
Theoretical Insight on the Formation Mechanism of a Trisubstituted Derivative of Closo-Decaborate Anion [B10H7O2CCH3(NCCH3)]0


 ,
,  , ,
, ,  and
and
Abstract
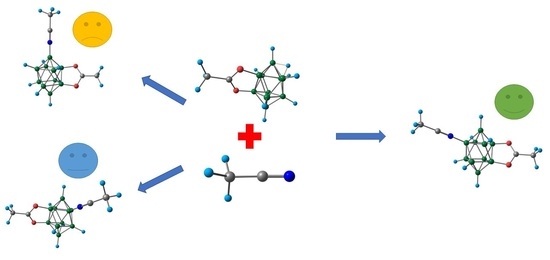
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hagemann, H. Boron hydrogen compounds: Hydrogen storage and battery applications. Molecules 2021, 26, 7425. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Uchman, M. Boron cluster compounds as new chemical leads for antimicrobial therapy. Coord. Chem. Rev. 2021, 431, 213684. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Z.; Chen, H.; Wang, Z.; Zhou, X.; Zhang, H. Progress in three-dimensional aromatic-like closo-dodecaborate. Coord. Chem. Rev. 2021, 444, 214042. [Google Scholar] [CrossRef]
- Gigante, A.; Duchêne, L.; Moury, R.; Pupier, M.; Remhof, A.; Hagemann, H. Direct Solution-Based Synthesis of Na4(B12H12)(B10H10) Solid Electrolyte. ChemSusChem 2019, 12, 4832–4837. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, L.; Lunghammer, S.; Burankova, T.; Liao, W.-C.; Embs, J.P.; Copéret, C.; Wilkening, H.M.R.; Remhof, A.; Hagemann, H.; Battaglia, C. Ionic Conduction Mechanism in the Na2(B12H12)0.5(B10H10)0.5 Closo-Borate Solid-State Electrolyte: Interplay of Disorder and Ion–Ion Interactions. Chem. Mater. 2019, 31, 3449–3460. [Google Scholar] [CrossRef]
- Laskova, J.; Serdyukov, A.; Kosenko, I.; Ananyev, I.; Titova, E.; Druzina, A.; Sivaev, I.; Antonets, A.A.; Nazarov, A.A.; Bregadze, V.I. New Azido Coumarins as Potential Agents for Fluorescent Labeling and Their “Click” Chemistry Reactions for the Conjugation with closo-Dodecaborate Anion. Molecules 2022, 27, 8575. [Google Scholar] [CrossRef] [PubMed]
- Diab, M.; Mateo, A.; El Cheikh, J.; El Hajj, Z.; Haouas, M.; Ranjbari, A.; Guérineau, V.; Touboul, D.; Leclerc, N.; Cadot, E.; et al. Grafting of Anionic Decahydro-Closo-Decaborate Clusters on Keggin and Dawson-Type Polyoxometalates: Syntheses, Studies in Solution, DFT Calculations and Electrochemical Properties. Molecules 2022, 27, 7663. [Google Scholar] [CrossRef]
- Dou, D.; Mavunkal, I.J.; Bauer, J.A.K.; Knobler, C.B.; Hawthorne, M.F.; Shore, S.G. Synthesis and Structure of Triethylammonium 2-(Acetonitrile)nonahydro-closo-decaborate(1-). Inorg. Chem. 1994, 33, 6432–6434. [Google Scholar] [CrossRef]
- Bayer, M.J.; Hawthorne, M.F. An Improved Method for the Synthesis of [closo-B12(OH)12]−2. Inorg. Chem. 2004, 43, 2018–2020. [Google Scholar] [CrossRef]
- Al-Joumhawy, M.; Cendoya, P.; Shmalko, A.; Marei, T.; Gabel, D. Improved synthesis of halo- and oxonium derivatives of dodecahydrido-closo-dodecaborate(2-). J. Organomet. Chem. 2021, 949, 121967. [Google Scholar] [CrossRef]
- Semioshkin, A.A.; Sivaev, I.B.; Bregadze, V.I. Cyclic oxonium derivatives of polyhedral boron hydrides and their synthetic applications. Dalton Trans. 2008, 11, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.-C.; Xing, Y.-Y.; Sun, Y.; Duttwyler, S.; Hong, X. Directed B–H functionalization of the closo -dodecaborate cluster via concerted iodination–deprotonation: Reaction mechanism and origins of regioselectivity. Org. Chem. Front. 2020, 7, 3648–3655. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Zhang, Y.; Liu, J.; van der Veen, S.; Duttwyler, S. The closo -Dodecaborate Dianion Fused with Oxazoles Provides 3D Diboraheterocycles with Selective Antimicrobial Activity. Chem.-A Eur. J. 2018, 24, 10364–10371. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Zhang, C.-Y.; Xu, T.-T.; Wu, J.; Wen, X.-Y.; Jiang, W.-J.; Chen, M.; Yang, J. Synthesis of Polyhedral Borane Cluster Fused Heterocycles via Transition Metal Catalyzed B-H Activation. Molecules 2020, 25, 391. [Google Scholar] [CrossRef] [PubMed]
- Shelly, K.; Knobler, C.B.; Hawthorne, F. Synthesis of Monosubstituted Derivatives of closo -Decahydrodecaborate(2-). X-ray Crystal Structures of [closo-2-B10H9CO]− and [closo-2- B10H9NCO]−. Inorg. Chem. 1992, 31, 2889–2892. [Google Scholar] [CrossRef]
- Laskova, J.; Ananiev, I.; Kosenko, I.; Serdyukov, A.; Stogniy, M.; Sivaev, I.; Grin, M.; Semioshkin, A.; Bregadze, V.I. Nucleophilic addition reactions to nitrilium derivatives [B12H11NCCH3]− and [B12H11NCCH2CH3]−. Synthesis and structures of closo -dodecaborate-based iminols, amides and amidines. Dalton Trans. 2022, 51, 3051–3059. [Google Scholar] [CrossRef]
- Kaszyński, P.; Ringstrand, B. Functionalization of closo-borates via iodonium zwitterions. Angew. Chem.-Int. Ed. 2015, 54, 6576–6581. [Google Scholar] [CrossRef]
- Laila, Z.; Yazbeck, O.; Ghaida, F.A.; Diab, M.; El Anwar, S.; Srour, M.; Mehdi, A.; Naoufal, D. Clean-activation of the B–H bond in closo-decahydrodecaborate [B10H10]2− anion via soft-route. J. Organomet. Chem. 2020, 910, 121132. [Google Scholar] [CrossRef]
- Knoth, W.H.; Sauer, J.C.; England, D.C.; Hertler, W.R.; Muetterties, E.L. Chemistry of Boranes. XIX. 1 Derivative Chemistry of B10H10−2 and B12H12−2. J. Am. Chem. Soc. 1964, 86, 3973–3983. [Google Scholar] [CrossRef]
- Miller, H.C.; Miller, N.E.; Muetterties, E.L. Chemistry of Boranes. XX. Syntheses of Polyhedral Boranes. Inorg. Chem. 1964, 3, 1456–1463. [Google Scholar] [CrossRef]
- Jelinek, T.; Štibr, B.; Mareš, F.; Plešek, J.; Heřmánek, S. Halogenation of 4,5-dicarba-arachno- nonaborane(13),4,5-C2B7H13. Polyhedron 1987, 6, 1737–1740. [Google Scholar] [CrossRef]
- Frank, R.; Adhikari, A.K.; Auer, H.; Hey-Hawkins, E. Electrophile-induced nucleophilic substitution of the nido- dicarbaundecaborate anion nido-7,8-C2B9H12 by conjugated heterodienes. Chem.-A Eur. J. 2014, 20, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Mahfouz, N.; Ghaida, F.A.; El Hajj, Z.; Diab, M.; Floquet, S.; Mehdi, A.; Naoufal, D. Recent Achievements on Functionalization within closo-Decahydrodecaborate [B10H10]2− Clusters. ChemistrySelect 2022, 7, e202200770. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Kulikova, N.Y.; Nizhnik, E.A.; Vichuzhanin, M.V.; Starikova, Z.A.; Semioshkin, A.A.; Bregadze, V.I. Practical synthesis of 1,4-dioxane derivative of the closo-dodecaborate anion and its ring opening with acetylenic alkoxides. J. Organomet. Chem. 2008, 693, 519–525. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Erokhina, S.A.; Sivaev, I.B.; Bregadze, V.I. Nitrilium derivatives of polyhedral boron compounds (boranes, carboranes, metallocarboranes): Synthesis and reactivity. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 983–988. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Prikaznov, A.V.; Naoufal, D. Fifty years of the closo-decaborate anion chemistry. Collect. Czechoslov. Chem. Commun. 2010, 75, 1149–1199. [Google Scholar] [CrossRef]
- Sivaev, I.B.; Bregadze, V.I.; Sjöberg, S. Chemistry of closo-Dodecaborate Anion [B12H12]2−: A Review. Collect. Czechoslov. Chem. Commun. 2002, 67, 679–727. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. The Mechanism of Halogenation of Decahydro-closo-Decaborate Dianion by Hydrogen Chloride. Russ. J. Inorg. Chem. 2021, 66, 1639–1648. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. The reaction of hydrogen halides with tetrahydroborate anion and hexahydro-closo-hexaborate dianion. Molecules 2021, 26, 3754. [Google Scholar] [CrossRef]
- Kochnev, V.K.; Avdeeva, V.V.; Malinina, E.A.; Kuznetsov, N.T. Theoretical study of H2 Elimination from [BnHn+1] monoanions (n = 6–9, 11). Russ. J. Inorg. Chem. 2014, 59, 1268–1275. [Google Scholar] [CrossRef]
- Klyukin, I.N.; Kubasov, A.S.; Limarev, I.P.; Zhdanov, A.P.; Matveev, E.Y.; Polyakova, I.N.; Zhizhin, K.Y.; Kuznetsov, N.T. The new approach to formation of exo boron-oxygen bonds from the decahydro-closo-decaborate(2-) anion. Polyhedron 2015, 101, 215–222. [Google Scholar] [CrossRef]
- Ezhov, A.V.; Vyal’ba, F.Y.; Kluykin, I.N.; Zhdanova, K.A.; Bragina, N.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Mironov, A.F.; Kuznetsov, N.T. Synthesis of New Bioinorganic Systems Based on Nitrilium Derivatives of closo-Decaborate Anion and meso-Arylporphyrins with Pendant Amino Groups. Macroheterocycles 2017, 10, 505–509. [Google Scholar] [CrossRef]
- Klyukin, I.N.; Zhdanov, A.P.; Matveev, E.Y.; Razgonyaeva, G.A.; Grigoriev, M.S.; Zhizhin, K.Y.; Kuznetsov, N.T. Synthesis and reactivity of closo-decaborate anion derivatives with multiple carbon-oxygen bonds. Inorg. Chem. Commun. 2014, 50, 28–30. [Google Scholar] [CrossRef]
- Klyukin, I.N.; Kolbunova, A.V.; Novikov, A.S.; Nelyubin, A.V.; Zhdanov, A.P.; Kubasov, A.S.; Selivanov, N.A.; Bykov, A.Y.; Zhizhin, K.Y.; Kuznetsov, N.T. Synthesis of Disubstituted Carboxonium Derivatives of Closo-Decaborate Anion [2,6-B10H8O2CC6H5]−: Theoretical and Experimental Study. Molecules 2023, 28, 1757. [Google Scholar] [CrossRef]
- Klyukin, I.N.; Kolbunova, A.V.; Novikov, A.S.; Nelyubin, A.V.; Selivanov, N.A.; Bykov, A.Y.; Klyukina, A.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Kuznetsov, N.T. Protonation of Borylated Carboxonium Derivative [2,6-B10H8O2CCH3]−: Theoretical and Experimental Investigation. Int. J. Mol. Sci. 2022, 23, 4190. [Google Scholar] [CrossRef]
- Kochnev, V.K.; Kuznetsov, N.T. Theoretical study of protonation of the B10H102− anion and subsequent hydrogen removal due to substitution reaction in acidic medium. Comput. Theor. Chem. 2016, 1075, 77–81. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracyElectronic supplementary information. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, V.; Cheshmedzhieva, D.; Ilieva, S.; Galabov, B. Atomic Charges in Describing Properties of Aromatic Molecules. J. Org. Chem. 2019, 84, 1908–1915. [Google Scholar] [CrossRef]
- Oller, J.; Pérez, P.; Ayers, P.W.; Vöhringer-Martinez, E. Global and local reactivity descriptors based on quadratic and linear energy models for α, β -unsaturated organic compounds. Int. J. Quantum Chem. 2018, 118, e25706. [Google Scholar] [CrossRef]
- Cheriet, M.; Madi, F.; Nouar, L.; Lafifi, I.; Himri, S.; Merabet, N.; Khatmi, D. A DFT study of inclusion complexes of the antituberculosis drugs pyrazinamide and isoniazid with cucurbit[7]uril. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 127–136. [Google Scholar] [CrossRef]
- Kee, C.W. Molecular Understanding and Practical In Silico Catalyst Design in Computational Organocatalysis and Phase Transfer Catalysis—Challenges and Opportunities. Molecules 2023, 28, 1715. [Google Scholar] [CrossRef] [PubMed]
- Basdogan, Y.; Groenenboom, M.C.; Henderson, E.; De, S.; Rempe, S.B.; Keith, J.A. Machine Learning-Guided Approach for Studying Solvation Environments. J. Chem. Theory Comput. 2020, 16, 633–642. [Google Scholar] [CrossRef]
- Xavier, N.F.; Da Silva, A.M.; Bauerfeldt, G.F. Supercell calculations of the geometry and lattice energy of α-glycine crystal. J. Mol. Model. 2019, 25, 244. [Google Scholar] [CrossRef]
- Neumolotov, N.K.; Selivanov, N.A.; Bykov, A.Y.; Klyukin, I.N.; Novikov, A.S.; Zhdanov, A.P.; Zhizin, K.Y.; Kuznetsov, N.T. New Methods for Preparation of the Monofluorosubstituted Derivative of the closo-Borate Anion [2-B10H9F]2−, Its Properties, and Analysis of Its Reactivity. Russ. J. Inorg. Chem. 2022, 67, 1583–1590. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.; Legare, D. Properties of atoms in molecules: Structures and reactivities of boranes and carboranes. Can. J. Chem. 1992, 70, 657–677. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comp. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; ISBN 9781118229101. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Available online: https://www.chemcraftprog.com (accessed on 27 April 2023).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Species | DFT | DLPNO-CCSDT |
|---|---|---|
| [B10H8O2CCH3(Hfac)] + CH3CN | 0.0 | 0.0 |
| Formation of Tris_B1 | ||
| TSmigr_B1 + CH3CN | 46.6 | 53.7 |
| H2_B1 + CH3CN | 43.1 | 52.3 |
| TSelim_B1 | 195.9 | 157.8 |
| Tris_B1 + H2 | −81.1 | −97.6 |
| Formation of Tris_B3 | ||
| Tsmigr_B3 + CH3CN | 38.6 | 48.2 |
| H2_B3 + CH3CN | 27.6 | 39.7 |
| Tselim_B3 | 151.0 | 115.1 |
| Tris_B3 + H2 | −83.4 | −100.9 |
| Formation of Tris_B4 | ||
| TSmigr_B4 + CH3CN | 33.1 | 43.6 |
| H2_B4 + CH3CN | 19.9 | 32.5 |
| TSelim_B4 | 145.2 | 109.5 |
| Tris_B4 + H2 | −88.5 | −106.5 |
| [B10H11]− + CH3CN | 0.0 | 0.0 |
| Formation of [1-B10H9NCCH3]− | ||
| B10_TSmigr_ap + CH3CN | 49.1 | 57.2 |
| B10_H2_ap + CH3CN | 41.5 | 52.0 |
| B10_TSelim_ap | 195.7 | 159.1 |
| [1-B10H9NCCH3]− + H2 | −57.9 | −74.5 |
| Formation of [2-B10H9NCCH3]− | ||
| B10_TSmigr_eq + CH3CN | 59.1 | 68.2 |
| B10_H2_eq + CH3CN | 29.8 | 42.2 |
| B10_TSelim_eq | 139.9 | 104.2 |
| [2-B10H9NCCH3]− +H2 | −58.9 | −75.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klyukin, I.N.; Kolbunova, A.V.; Novikov, A.S.; Zhdanov, A.P.; Zhizhin, K.Y.; Kuznetsov, N.T. Theoretical Insight on the Formation Mechanism of a Trisubstituted Derivative of Closo-Decaborate Anion [B10H7O2CCH3(NCCH3)]0. Inorganics 2023, 11, 201. https://doi.org/10.3390/inorganics11050201
Klyukin IN, Kolbunova AV, Novikov AS, Zhdanov AP, Zhizhin KY, Kuznetsov NT. Theoretical Insight on the Formation Mechanism of a Trisubstituted Derivative of Closo-Decaborate Anion [B10H7O2CCH3(NCCH3)]0. Inorganics. 2023; 11(5):201. https://doi.org/10.3390/inorganics11050201
Chicago/Turabian StyleKlyukin, Ilya N., Anastasia V. Kolbunova, Alexander S. Novikov, Andrey P. Zhdanov, Konstantin Yu. Zhizhin, and Nikolay T. Kuznetsov. 2023. "Theoretical Insight on the Formation Mechanism of a Trisubstituted Derivative of Closo-Decaborate Anion [B10H7O2CCH3(NCCH3)]0" Inorganics 11, no. 5: 201. https://doi.org/10.3390/inorganics11050201
APA StyleKlyukin, I. N., Kolbunova, A. V., Novikov, A. S., Zhdanov, A. P., Zhizhin, K. Y., & Kuznetsov, N. T. (2023). Theoretical Insight on the Formation Mechanism of a Trisubstituted Derivative of Closo-Decaborate Anion [B10H7O2CCH3(NCCH3)]0. Inorganics, 11(5), 201. https://doi.org/10.3390/inorganics11050201