

Precipitative Coating of Calcium Phosphate on Microporous Silica–Titania Hybrid Particles in Simulated Body Fluid


 ,
,
Abstract
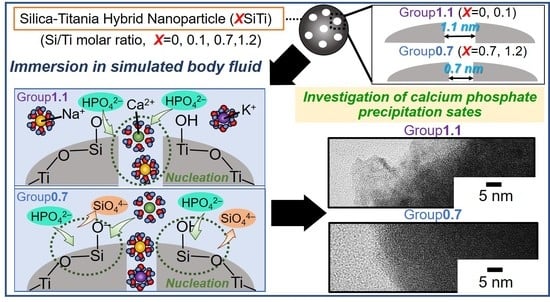
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
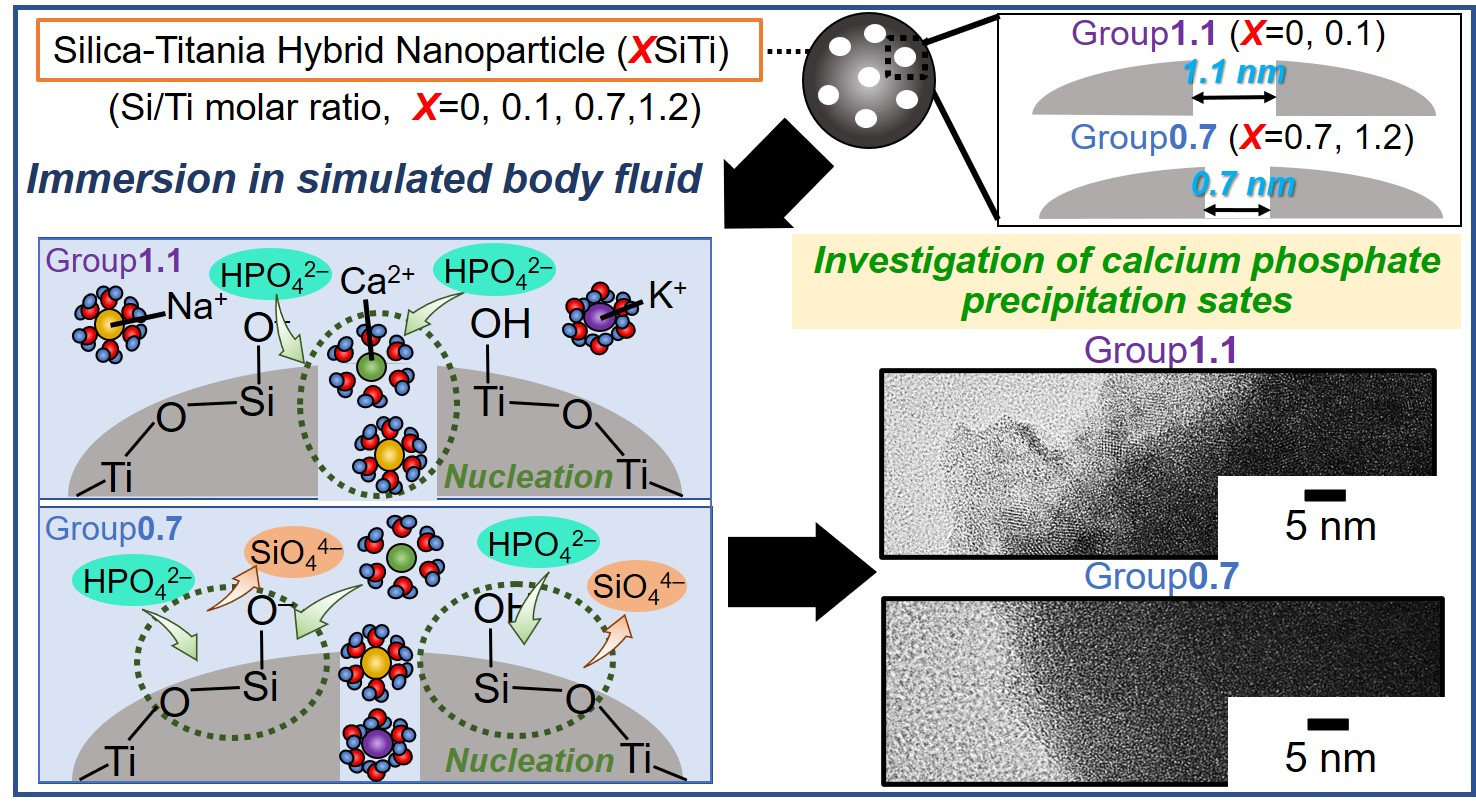
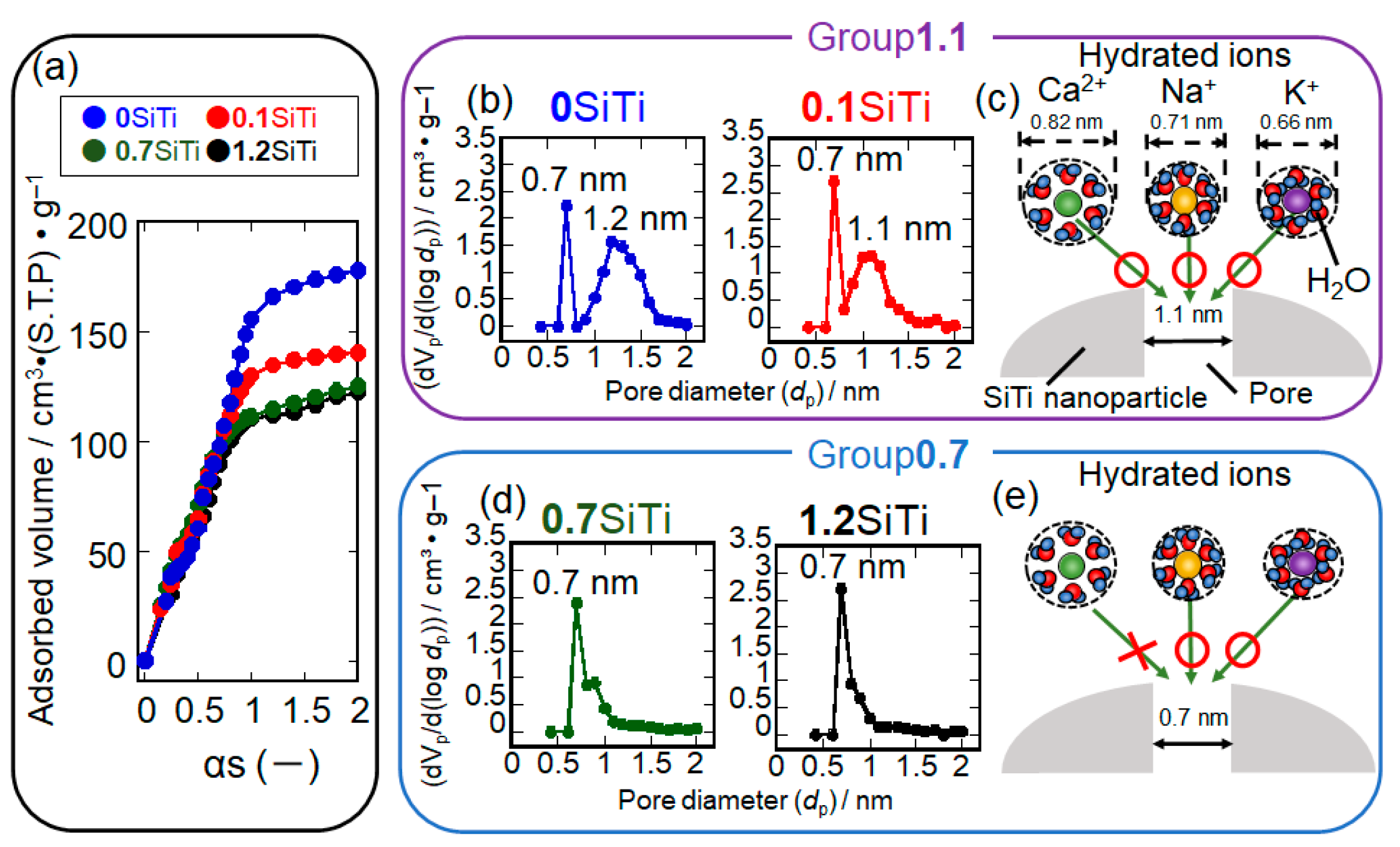
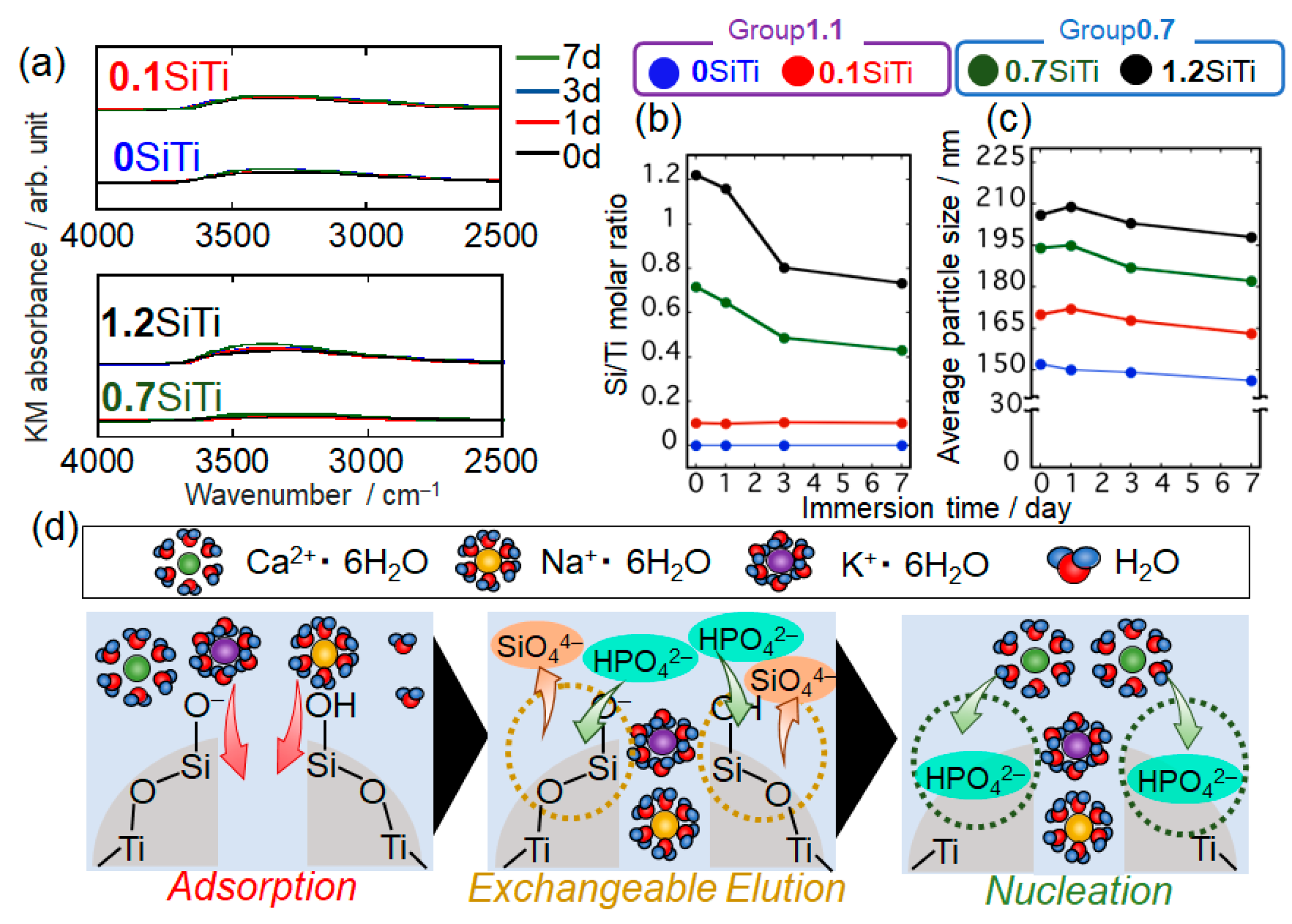
2. Results and Discussion
2.1. Synthesis Result of XSiTi Nanoparticles
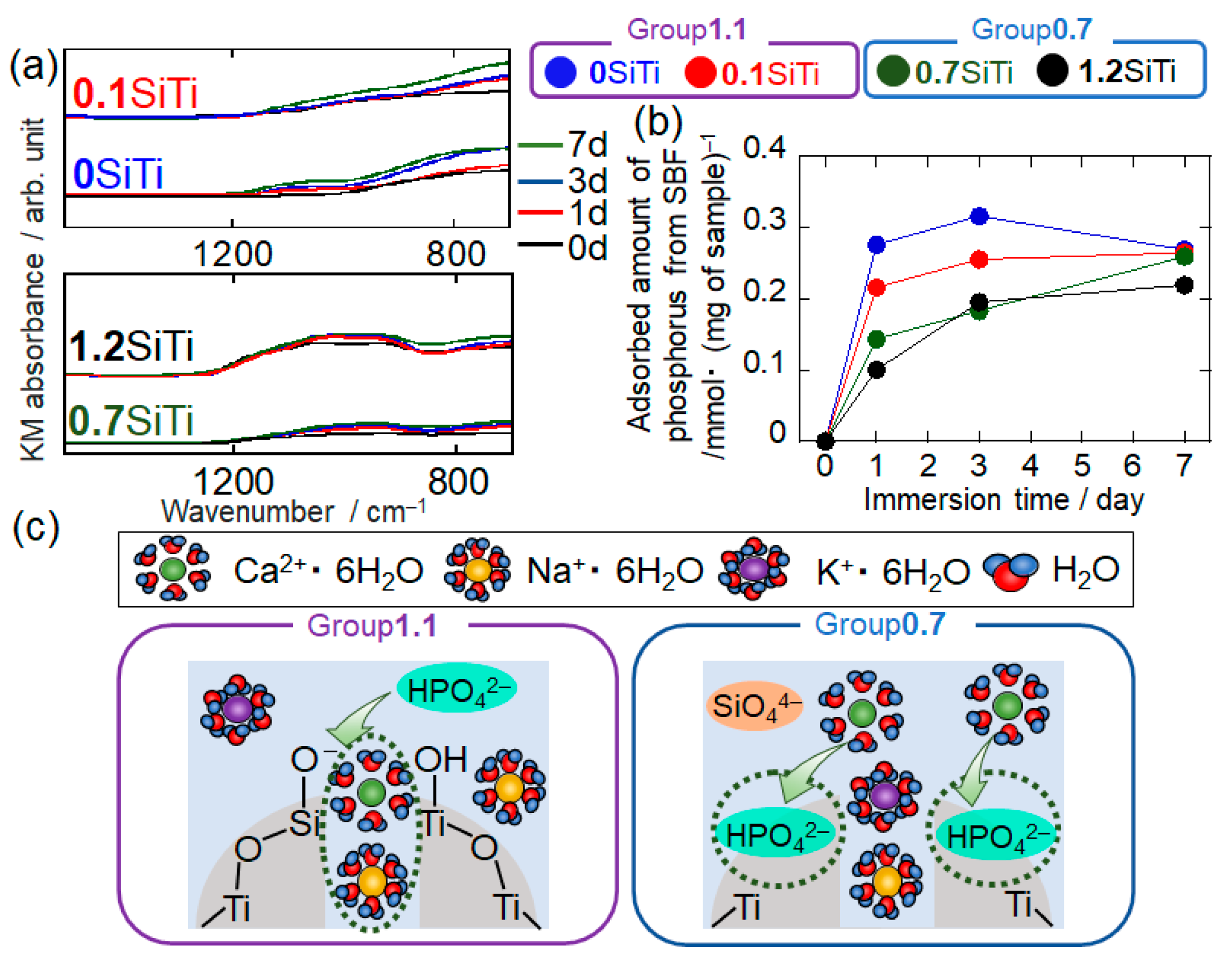
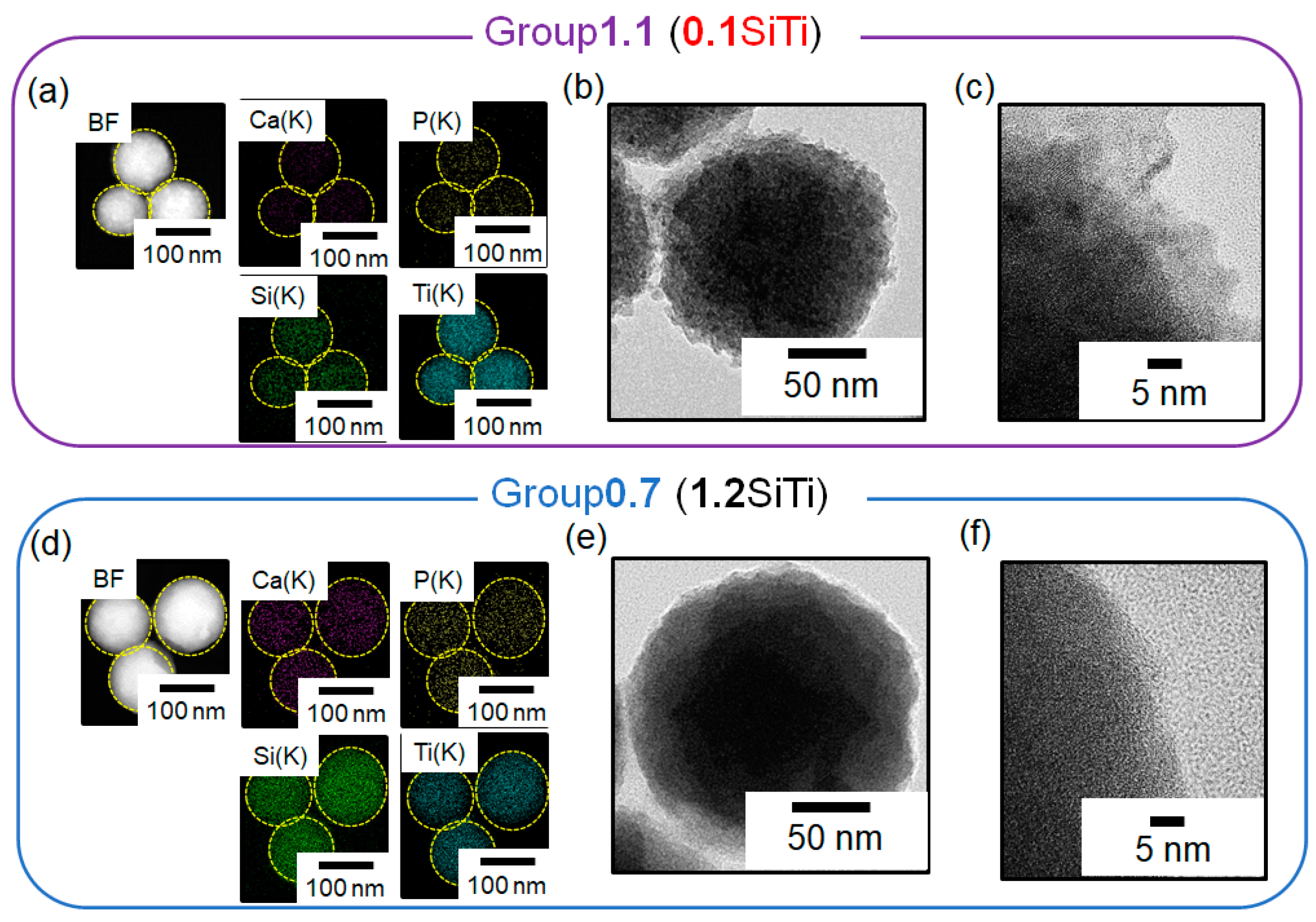
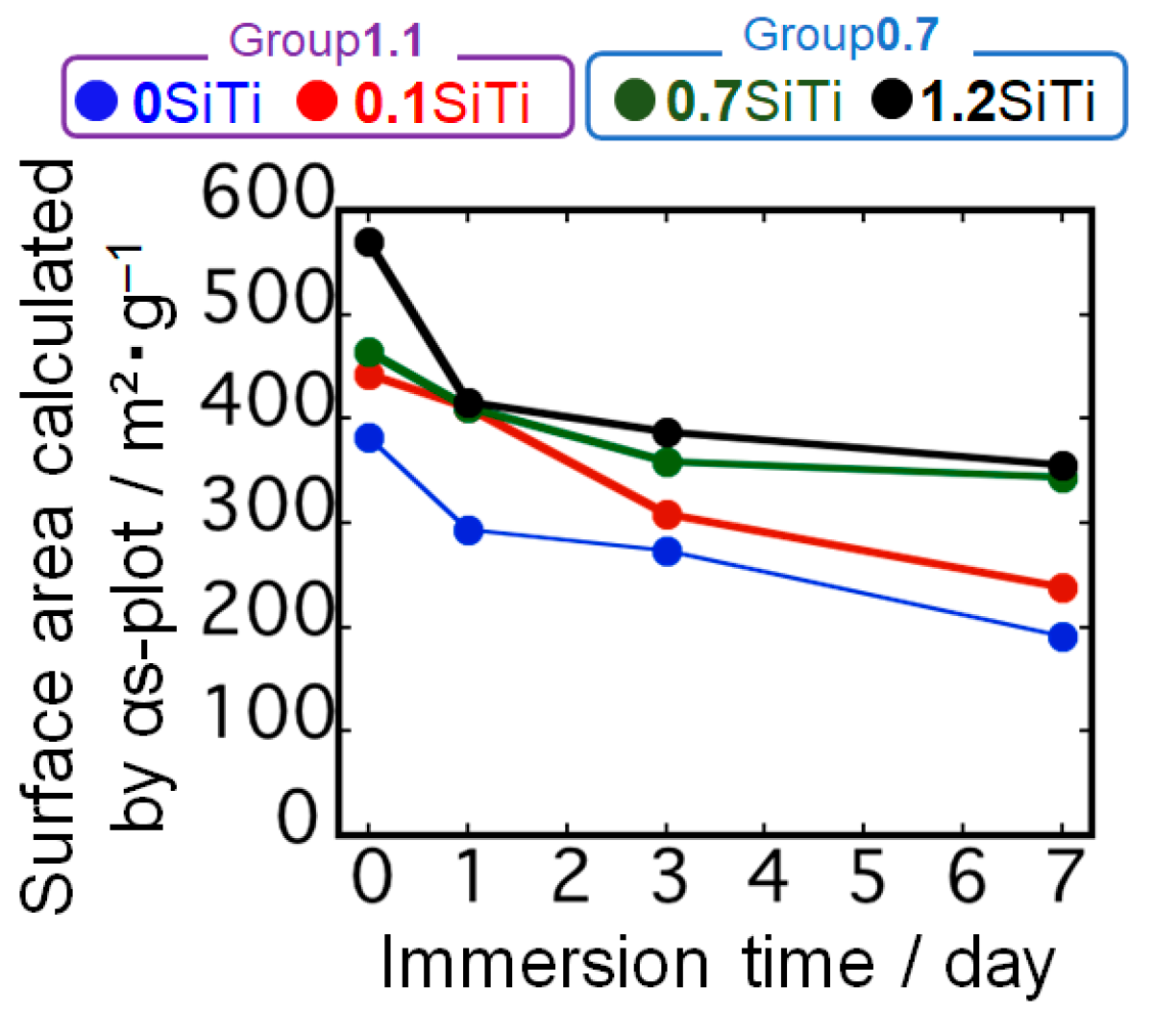
2.2. Results Regarding the SBF-Immersed SiTi Nanoparticles
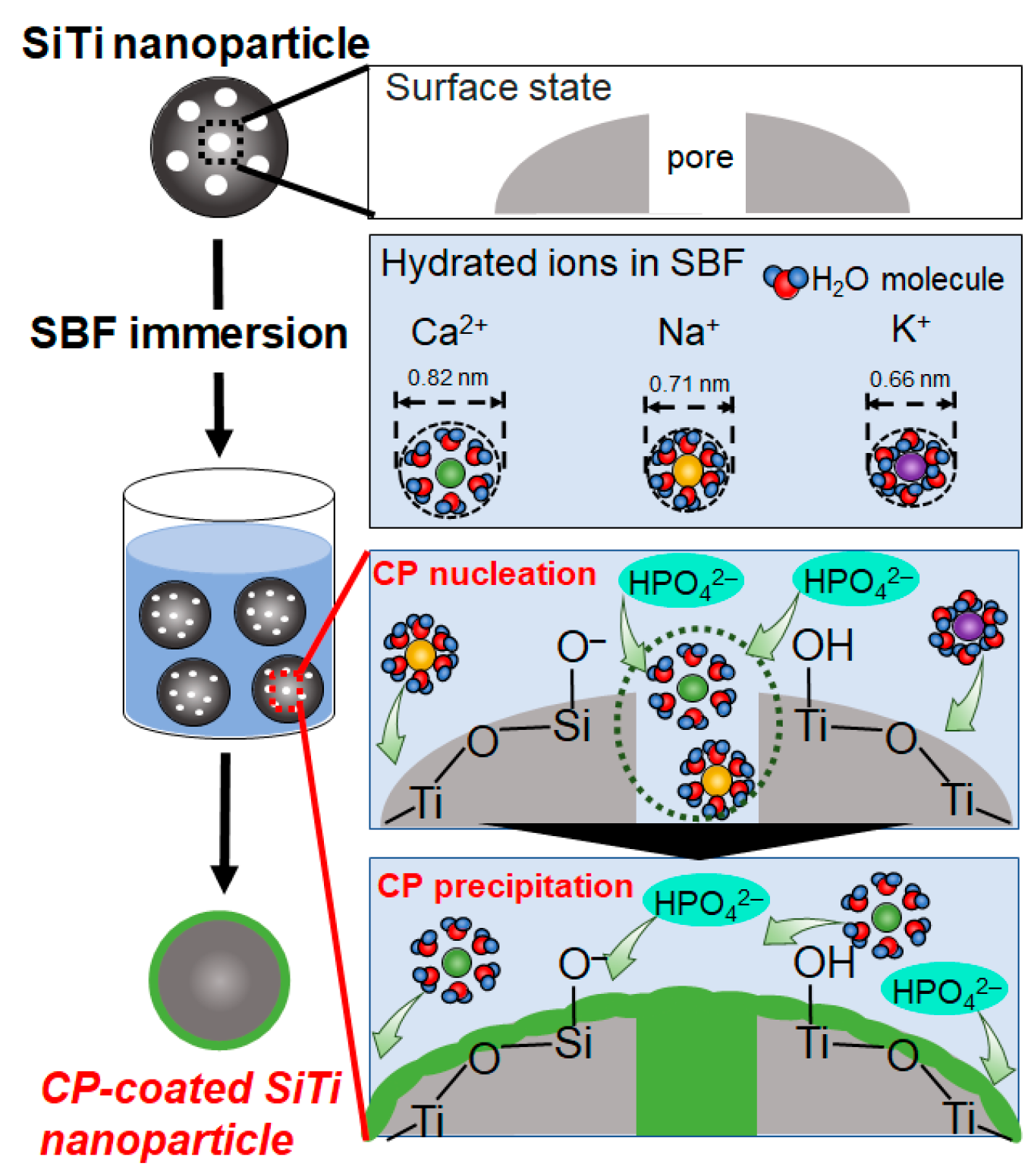
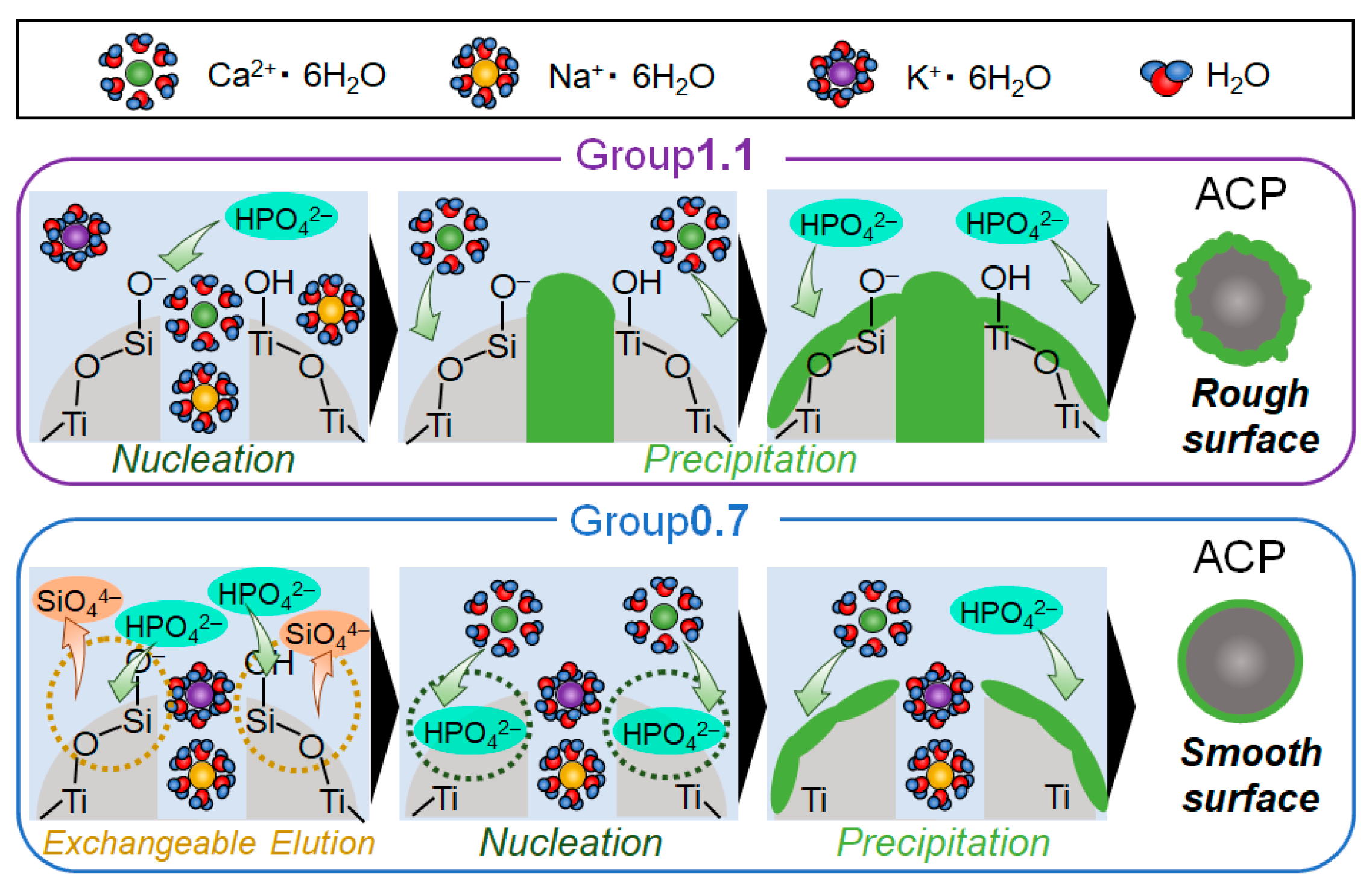
2.3. Mechanism of ACP Precipitation on XSiTi Nanoparticles after Immersion
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis
3.2.1. Synthesis of SiTi Nanoparticles
3.2.2. Immersion of SiTi Nanoparticles into SBF
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, M.; Giri, K.; Garcia-Bennett, A.E. Antioxidant Properties of Probucol Released from Mesoporous Silica. Eur. J. Pharm. Sci. 2019, 138, 105038. [Google Scholar] [CrossRef] [PubMed]
- Marchi, J.; Ussui, V.; Delfino, C.S.; Bressiani, A.H.A.; Marques, M.M. Analysis in Vitro of the Cytotoxicity of Potential Implant Materials. I: Zirconia-Titania Sintered Ceramics. J. Biomed. Mater. Res. B. Appl. Biomater. 2010, 94, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Shimazu, R.; Nagai, H.; Tada, M.; Nakagawa, T.; Sandhu, A.; Handa, H.; Abe, M. Preparation of Spherical and Uniform-Sized Ferrite Nanoparticles with Diameters between 50 and 150 Nm for Biomedical Applications. J. Magn. Magn. Mater. 2009, 321, 1417–1420. [Google Scholar] [CrossRef]
- Rosenholm, J.M.; Peuhu, E.; Eriksson, J.E.; Sahlgren, C.; Lindén, M. Targeted Intracellular Delivery of Hydrophobic Agents Using Mesoporous Hybrid Silica Nanoparticles as Carrier Systems. Nano. Lett. 2009, 9, 3308–3311. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Shiba, K.; Nagata, S.; Yamada, I.; Chai, Y.; Tagaya, M. Preparation of Monodispersed Nanoporous Eu(III)/Titania Loaded with Ibuprofen: Optimum Loading, Luminescence, and Sustained Release. Inorg. Chem. 2021, 60, 8765–8776. [Google Scholar] [CrossRef] [PubMed]
- Chiavaioli, F.; Biswas, P.; Trono, C.; Jana, S.; Bandyopadhyay, S.; Basumallick, N.; Giannetti, A.; Tombelli, S.; Bera, S.; Mallick, A.; et al. Sol-Gel-Based Titania-Silica Thin Film Overlay for Long Period Fiber Grating-Based Biosensors. Anal. Chem. 2015, 87, 12024–12031. [Google Scholar] [CrossRef]
- Arcos, D.; Vallet-Regí, M. Sol-Gel Silica-Based Biomaterials and Bone Tissue Regeneration. Acta. Biomater. 2010, 6, 2874–2888. [Google Scholar] [CrossRef]
- Li, Z.; Hou, B.; Xu, Y.; Wu, D.; Sun, Y.; Hu, W.; Deng, F. Comparative Study of Sol-Gel-Hydrothermal and Sol-Gel Synthesis of Titania-Silica Composite Nanoparticles. J. Solid. State. Chem. 2005, 178, 1395–1405. [Google Scholar] [CrossRef]
- Aw, M.S.; Addai-Mensah, J.; Losic, D. A Multi-Drug Delivery System with Sequential Release Using Titania Nanotube Arrays. Chem. Comm. 2012, 48, 3348–3350. [Google Scholar] [CrossRef]
- Aw, M.S.; Losic, D. Ultrasound Enhanced Release of Therapeutics from Drug-Releasing Implants Based on Titania Nanotube Arrays. Int. J. Pharm. 2013, 443, 154–162. [Google Scholar] [CrossRef]
- Kasar, S.; Kumar, S.; Kar, A.S.; Godbole, S.V.; Tomar, B.S. Sorption of Eu(III) by Amorphous Titania, Anatase and Rutile: Denticity Difference in Surface Complexes. Col. Surf. A. Physicochem. Eng. Asp. 2013, 434, 72–77. [Google Scholar] [CrossRef]
- Ekka, B.; Sahu, M.K.; Patel, R.K.; Dash, P. Titania Coated Silica Nanocomposite Prepared via Encapsulation Method for the Degradation of Safranin-O Dye from Aqueous Solution: Optimization Using Statistical Design. Water. Resour. Ind. 2019, 22, 100071. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Qi, D.; Deng, C.; Zhang, X.; Zhao, D. Superparamagnetic High-Magnetization Microspheres with an Fe3O4@SiO2 Core and Perpendicularly Aligned Mesoporous SiO2 Shell for Removal of Microcystins. J. Am. Chem. Soc. 2008, 130, 28–29. [Google Scholar] [CrossRef] [PubMed]
- Jana, N.R.; Earhart, C.; Ying, J.Y. Synthesis of Water-Soluble and Functionalized Nanoparticles by Silica Coating. Chem. Mater. 2007, 19, 5074–5082. [Google Scholar] [CrossRef]
- Shiba, K.; Sato, S.; Ogawa, M. Preparation of Well-Defined Titania-Silica Spherical Particles. J. Mater. Chem. 2012, 22, 9963–9969. [Google Scholar] [CrossRef]
- He, Q.; Shi, J.; Zhu, M.; Chen, Y.; Chen, F. The Three-Stage in Vitro Degradation Behavior of Mesoporous Silica in Simulated Body Fluid. Microporous. Mesoporous. Mater. 2010, 131, 314–320. [Google Scholar] [CrossRef]
- Raula, J.; Eerikäinen, H.; Peltonen, L.; Hirvonen, J.; Kauppinen, E. Aerosol-Processed Polymeric Drug Nanoparticles for Sustained and Triggered Drug Release. J. Control. Release 2010, 148, e52–e53. [Google Scholar] [CrossRef]
- Zhao, C.; Zhuang, X.; He, P.; Xiao, C.; He, C.; Sun, J.; Chen, X.; Jing, X. Synthesis of Biodegradable Thermo- and PH-Responsive Hydrogels for Controlled Drug Release. Polymer 2009, 50, 4308–4316. [Google Scholar] [CrossRef]
- Paital, S.R.; Dahotre, N.B. Wettability and Kinetics of Hydroxyapatite Precipitation on a Laser-Textured Ca-P Bioceramic Coating. Acta. Biomater. 2009, 5, 2763–2772. [Google Scholar] [CrossRef]
- Barfeie, A.; Wilson, J.; Rees, J. Implant Surface Characteristics and Their Effect on Osseointegration. Br Dent J 2015, 218. [Google Scholar] [CrossRef]
- Shadanbaz, S.; Dias, G.J. Calcium Phosphate Coatings on Magnesium Alloys for Biomedical Applications: A Review. Acta. Biomater. 2012, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; De Groot, K.; Hunziker, E.B. Osteoinductive Implants: The Mise-En-Scène for Drug-Bearing Biomimetic Coatings. Ann. Biomed. Eng. 2004, 32, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Wolke, J.G.C.; Van Der Waerden, J.P.C.M.; Schaeken, H.G.; Jansen, J.A. In Vivo Dissolution Behavior of Various RF Magnetron-Sputtered Ca-P Coatings on Roughened Titanium Implants. Biomater. 2003, 24, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, S.; Wolke, J.; Schoonman, J.; Jansen, J. Electrostatic Spray Deposition (ESD) of Calcium Phosphate Coatings. J. Biomed. Mater. Res. A 2003, 66, 330–334. [Google Scholar] [CrossRef]
- Radin, S.R.; Ducheyne, P. Plasma Spraying Induced Changes of Calcium Phosphate Ceramic Characteristics and the Effect on in Vitro Stability. J. Mater. Sci. Mater. Med. 1992, 3, 33–42. [Google Scholar] [CrossRef]
- Shang, Y.; Yamada, S.; Chai, Y.; Tagaya, M. Synthesis of Spherical Phosphorus-Containing Mesoporous Silica for Improving Their Reaction Behavior in Simulated Body Fluid. Key. Eng. Mater. 2018, 782 KEM, 59–64. [Google Scholar] [CrossRef]
- Kataoka, T.; Shiba, K.; Wang, L.Y.; Yamada, S.; Tagaya, M. Hybrid Preparation of Terbium(III)-Doped Mesoporous Silica Particles with Calcium Phosphates. RSC Adv. 2017, 7, 19479–19485. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; De Groot, K.; Wang, D.; Hu, Q.; Wismeijer, D.; Liu, Y. A Review Paper on Biomimetic Calcium Phosphate Coatings. Open Biomed. Eng. J. 2015, 9, 56–64. [Google Scholar] [CrossRef]
- Li, F.; Feng, Q.L.; Cui, F.Z.; Li, H.D.; Schubert, H. A Simple Biomimetic Method for Calcium Phosphate Coating. Surf. Coat. Technol. 2002, 154, 88–93. [Google Scholar] [CrossRef]
- Leeuwenburgh, S.; Layrolle, P.; Barrre, F.; De Bruijn, J.; Schoonman, J.; Van Blitterswijk, C.A.; De Groot, K. Osteoclastic Resorption of Biomimetic Calcium Phosphate Coatings in Vitro. J. Biomed. Mater. Res. 2001, 56, 208–215. [Google Scholar] [CrossRef]
- Cao, J.; Lian, R.; Jiang, X.; Liu, X. Formation of Porous Apatite Layer after Immersion in SBF of Fluorine-Hydroxyapatite Coatings by Pulsed Laser Deposition Improved in Vitro Cell Proliferation. ACS. Appl. Bio. Mater. 2020, 3, 3698–3706. [Google Scholar] [CrossRef]
- Tanahashi, M.; Matsuda, T. Surface Functional Group Dependence on Apatite Formation on Self- Assembled Monolayers in a Simulated Body Fluid. J. Biomed. Mater. Res. 1997, 34, 305–315. [Google Scholar] [CrossRef]
- Gu, Y.W.; Khor, K.A.; Cheang, P. In Vitro Studies of Plasma-Sprayed Hydroxyapatite/Ti-6Al-4V Composite Coatings in Simulated Body Fluid (SBF). Biomater. 2003, 24, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Beniash, E.; Gawalt, E.; Xu, Z.; Sfeir, C. Biomimetic Coating of Magnesium Alloy for Enhanced Corrosion Resistance and Calcium Phosphate Deposition. Acta. Biomater. 2013, 9, 8650–8659. [Google Scholar] [CrossRef]
- Bigi, A.; Boanini, E.; Bracci, B.; Facchini, A.; Panzavolta, S.; Segatti, F.; Sturba, L. Nanocrystalline Hydroxyapatite Coatings on Titanium: A New Fast Biomimetic Method. Biomater. 2005, 26, 4085–4089. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.; Pochert, A.; Beck, M.; Fiedler, R.; Gruber, J.; Lindén, M. Dissolution Kinetics of Mesoporous Silica Nanoparticles in Different Simulated Body Fluids. J. Solgel. Sci. Technol. 2016, 79, 319–327. [Google Scholar] [CrossRef]
- Chai, Y.; Maruko, Y.; Liu, Z.; Tagaya, M. Design of Oriented Mesoporous Silica Films for Guiding Protein Adsorption States. J. Mater. Chem. B. 2021, 9, 2054–2065. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, J.H.; Kim, Y.T.; Riu, D.H.; Jung, S.J.; Lee, Y.J.; Chung, S.C.; Kim, Y.H. Synthesis of Si, Mg Substituted Hydroxyapatites and Their Sintering Behaviors. Biomaterials 2003, 24, 1389–1398. [Google Scholar] [CrossRef]
- Eliad, L.; Salitra, G.; Soffer, A.; Aurbach, D. Ion Sieving Effects in the Electrical Double Layer of Porous Carbon Electrodes: Estimating Effective Ion Size in Electrolytic Solutions. J. Phy. Chem. B. 2001, 105, 6880–6887. [Google Scholar] [CrossRef]
- Banda, H.; Daffos, B.; Périé, S.; Chenavier, Y.; Dubois, L.; Aradilla, D.; Pouget, S.; Simon, P.; Crosnier, O.; Taberna, P.L.; et al. Ion Sieving Effects in Chemically Tuned Pillared Graphene Materials for Electrochemical Capacitors. Chem. Mater. 2018, 30, 3040–3047. [Google Scholar] [CrossRef]
- Porter, D.W.; Wu, N.; Hubbs, A.F.; Mercer, R.R.; Funk, K.; Meng, F.; Li, J.; Wolfarth, M.G.; Battelli, L.; Friend, S.; et al. Differential Mouse Pulmonary Dose and Time Course Responses to Titanium Dioxide Nanospheres and Nanobelts. Toxicol. Sci. 2013, 131, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Oberdorster, G.; Ferin, J.; Gelein, R.; Soderholm, S.C.; Finkelstein, J. Role of the Alveolar Macrophage in Lung Injury: Studies with Ultrafine Particles. Environ. Health Perspect. 1992, 97, 193–199. [Google Scholar] [PubMed] [Green Version]
- Yu, J.C.; Zhang, L.; Zheng, Z.; Zhao, J. Synthesis and Characterization of Phosphated Mesoporous Titanium Dioxide with High Photocatalytic Activity. Chem. Mater. 2003, 15, 2280–2286. [Google Scholar] [CrossRef]
- Huang, X.; Wang, P.; Yin, G.; Zhang, S.; Zhao, W.; Wang, D.; Bi, Q.; Huang, F. Removal of Volatile Organic Compounds Driven by Platinum Supported on Amorphous Phosphated Titanium Oxide. Wuji Cailiao Xuebao/J. Inorg. Mater. 2020, 35, 482–490. [Google Scholar]
- Aizawa, M.; Nosaka, Y.; Fujii, N. FT-IR Liquid Attenuated Total Reflection Study of TiO2-SiO2 Sol-Gel Reaction. J. Non-Cryst. Solids 1991, 128, 77–85. [Google Scholar] [CrossRef]
- Schraml, M.; Walther, K.; Wokaun, A.; Handy, B.; Baiker, A. Porous Silica Gels and TiO2/SiO2 Mixed Oxides Prepared via the Sol-Gel Process: Characterization by Spectroscopic Techniques. J. Non-Cryst. Solids 1992, 143, 93–111. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure. Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitsugi, T.; Yamamuro, T. Solutions Able to Reproduce in Vivo Surface-Structure Changes in Bioactive Glass-Ceramic A-W3. J. Biomed. Mater. Res. 1990, 24, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, D.; McLeod, A.I.; Sing, K.S.W. Adsorptive Properties of Microporous Carbons: Primary and Secondary Micropore Filling. J. Chim. Phys. 1984, 81, 791–794. [Google Scholar] [CrossRef]
- Mikhail, R.S.; Brunauer, S.; Bodor, E.E. Investigations of a Complete Pore Structure Analysis I. Anal. Micropores; J. coll. Interf Sci. 1968, 26, 45–53. [Google Scholar] [CrossRef]
- Setoyama, N.; Suzuki, T.; Kaneko, K. A Molecular Simulation Study on Empirical Determination Method of Pore Structures of Activated Carbons. Tanso 1997, 1997, 159–166. [Google Scholar] [CrossRef] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura, R.; Shiba, K.; Fujiwara, K.; Zhou, Y.; Yamada, I.; Tagaya, M. Precipitative Coating of Calcium Phosphate on Microporous Silica–Titania Hybrid Particles in Simulated Body Fluid. Inorganics 2023, 11, 235. https://doi.org/10.3390/inorganics11060235
Kimura R, Shiba K, Fujiwara K, Zhou Y, Yamada I, Tagaya M. Precipitative Coating of Calcium Phosphate on Microporous Silica–Titania Hybrid Particles in Simulated Body Fluid. Inorganics. 2023; 11(6):235. https://doi.org/10.3390/inorganics11060235
Chicago/Turabian StyleKimura, Reo, Kota Shiba, Kanata Fujiwara, Yanni Zhou, Iori Yamada, and Motohiro Tagaya. 2023. "Precipitative Coating of Calcium Phosphate on Microporous Silica–Titania Hybrid Particles in Simulated Body Fluid" Inorganics 11, no. 6: 235. https://doi.org/10.3390/inorganics11060235
APA StyleKimura, R., Shiba, K., Fujiwara, K., Zhou, Y., Yamada, I., & Tagaya, M. (2023). Precipitative Coating of Calcium Phosphate on Microporous Silica–Titania Hybrid Particles in Simulated Body Fluid. Inorganics, 11(6), 235. https://doi.org/10.3390/inorganics11060235