into spodium–π bonding characteristics of the MX2⋯π (M = Zn, Cd and Hg; X = Cl, Br and I) complexes—A theoretical study. Molecules 2022, 27, 2885. [Google Scholar] [CrossRef] [PubMed]
- Rozhkov, A.V.; Katlenok, E.A.; Zhmykhova, M.V.; Kuznetsov, M.L.; Khrustalev, V.N.; Tugashov, K.I.; Bokach, N.A.; Kukushkin, V.Y. Spodium bonding to anticrown-Hg3 boosts phosphorescence of cyclometalated-PtII complexes. Inorg. Chem. Front. 2023, 10, 493–510. [Google Scholar] [CrossRef]
- Wu, Q.; McDowell, S.A.C.; Li, Q. Single-electron spodium bonds: Substituent effects. Appl. Organomet. Chem. 2023, 37, e7052. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P.J. σ-hole bonding: Molecules containing group VI atoms. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer modeling of halogen bonds and other σ-hole interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-Hole interactions: Perspectives and misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. Interaction and polarization energy relationships in σ-hole and π-hole bonding. Crystals 2020, 10, 76. [Google Scholar] [CrossRef]
- Jotani, M.M.; Tan, Y.S.; Tiekink, E.R.T. Bis[bis(N-2-hydroxyethyl,N-isopropyldithiocarbamato)mercury(II)]2: Crystal structure and Hirshfeld surface analysis. Z. Kristallogr.—Cryst. Mater. 2016, 231, 403–413. [Google Scholar] [CrossRef]
- Winter, G. Inorganic xanthates. Rev. Inorg. Chem. 1980, 2, 253–342. [Google Scholar]
- Haiduc, I.; Sowerby, D.B. Stereochemical aspects of phosphor-1,1-dithiolato metal complexes: Coordination patterns, molecular structures and supramolecular associations in dithiophosphinates and related compounds. Polyhedron 1996, 15, 2469–2521. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular association via Hg···S secondary-bonding interactions in crystals of organomercury(II) species: A survey of the Cambridge Structure Database. Crystals 2023, 13, 385. [Google Scholar] [CrossRef]
- Gomila, R.M.; Frontera, A.; Tiekink, E.R.T. Supramolecular aggregation featuring Hg···S secondary-bonding interactions in crystals of mercury(II) species augmented by computational chemistry calculations. CrystEngComm 2023, 25, 5262–5285. [Google Scholar] [CrossRef]
- Iwasaki, H. The crystal structures of dimeric and monoclinic forms of mercury(II) N,N-diethyldithiocarbamate, Hg2(S2CNEt2)4 and Hg(S2CNEt2)2. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 1973, 29, 2115–2124. [Google Scholar] [CrossRef]
- Lai, C.S.; Tiekink, E.R.T. Refinement of the crystal structure of bis[(N,N-diethyldithiocarbamato)mercury(II)], [Hg(S2CNEt2)2]2. Z. Kristallogr.—New Cryst. Struct. 2002, 217, 593–594. [Google Scholar] [CrossRef]
- Cox, M.J.; Tiekink, E.R.T. Structural diversity in the mercury(II) bis(N,N-dialkyldithiocarbamate) compounds: An example of the importance of considering crystal structure when rationalising molecular structure. Z. Kristallogr.—Cryst. Mater. 1999, 214, 571–579. [Google Scholar] [CrossRef]
- Cox, M.J.; Tiekink, E.R.T. The crystal structure of monomeric bis(dicyclohexyldithiocarbamato)mercury(II). Main Group Met. Chem. 2000, 23, 793–794. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maglic, J.B.; Lavendomme, R. MoloVol: An easy-to-use program for analyzing cavities, volumes and surface areas of chemical structures. J. Appl. Cryst. 2022, 55, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Berndt, M. DIAMOND, Version 3.2k; GbR: Bonn, Germany, 2006.
- Spackman, P.R.; Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 308–318. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The programsystem turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. Consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Karafiloglou, P.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2018. [Google Scholar]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Grdenić, D. The structural chemistry of mercury. Q. Rev. Chem. Soc. 1965, 19, 303–328. [Google Scholar] [CrossRef]
- Lobana, T.S. Coordination chemistry of organomercury(II) involving phenanthrolines, bipyridines, tertiary phosphines/arsines and some related ligands. Coord. Chem. Rev. 1985, 63, 161–215. [Google Scholar] [CrossRef]
- House, D.A.; Robinson, W.T.; McKee, V. Chloromercury(II) anions. Coord. Chem. Rev. 1994, 135–136, 533–586. [Google Scholar] [CrossRef]
- Morsali, A.; Masoomi, M.Y. Structures and properties of mercury(II) coordination polymers. Coord. Chem. Rev. 2009, 253, 1882–1905. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Mercurophilic interactions. Organometallics 2015, 34, 2048–2066. [Google Scholar] [CrossRef]
- Sarker, J.C.; Hogarth, G. Dithiocarbamate complexes as single source precursors to nanoscale binary, ternary and quaternary metal sulfides. Chem. Rev. 2021, 121, 6057–6123. [Google Scholar] [CrossRef]
- Rodina, T.A.; Loseva, O.V.; Smolentsev, A.I.; Antzutkin, O.N.; Ivanov, A.V. Crystal structure, solid-state 13C and 15N NMR characterisation, chemisorption activity and thermal behaviour of new mercury(II) dipropyldithiocarbamate: Binuclear, pseudo-binuclear and heteronuclear complexes of [Hg2(PrDtc)4], [Hg(PrDtc)2]2 and [Au(PrDtc)2]2[Hg2Cl6]. Inorg. Chim. Acta 2020, 508, 119630. [Google Scholar] [CrossRef]
- Loseva, O.V.; Rodina, T.A.; Smolentsev, A.I.; Ivanov, A.V. A new polymorphic modification and chemisorption activity of mercury(II) N,N-di-iso-propyldithiocarbamate: Synthesis and characterisation of the heteronuclear double complex of ([Au{S2CN(iso-C3H7)2}2]2[Hg2Cl6]·OC(CH3)2)n. Polyhedron 2017, 134, 238–245. [Google Scholar] [CrossRef]
- Gomathi, G.; Dar, S.H.; Thirumaran, S.; Ciattini, S.; Selvanayagam, S. Bis(N-benzyl-N-furfuryldithiocarbamato-S,S′)mercury(II) as a precursor for the preparation of mercury sulfide nanoparticles. C. R. Chim. 2015, 18, 499–510. [Google Scholar] [CrossRef]
- Onwudiwe, D.C.; Ajibade, P.A. Synthesis, characterization and thermal studies of Zn(II), Cd(II) and Hg(II) complexes of N-methyl-N-phenyldithiocarbamate: The single crystal structure of [(C6H5)(CH3)NCS2]4Hg2. Int. J. Mol. Sci. 2011, 12, 1964–1978. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Prince, P.; Gardener, M.; Steed, J. Mercury(II) N,N′-methyl-phenylethyl-dithiocarbamate and its use as a precursor for the room-temperature solution deposition of β-HgS thin films. Adv. Mater. 2004, 16, 994–996. [Google Scholar] [CrossRef]
- Onwudiwe, D.C.; Ajibade, P.A. Synthesis and crystal structure of bis(N-alkyl-N-phenyl dithiocarbamato)mercury(II). J. Chem. Crystallogr. 2011, 41, 980–985. [Google Scholar] [CrossRef]
- Ajibade, P.A.; Onwudiwe, D.C. Synthesis and characterization of group 12 complexes of N,N-methylphenyl-N,N-butyl phenyl dithiocarbamate. J.Coord. Chem. 2011, 63, 2964–2973. [Google Scholar] [CrossRef]
- Ondrušová, D.; Pajtášová, M.; Jóna, E.; Koman, M. Structural properties of Co(III), Hg(II) and Pb(II) N-ethyl-N-phenyl-dithiocarbamates and their application in the rubber industry. Solid State Phenom. 2003, 90–91, 383–388. [Google Scholar] [CrossRef]
- Gurumoorthy, G.; Thirumaran, S.; Ciattini, S. Unusual octahedral Hg(II) dithiocarbamate: Synthesis, spectral and structural studies on Hg(II) complexes with pyrrole based dithiocarbamates and their utility for the preparation of α- and β-HgS. Polyhedron 2016, 118, 143–153. [Google Scholar] [CrossRef]
- Guzmán-Percástegui, E.; Zakharov, L.N.; Alvarado-Rodríguez, J.G.; Carnes, M.E.; Johnson, D.W. Synthesis of a self-assembled Hg(II)-dithiocarbamate metallomacrocycle. Cryst. Growth Des. 2014, 14, 2087–2091. [Google Scholar] [CrossRef]
- Yadav, R.; Trivedi, M.; Kociok-Köhn, G.; Chauhan, R.; Kumar, A.; Gosavi, S.W. Ferrocenyl dithiocarbamate based d10 transition-metal complexes as potential co-sensitizers in dye-sensitized solar cells. Eur. J. Inorg. Chem. 2016, 2016, 1013–1021. [Google Scholar] [CrossRef]
- Dar, S.H.; Thirumaran, S.; Selvanayagam, S. Synthesis, spectral and X-ray structural studies on Hg(II) dithiocarbamate complexes: A new precursor for HgS nanoparticles. Polyhedron 2015, 96, 16–24. [Google Scholar] [CrossRef]
- Kumar, V.; Manar, K.K.; Gupta, A.N.; Singh, V.; Drew, M.G.B.; Singh, N. Impact of ferrocenyl and pyridyl groups attached to dithiocarbamate moieties on crystal structures and luminescent characteristics of group 12 metal complexes. J. Organomet. Chem. 2016, 820, 62–69. [Google Scholar] [CrossRef]
- Altaf, M.; Stoeckli-Evans, H.; Batool, S.S.; Isab, A.A.; Ahmad, S.; Saleem, M.; Awan, S.A.; Shaheen, M.A. Mercury(II) complexes of pyrrolidinedithiocarbamate, crystal structure of bis{[μ2-(pyrrolidinedithiocarbamato-S,S′)(pyrrolidinedithiocarbamato-S,S′)mercury(II)]}. J. Coord. Chem. 2010, 63, 1176–1185. [Google Scholar] [CrossRef]
- Loseva, O.V.; Rodina, T.A.; Antzutkin, O.N.; Ivanov, A.V. Chemisorption activity of mercury(II) cyclopentamethylenedithiocarbamate: Synthesis, structure, and thermal behavior of the [Hg2{S2CN(CH2)5}4] and [Au3{S2CN(CH2)5}6][Au{S2CN(CH2)5}2][Hg2Cl6]2 complexes. Russ. J. Gen. Chem. 2018, 88, 2540–2549. [Google Scholar] [CrossRef]
- Ivanov, A.V.; Korneeva, E.V.; Bukvetskii, B.V.; Goryan, A.S.; Antsutkin, O.N.; Forshling, W. Structural organization of mercury(II) and copper(II) dithiocarbamates from EPR and 13C and 15N MAS NMR spectra and X-ray diffraction analysis. Russ. J. Coord. Chem. 2008, 34, 59–69. [Google Scholar] [CrossRef]
- Benedetti, A.; Fabretti, A.C.; Preti, C. Structure, IR, and NMR spectra of tetrakis(4-methyl piperidinedithiocarbamate) dimercury(II). J. Crystallogr. Spectrosc. Res. 1988, 18, 685–692. [Google Scholar] [CrossRef]
- Khan, A.; Hayat, F.; Butler, I.S.; Tahir, M.N.; ur Rehman, Z. Mercury(II) dithiocarbamates: Structural aspects and their use as single-source precursors for shape-controlled facile synthesis of HgS nanoparticles. Polyhedron 2021, 193, 114876. [Google Scholar] [CrossRef]
- Srinivasan, N.; Thirumaran, S.; Ciattini, S. Effect of co-crystallization of ethanol, pyridine and 2,2′-bipyridine on molecular aggregation in bis(1,2,3,4-tetrahydroquinolinedithiocarbamato-S,S′)mercury(II) and synthesis of HgS nanoparticles. RSC Adv. 2014, 4, 22971–22979. [Google Scholar] [CrossRef]
- Onwudiwe, D.C.; Ajibade, P.A. Synthesis and characterization of Zn(II), Cd(II), and Hg(II) alkyl-aryl dithiocarbamate: X-ray crystal structure of [(C6H5N(et)CS2)Hg(C6H5N(butyl)CS2)]. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2010, 40, 279–284. [Google Scholar] [CrossRef]
- Marimuthu, G.; Ramalingam, K.; Rizzoli, C.; Arivanandhan, M. Solvothermal preparation of nano-β-HgS from a precursor, bis(dibenzyldithiocarbamato)mercury(II). J. Nanopart. Res. 2012, 14, 710. [Google Scholar] [CrossRef]
- Yadav, M.K.; Rajput, G.; Gupta, A.N.; Kumar, V.; Drew, M.G.B.; Singh, N. Exploring the coordinative behaviour and molecular architecture of new PhHg(II)/Hg(II) dithiocarbamate complexes. Inorg. Chim. Acta 2014, 421, 210–217. [Google Scholar] [CrossRef]
- Kumar, A.; Chauhan, R.; Molloy, K.C.; Kociok-Köhn, G.; Bahadur, L.; Singh, N. Synthesis, structure and light-harvesting properties of some new transition-metal dithiocarbamates involving ferrocene. Chem.-Eur. J. 2010, 16, 4307–4314. [Google Scholar] [CrossRef]
- Marimuthu, G.; Ramalingam, K.; Rizzoli, C. Predominant ionic interactions in CdS4N2 and HgS4 coordination environments. J. Coord. Chem. 2013, 66, 699–711. [Google Scholar] [CrossRef]
- Ito, M.; Iwasaki, H. The structure of the monomeric form of mercury(II) N,N-diisopropyldithiocarbamate [bis(N,N-diisopropyldithiocarbamato)mercury(II)]. Acta Crystallogr. B 1979, 35, 2720–2721. [Google Scholar] [CrossRef]
- Angeloski, A.; Rawal, A.; Bhadbhade, M.; Hook, J.M.; Schurko, R.W.; McDonagh, A.M. An unusual mercury(II) diisopropyldithiocarbamate coordination polymer. Cryst. Growth Des. 2019, 19, 1125–1133. [Google Scholar] [CrossRef]
- Singh, R.P.; Maurya, V.K.; Maiti, B.; Siddiqui, K.A.; Prasad, L.B. Synthesis, structure and thermogravimetric analysis of novel dithiocarbamate based Zn(II),Cd(II) and Hg(II) complexes. J. Molec. Struct. 2019, 1198, 126912. [Google Scholar] [CrossRef]
- Rajput, G.; Yadav, M.K.; Thakur, T.S.; Drew, M.G.B.; Singh, N. Versatile coordination environment and interplay of metal assisted secondary interactions in the organization of supramolecular motifs in new Hg(II)/PhHg(II) dithiolates. Polyhedron 2014, 69, 225–233. [Google Scholar] [CrossRef]
- Lai, C.S.; Tiekink, E.R.T. Crystallographic report: Bis(pyrrolinedithiocarbamato)mercury(II). Appl. Organomet. Chem. 2003, 17, 143. [Google Scholar] [CrossRef]
- Oladipo, S.D.; Omondi, B. N,N′-diarylformamidine dithiocarbamates as single-source precursors for the preparation of oleylamine-capped HgS nanoparticles. Transit. Met. Chem. 2020, 45, 391–402. [Google Scholar] [CrossRef]
- Singh, V.; Kumar, V.; Gupta, A.N.; Drew, M.G.B.; Singh, N. Effect of pyridyl substituents leading to the formation of green luminescent mercury(II) coordination polymers, zinc(II) dimers and a monomer. New J. Chem. 2014, 38, 3737–3748. [Google Scholar] [CrossRef]
- Healy, P.C.; White, A.H. Crystal structure of bis(NN-diethyldithiocarbamato)mercury(II). J. Chem. Soc. Dalton Trans. 1973, 284–287. [Google Scholar] [CrossRef]
- Howie, R.A.; Tiekink, E.R.T.; Wardell, J.L.; Wardell, S.M.S.V. Complementary Supramolecular Aggregation via O–H···O Hydrogen-bonding and Hg···S Interactions in Bis[N,N′-di(2-hydroxyethyl)-dithiocarbamato-S,S′]mercury(II): Hg[S2CN(CH2CH2OH)2]2. J. Chem. Crystallogr. 2009, 39, 293–298. [Google Scholar] [CrossRef]
- Singh, V.; Kumar, A.; Prasad, R.; Rajput, G.; Drew, M.G.B.; Singh, N. The interplay of secondary Hg⋯S, Hg⋯N and Hg⋯π bonding interactions in supramolecular structures of phenylmercury(II) dithiocarbamates. CrystEngComm 2011, 13, 6817–6826. [Google Scholar] [CrossRef]
- Loseva, O.V.; Rodina, T.A.; Ivanov, A.V. Pseudo-polymeric mercury(II) morpholinedithiocarbamate [Hg{S2CN(CH2)4O}2]n: Supramolecular structure (a role of secondary Hg···S Bonds), 13C and 15N CP-MAS NMR Spectra, and thermal behavior. Coord. Chem. 2019, 45, 22–29. [Google Scholar] [CrossRef]
- Cox, M.J.; Tiekink, E.R.T. Structural variations in the mercury(II) bis(1,1-dithiolate)s: The crystal and molecular structure of [Hg(S2CNMe2)2]. Z. Kristallogr. Cryst. Mater. 2010, 212, 542–544. [Google Scholar] [CrossRef]
- Chieh, C.; Cheung, S.K. A crystallographic and spectroscopic study of mercury(II) dithiocarbamate. Can. J. Chem. 1981, 59, 2746–2749. [Google Scholar] [CrossRef]
- Konarev, D.V.; Khasanov, S.S.; Lopatin, D.V.; Rodaev, V.V.; Lyubovskaya, R.N. Fullerene complexes with divalent metal dithiocarbamates: Structures, magnetic properties, and photoconductivity. Russ. Chem. Bull. 2007, 56, 2145–2161. [Google Scholar] [CrossRef]
- Ajibade, P.A.; Mbese, J.Z.; Omondi, B. Group 12 dithiocarbamate complexes: Synthesis, characterization, and X-ray crystal structures of Zn (II) and Hg (II) complexes and their use as precursors for metal sulfide nanoparticles. Inorg. Nano-Met. Chem. 2017, 47, 202–212. [Google Scholar] [CrossRef]
- Moulton, B.; Zaworotko, M.J. From molecules to crystal engineering: Supramolecular isomerism and polymorphism in network solids. Chem. Rev. 2001, 101, 1629–1658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-P.; Huang, X.-C.; Chen, X.-M. Supramolecular isomerism in coordination polymers. Chem. Soc. Rev. 2009, 38, 2385–2396. [Google Scholar] [CrossRef]
- Tan, Y.S.; Halim, S.N.A.; Tiekink, E.R.T. Exploring the crystallization landscape of cadmium bis(N-hydroxyethyl, N-isopropyl-dithiocarbamate), Cd[S2CN(iPr)CH2CH2OH]2. Z. Kristallogr. 2016, 231, 113–126. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, C.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Daltons Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Grimme, S.; Mück-Lichtenfeld, C.; Erker, G.; Kehr, G.; Wang, H.; Beckers, H.; Willner, H. When do interacting atoms form a chemical bond? Spectroscopic measurements and theoretical analyses of dideuteriophenanthrene. Angew. Chem. Int. Ed. 2009, 48, 2592–2595. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Shahbazian, S.; Marek, R. Toward a consistent interpretation of the QTAIM: Tortuous link between chemical bonds, interactions, and bond/line paths. Chem.–Eur. J. 2014, 20, 10140–10152. [Google Scholar] [CrossRef]
- Spackman, M.A. How reliable are intermolecular interaction energies estimated from topological analysis of experimental electron densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. J. Mol. Model. 2018, 31, 142. [Google Scholar] [CrossRef]
- Scheiner, S. Quantum chemical analysis of noncovalent bonds within crystals. Concepts and concerns. CrystEngComm 2023, 25, 5060–5071. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists. Coord. Chem. Rev. 2017, 345, 209–228. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef]
- Hu, S.-Z.; Zhou, Z.-H.; Xie, Z.-X.; Robertson, B.E. A comparative study of crystallographic van der Waals radii. Z. Kristallogr.—Cryst. Mater. 2014, 229, 517–523. [Google Scholar] [CrossRef]
- Balmohammadi, Y.; Khavasi, H.R.; Naghavi, S.S. Existence of untypical halogen-involving interactions in crystal packings: A statistical and first-principles study. CrystEngComm 2020, 22, 2756–2765. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The use and misuse of van der Waals radii. Struct. Chem. 2021, 32, 623–629. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Substituent | Second Substituent | Recode | Ref. |
|---|---|---|---|
| Binuclear motif: | |||
| R = Et | R = Et | HGETCB13 | [41] |
| R = nPr | R = nPr | HUKCAU | [67] |
| R = iPr | R = iPr | ZAVYED | [68] |
| R = nBu | R = nBu | CAZRAW | [42] |
| R = CH2(2-furyl) | R = CH2(2-furyl) | ROVTED | [69] |
| R = Me | R’ = Ph | HAKKIP | [70] |
| R = Me | R’ = CH2CH2Ph | YABJIV | [71] |
| R = Me a R = Et | R’ = Ph R’= Ph | OROHEJ | [72] |
| R = Me a R = nBu | R’ = Ph R’ = Ph | HAGLOS | [73] |
| R = Et | R’ = Cy | CAZQUP | [42] |
| R = iPr | R’ = CH2CH2OH | OWOHUF | [35] |
| R = Et | R’ = Ph | YEDQEE | [74] |
| R = nBu | R’= CH2(2-pyrrolyl) | ITEGUL | [75] |
| R = nBu | R’ = R1 b | XOHBAZ | [76] |
| R = CH2CH2OH | R’ = CH2(Fc) c | EJAYOF | [77] |
| R = CH2Ph | R’ = CH2(2-furyl) | ROVVAB | [69] |
| R = CH2CH2Ph | R’ = CH2(2-furyl) | TUMDOW | [78] |
| R = CH2(2-furyl) | R’ = CH2CH2(2-thienyl) | TULJIV | [78] |
| R = CH2(3-pyridyl) | R’ = CH2(Fc) c | UTEKEL | [79] |
| NRR’ = N(CH2)4 | DUWSIY | [80] | |
| NRR’ = N(CH2)5 | POGSUC | [81] | |
| NRR’ = N(CH2)6 | VOHKUZ | [82] | |
| NRR’ = 4-methylpiperidine | KAFFIG | [83] | |
| NRR’ = 4-benzylpiperidine | QAJNAU | [84] | |
| NRR’ = 1,2,3,4-dihydroquinoline | SODNEG | [85] | |
| Quasi-dimeric motif: | |||
| R = Et a R =nBu | R = Ph R = Ph | YEQFEI | [86] |
| R = nPr | R = nPr | HUKCAU | [67] |
| R = iBu | R = iBu | CAZQID | [42] |
| R = CH2Ph | R = CH2Ph | ATADEE | [87] |
| R = iPr | R’ = Cy | CAZQOJ | [42] |
| R = nBu | R’= CH2(2-pyrrolyl) | ITEGOF | [75] |
| R = CH2Ph | R’ = CH2(N-methyl-pyrrol-2-yl) | YOMYUW | [88] |
| R = CH2Ph | R’ = CH2(Fc) c | MUYXOU | [89] |
| NRR’ = 4-(3-phenylprop-2-en-1-yl)piperazine | LIFFEN | [90] | |
| Tetrahedral motif: | |||
| R = iPr | R = iPr | IPTCHG | [91,92] |
| R = Cy | R = Cy | ROPQIW | [43] |
| R = Et | R’= CH2C6H2(OMe)3-3,4,5 | NIMWEO | [93] |
| R = CH2(3-pyridyl) | R’ = CH2(N-methyl-pyrrol-2-yl) | XOBCEY | [94] |
| R = CH2(4-pyridyl) | R’ = CH2(N-methyl-pyrrol-2-yl) | YOMYOQ | [88] |
| NRR’ = N(CH2)4 | MUWDOX | [95] | |
| Square-planar motif: | |||
| R = C6H3(iPr)2-2,6 | R’ = C(H)=NC6H3(iPr)2-2,6 | VUWLUX | [96] |
| One dimensional: zigzag: | |||
| R = CH2CH2Ph | R’ = CH2(3-pyridyl) | FODROH | [97] |
| One dimensional: linear: | |||
| R = Et | R = Et | HGETCB01 | [98] |
| R = CH2CH2OH | R = CH2CH2OH | FOPWAJ | [99] |
| R = CH2Ph | R’ = CH2(3-pyridyl) | FODSAU | [97] |
| R = CH2Ph | R’ = CH2(4-pyridyl) | EBUTAY | [100] |
| NRR’ = N(CH2)4 | POLNEM | [101] | |
| One-dimensional: twisted: | |||
| R = Me | R = Me | ROQNEQ | [102] |
| R = CH2(3-pyridyl) | R’ = CH2(3-pyridyl) | YOMYIK | [88] |
| R = C6H3Me2-2,5 | R’ = C(H)=N(C6H3Me2-2,5) | VUWLOR | [96] |
| NRR’ = 1,2,3,4-dihydroquinoline | SODNIK | [85] | |
| Two-dimensional: | |||
| R = H | R = H | BAWWOL | [103] |
| Multicomponent crystals: | |||
| R = Et d | R = Et | QIYTOI | [104] |
| R = Et e | R’ = Ph | AXIQEF | [105] |
| R = C6H3Me3-2,4,6 f | R’ = C(H)=NC6H3Cl2-2,5 | VUWMAE | [96] |
| NRR’ = N(CH2CH2)NPh2 g | QAJMUN | [84] | |
| NRR’ = 1,2,3,4-dihydroquinoline h | SODNEG | [85] | |
| NRR’ = 1,2,3,4-dihydroquinoline i | SODNIK | [85] | |
| NRR’ = 1,2,3,4-dihydroquinoline j | SODNAC | [85] |
| Parameter | 1 | 2 | 3 a |
|---|---|---|---|
| Hg–S1 | 2.4216(11) | 2.710(3) | 2.527(3) |
| Hg–S2 | 3.1266(9) | 2.438(2) | 2.536(4) |
| Hg–S3 | 2.5183(10) | 2.417(3) | 2.527(3) |
| Hg–S4 | 2.6408(10) | 2.714(3) | 2.536(4) |
| S2–Hg i | 2.6725(10) | 3.727(4) | - |
| S1–Hg–S2 | 63.87(3) | 70.43(9) | 71.69(10) |
| S1–Hg–S3 | 144.88(3) | 126.57(9) | 129.87(16) |
| S1–Hg–S4 | 122.28(3) | 105.96(9) | 126.07(9) |
| S2–Hg–S3 | 92.98(3) | 153.92(8) | 126.07(9) |
| S2–Hg–S4 | 157.70(3) | 127.92(9) | 142.18(18) |
| S3–Hg–S4 | 70.40(3) | 70.58(9) | 71.69(10) |
| S1–Hg–S2 i | 107.96(3) | 131.64(7) | - |
| S2–Hg–S2 i | 100.81(3) | 76.04(6) | - |
| S3–Hg–S2 i | 101.89(3) | 78.20(6) | - |
| S4–Hg–S2 i | 97.22(3) | 122.14(7) | - |
| Hg–S2 i–Hg i | 79.19(3) | 103.96(6) | - |
| Symmetry | |||
| operation i | −x, −y, 1 − z | ½ − x, ½ − y, −z | - |
| Parameter | 1 | 2 | 3 a |
|---|---|---|---|
| van der Waals volume a | 588.2/294.1 | 431.3 | 814.8 |
| Probe-excluded void volume a | 59.4/29.7 | 28.5 | 17.9 |
| Molecular volume a | 647.6/323.8 | 459.7 | 832.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomila, R.M.; Tiekink, E.R.T.; Frontera, A. A Computational Chemistry Investigation of the Influence of Steric Bulk of Dithiocarbamato-Bound Organic Substituents upon Spodium Bonding in Three Homoleptic Mercury(II) Bis(N,N-dialkyldithiocarbamato) Compounds for Alkyl = Ethyl, Isobutyl, and Cyclohexyl. Inorganics 2023, 11, 468. https://doi.org/10.3390/inorganics11120468
Gomila RM, Tiekink ERT, Frontera A. A Computational Chemistry Investigation of the Influence of Steric Bulk of Dithiocarbamato-Bound Organic Substituents upon Spodium Bonding in Three Homoleptic Mercury(II) Bis(N,N-dialkyldithiocarbamato) Compounds for Alkyl = Ethyl, Isobutyl, and Cyclohexyl. Inorganics. 2023; 11(12):468. https://doi.org/10.3390/inorganics11120468
Chicago/Turabian StyleGomila, Rosa M., Edward R. T. Tiekink, and Antonio Frontera. 2023. "A Computational Chemistry Investigation of the Influence of Steric Bulk of Dithiocarbamato-Bound Organic Substituents upon Spodium Bonding in Three Homoleptic Mercury(II) Bis(N,N-dialkyldithiocarbamato) Compounds for Alkyl = Ethyl, Isobutyl, and Cyclohexyl" Inorganics 11, no. 12: 468. https://doi.org/10.3390/inorganics11120468
APA StyleGomila, R. M., Tiekink, E. R. T., & Frontera, A. (2023). A Computational Chemistry Investigation of the Influence of Steric Bulk of Dithiocarbamato-Bound Organic Substituents upon Spodium Bonding in Three Homoleptic Mercury(II) Bis(N,N-dialkyldithiocarbamato) Compounds for Alkyl = Ethyl, Isobutyl, and Cyclohexyl. Inorganics, 11(12), 468. https://doi.org/10.3390/inorganics11120468