
Exploring the Nature of Ag–Ag Interactions in Different Tellurides by Means of the Crystal Orbital Bond Index (COBI)


Abstract

1. Introduction
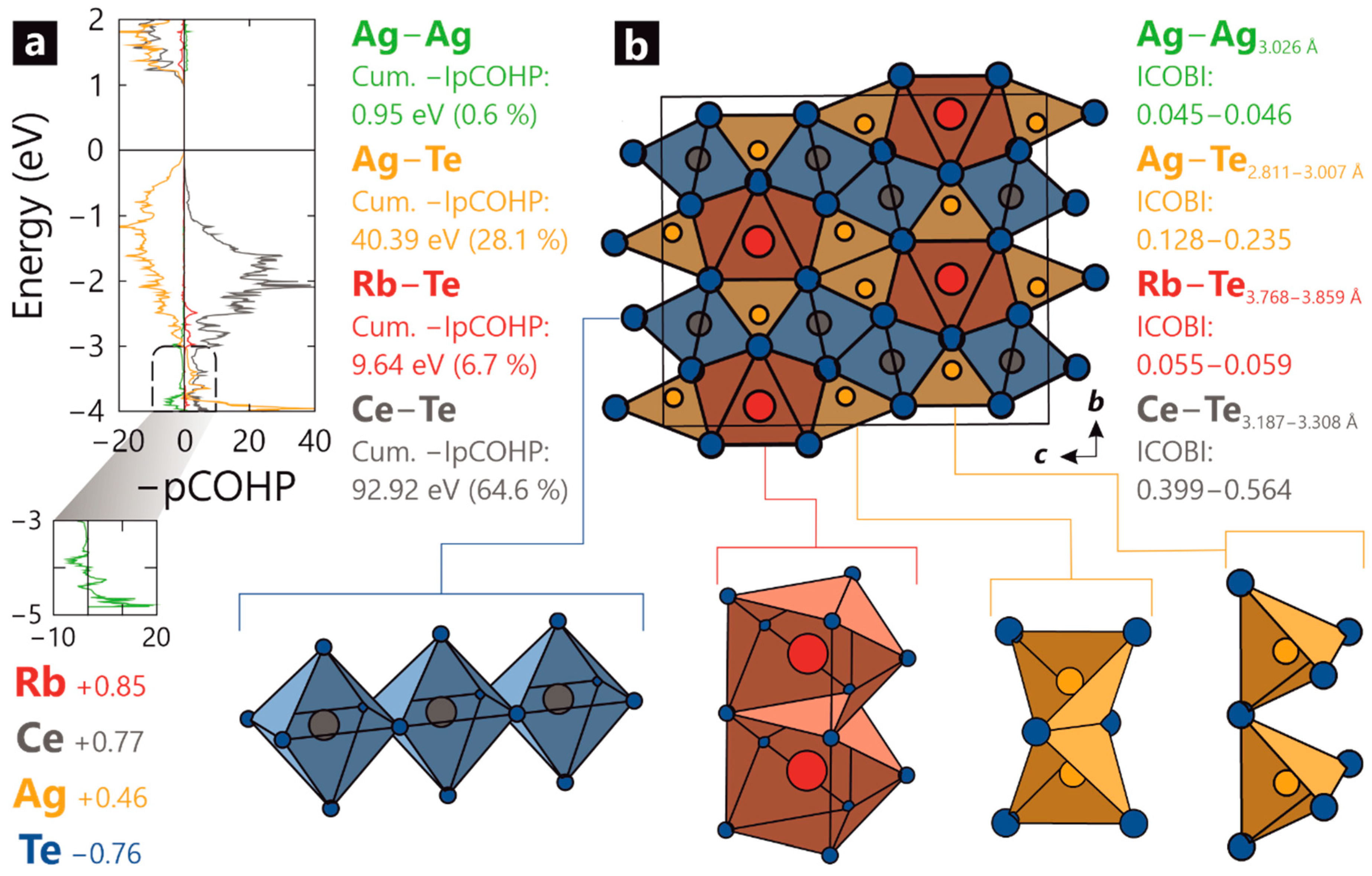
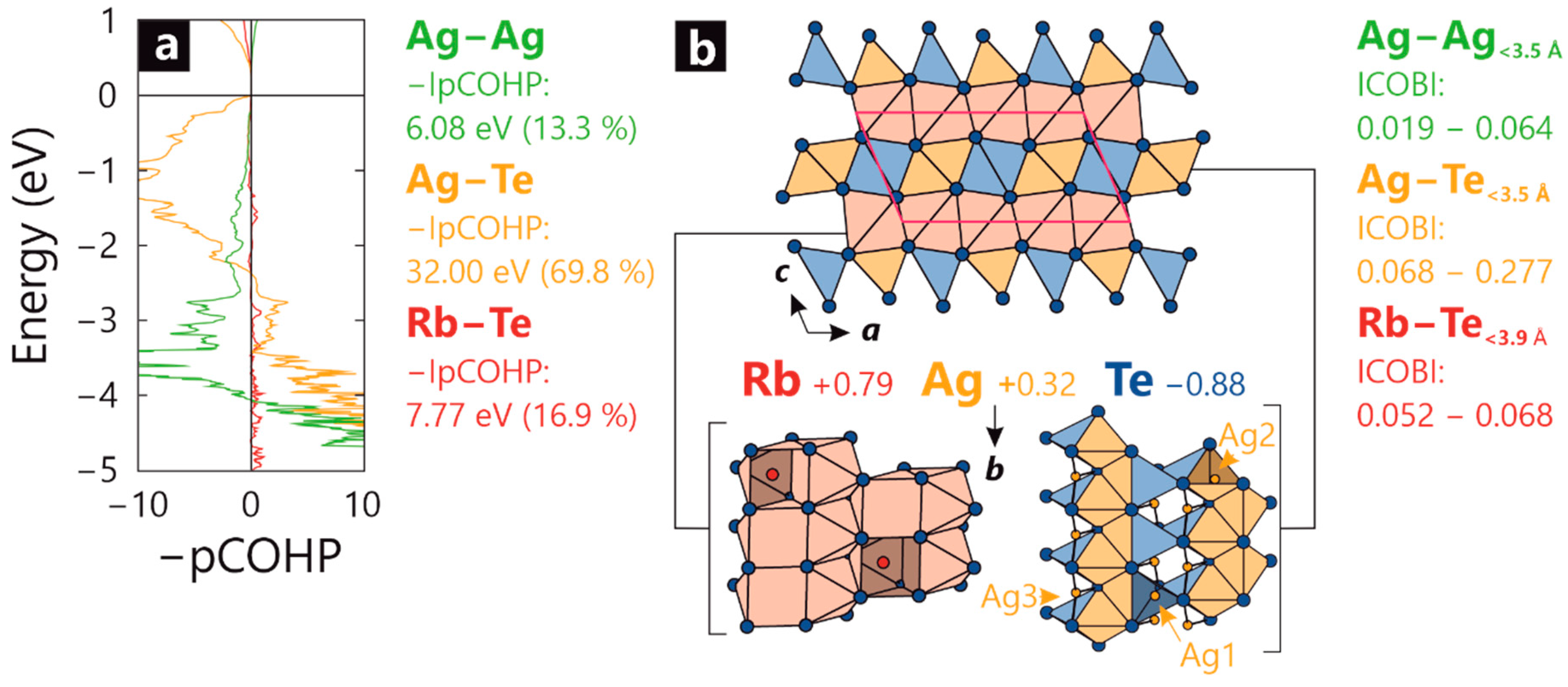
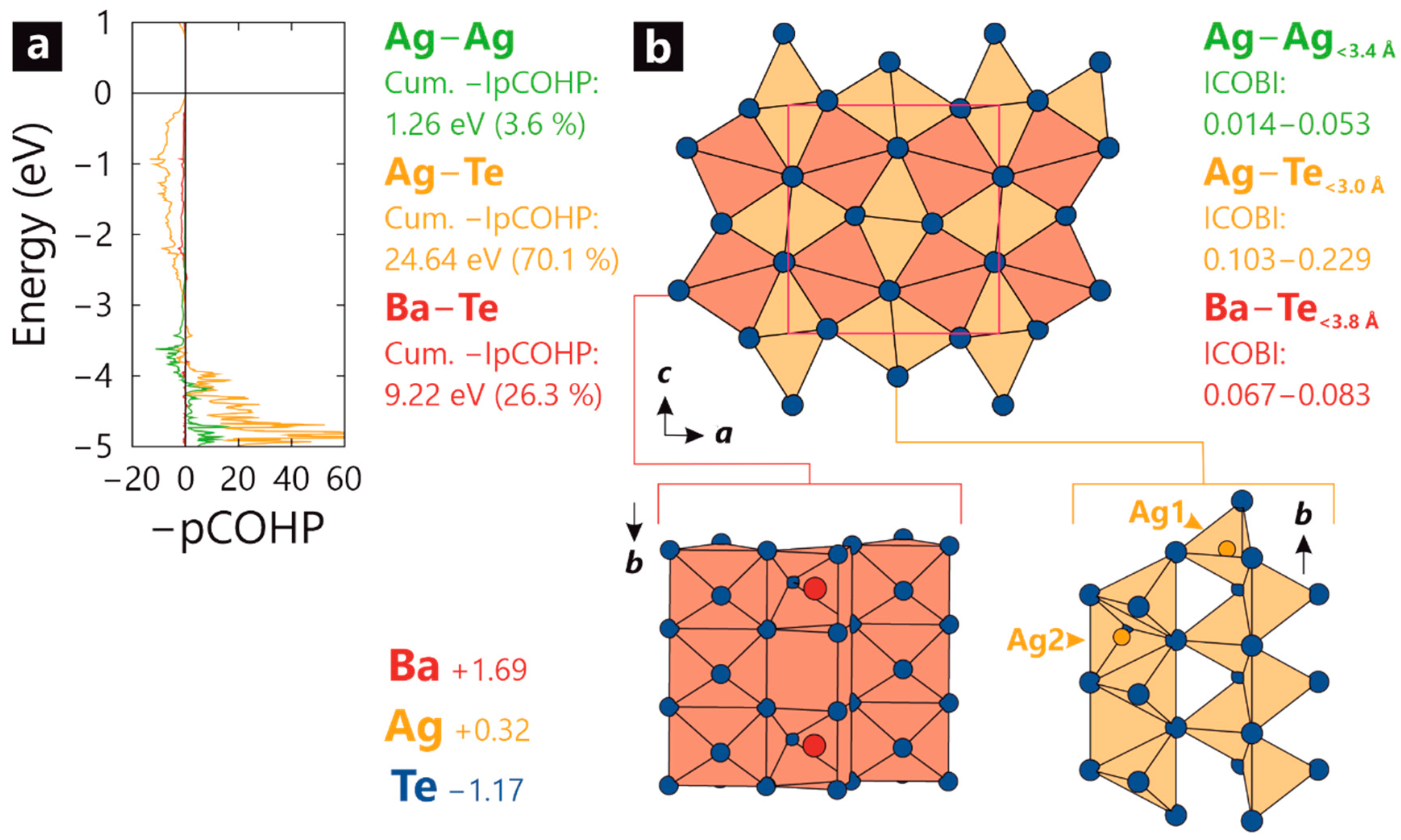
2. Results
3. Discussion
4. Experimental Section
4.1. Synthesis
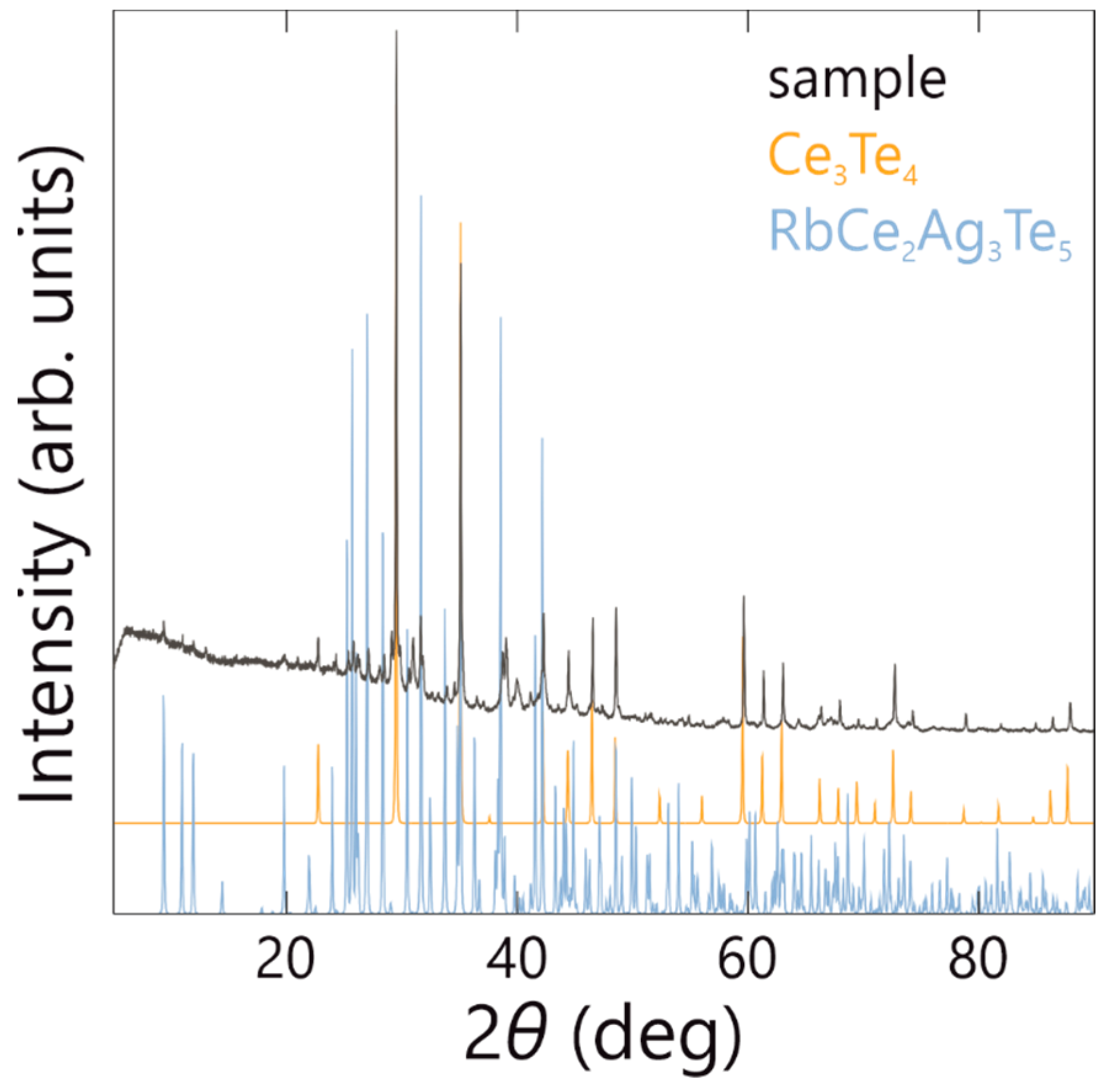
4.2. X-ray Diffraction Experiments
4.3. Computational Details
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruedenberg, K. The Physical Nature of the Chemical Bond. Rev. Mod. Phys. 1962, 34, 326–376. [Google Scholar] [CrossRef]
- Frenking, G.; Shaik, S. The Chemical Bond—Fundamental Aspects of Chemical Bonding; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Miller, G.J. The “Coloring Problem” in Solids: How It Affects Structure, Composition and Properties. Eur. J. Inorg. Chem. 1998, 1998, 523–536. [Google Scholar] [CrossRef]
- Lin, Q.; Miller, G.J. Electron-Poor Polar Intermetallics: Complex Structures, Novel Clusters, and Intriguing Bonding with Pronounced Electron Delocalization. Acc. Chem. Res. 2018, 51, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.J.; Zhang, Y.; Wagner, F.R. Chemical Bonding in Solids. In Handbook of Solid State Chemistry; Dronskowski, R., Kikkawa, S., Stein, A., Eds.; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Dronskowski, R. Chemical Bonding—From Plane Waves via Atomic Orbitals; Walter de Gruyter: Berlin, Germany; Boston, MA, USA, 2023. [Google Scholar]
- Wagner, F.R.; Grin, Y. Chemcial bonding analysis in position space. In Comprehensive Inorganic Chemistry III; Reedijk, J., Poeppelmeier, K.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 3, pp. 222–237. [Google Scholar]
- Jones, R.O.; Elliott, S.R.; Dronskowski, R. The Myth of “Metavalency” in Phase-Change Materials. Adv. Mater. 2023, 35, 2300836. [Google Scholar] [CrossRef] [PubMed]
- Wuttig, M.; Schön, C.-F.; Kim, D.; Golub, P.; Raty, J.-Y.; Kooi, B.J.; Pendás, A.M.; Arora, R.; Waghmare, U.V. Metavalent or Hypervalent Bonding: Is There a Chance for Reconciliation? Adv. Sci. 2024, 11, 2308578. [Google Scholar] [CrossRef]
- Müller, P.C.; Elliott, S.R.; Dronskowski, R.; Jones, R.O. Chemical bonding in phase-change chalcogenides. J. Phys. Condens. Matter, 2024; accepted. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Strong Closed-Shell Interactions in Inorganic Chemistry. Chem. Rev. 1997, 97, 597–636. [Google Scholar] [CrossRef]
- Jansen, M. Über Die Silberteilstruktren in Silberreichen Oxiden. J. Less-Comm. Met. 1980, 76, 285–292. [Google Scholar] [CrossRef]
- Jansen, M. Homoatomic d10-d10 Interactions: Their Effects on Structure and Chemical and Physical Properties. Angew. Chem. Int. Ed. Engl. 1987, 26, 1098–1110. [Google Scholar] [CrossRef]
- Schmidbauer, H.; Schier, A. Argentophilic Interactions. Angew. Chem. Int Ed. 2015, 54, 746–784. [Google Scholar] [CrossRef]
- Sculfort, S.; Braunstein, P. Intramolecular d10–d10 interactions in heterometallic clusters of the transition metals. Chem. Soc. Rev. 2011, 40, 2741–2760. [Google Scholar] [CrossRef] [PubMed]
- Schier, A.; Schmidbauer, H. Aurophilic interactions as a subject of current research: An up-date. Chem. Soc. Rev. 2012, 41, 370–412. [Google Scholar]
- Göbgen, K.C.; Fries, K.S.; Gladisch, F.C.; Dronskowski, R.; Steinberg, S. Revealing the Nature of Chemical Bonding in an ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Telluride. Inorganics 2019, 7, 70. [Google Scholar] [CrossRef]
- Eickmeier, K.; Fries, K.S.; Gladisch, F.C.; Dronskowski, R.; Steinberg, S. Revisiting the Zintl-Klemm Concept for ALn2Ag3Te5-Type Alkaline-Metal (A) Lanthanide (Ln) Silver Tellurides. Crystals 2020, 10, 184. [Google Scholar] [CrossRef]
- Smid, S.; Steinberg, S. Probing the Validity of the Zintl Klemm Concept for Alkaline-Metal Copper Tellurides by Means of Quantum-Chemical Techniques. Materials 2020, 13, 2178. [Google Scholar] [CrossRef]
- Gladisch, F.C.; van Leusen, J.; Passia, M.T.; Kögerler, P.; Steinberg, S. Rb3Er4Cu5Te10: Exploring the Frontier between Polar Intermetallics and Zintl-Phases via Experimental and Quantumchemical Approaches. Eur. J. Inorg. Chem. 2021, 2021, 4946–4953. [Google Scholar] [CrossRef]
- Gladisch, F.C.; Pippinger, T.; Meyer, J.; Pries, J.; Richter, J.; Steinberg, S. Examination of a Structural Preference in Quaternary Alkali-Metal (A) Rare-Earth (R) Copper Tellurides by Combining Experimental and Quantum-chemical Means. Inorg. Chem. 2022, 61, 9269–9282. [Google Scholar] [CrossRef]
- Koch, P.; Steinberg, S. Exploring the subtle factors that control the structural preferences in Cu7Te4. J. Phys. Condens. Matter 2023, 35, 064003. [Google Scholar] [CrossRef] [PubMed]
- Göbgen, K.C.; Gladisch, F.C.; Steinberg, S. The Mineral Stützite: A Zintl-Phase or Polar Intermetallic? A Case Study Using Experimental and Quantum-Chemical Techniques. Inorg. Chem. 2018, 57, 412–421. [Google Scholar] [CrossRef]
- Zintl, E. Intermetallische Verbindungen. Angew. Chem. 1939, 52, 1–6. [Google Scholar] [CrossRef]
- Klemm, W. Metalloids and their Compounds with the Alkali Metals. Proc. Chem. Soc. 1958, 329–364. [Google Scholar] [CrossRef]
- Klemm, W.; Busmann, E. Volumeninkremente und Radien einiger einfach negativ geladener lonen. Z. Anorg. Allg. Chem. 1963, 319, 297–311. [Google Scholar] [CrossRef]
- Schäfer, H.; Eisenmann, B.; Müller, W. Zintl Phases: Transitions between Metallic and Ionic Bonding. Angew. Chem. Int. Ed. 1973, 12, 694–712. [Google Scholar] [CrossRef]
- Nesper, R. The Zintl-Klemm Concept—A Historical Survey. Z. Anorg. Allg. Chem. 2014, 640, 2639–2648. [Google Scholar] [CrossRef]
- Janka, O.; Kauzlarich, S. Zintl Compounds. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R.A., Ed.; Wiley-VCH: Weinheim, Germany, 2021. [Google Scholar]
- Pan, F.; Weinert, B.; Dehnen, S. Binary Zintl Anions Involving Group 13–15 (Semi-)Metal Atoms, and the Relationship of Their Structures to Electron Count. Struct. Bond. 2021, 188, 103–148. [Google Scholar]
- Pöttgen, R.; Johrendt, D. Intermetallics; Walter de Gruyter: Berlin, Germany; Boston, MA, USA, 2019. [Google Scholar]
- Böttcher, P. Tellurium-Rich Tellurides. Angew. Chem. Int. Ed. Engl. 1988, 27, 759–772. [Google Scholar] [CrossRef]
- Sheldrick, W.S. Polychalcogenide Anions: Structural Diversity and Ligand Versatility. Z. Anorg. Allg. Chem. 2012, 638, 2401–2424. [Google Scholar] [CrossRef]
- Müller, P.C.; Ertural, C.; Hempelmann, J.; Dronskowski, R. Crystal Orbital Bond Index: Covalent Bond Orders in Solids. J. Phys. Chem. C 2021, 125, 7959–7970. [Google Scholar] [CrossRef]
- Li, J.; Guo, H.-Y.; Zhang, X.; Kanatzides, M.G. CsAg5Te3: A new metal-rich telluride with a unique tunnel structure. J. Alloys Compds. 1995, 218, 1–4. [Google Scholar] [CrossRef]
- Eanes, M.E.; Schimek, G.L.; Kolis, J.W. Synthesis and structural characterization of CsAg5Se3 and RbAg3Te2. J. Chem. Crystallogr. 2000, 30, 223–226. [Google Scholar] [CrossRef]
- Assoud, A.; Cui, Y.; Thomas, S.; Sutherland, B.; Kleinke, H. Structure and physical properties of the new telluride BaAg2Te2 and its quaternary variants BaCudAg2–dTe2. J. Solid State Chem. 2008, 181, 2024–2030. [Google Scholar] [CrossRef]
- Mayasree, O.; Sankar, C.R.; Kleinke, K.M.; Kleinke, H. Cu clusters and chalcogen-chalcogen bonds in various copper polychalcogenides. Coord. Chem. Rev. 2012, 256, 1377–1383. [Google Scholar] [CrossRef]
- Meng, C.-Y.; Chen, H.; Wang, P. Syntheses, Structures, and Physical Properties of CsRE2Ag3Te5 (RE = Pr, Nd, Sm, Gd-Er) and RbRE2Ag3Te5 (RE = Sm, Gd-Dy). Inorg. Chem. 2014, 53, 6893–6903. [Google Scholar] [CrossRef] [PubMed]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2008, 2832–2838. [Google Scholar] [CrossRef]
- Cotton, S. Lanthanide and Actinide Chemistry; John Wiley & Sons Ltd.: Chichester, UK, 2006. [Google Scholar]
- Miller, G.J.; Schmidt, M.W.; Wang, F.; You, T.-S. Quantitative Advances in the Zintl-Klemm Formalism. Struct. Bond. 2011, 139, 1–55. [Google Scholar]
- Steinberg, S. Revisiting the frontier of the Zintl–Klemm approach for the examples of three Mo2FeB2-type intermetallics by means of quantumchemical techniques. Z. Anorg. Allg. Chem. 2023, 649, e202300113. [Google Scholar] [CrossRef]
- Simons, J.; Hempelmann, J.; Fries, K.S.; Müller, P.C.; Dronskowski, R.; Steinberg, S. Bonding diversity in rock salt-type tellurides: Examining the interdependence between chemical bonding and materials properties. RSC Adv. 2021, 11, 20679–20686. [Google Scholar] [CrossRef] [PubMed]
- Gladisch, F.C.; Steinberg, S. Revealing Tendencies in the Electronic Structures of Polar Intermetallic Compounds. Crystals 2018, 8, 80. [Google Scholar] [CrossRef]
- Steinberg, S.; Dronskowski, R. The Crystal Orbital Hamilton Population (COHP) Method as a Tool to Visualize and Analyze Chemical Bonding in Intermetallic Compounds. Crystals 2018, 8, 225. [Google Scholar] [CrossRef]
- Wu, Z.; Hoffmann, R.-D.; Johrendt, D.; Mosel, B.D.; Eckert, H.; Pöttgen, R. Electronic structure, physical properties and ionic mobility of LiAg2Sn. J. Mater. Chem. 2003, 13, 2561–2565. [Google Scholar] [CrossRef]
- Sebastian, C.P.; Eckert, H.; Fehse, C.; Wright, J.P.; Attfield, J.P.; Johrendt, D.; Rayaprol, S.; Hoffmann, R.-D.; Pöttgen, R. Structural, magnetic, and spectroscopic studies of YAgSn, TmAgSn, and LuAgSn. J. Solid State Chem. 2006, 179, 2376–2385. [Google Scholar] [CrossRef]
- Assoud, A.; Xu, J.; Kleinke, H. Structures and Physical Properties of New Semiconducting Polyselenides Ba2CuδAg4-δSe5 with Unprecedented Linear Se34− Units. Inorg. Chem. 2007, 46, 9906–9911. [Google Scholar] [CrossRef]
- Davaasuren, B.; Dashjav, E.; Rothenberger, A. Synthesis and Characterization of the Ternary Telluroargentate K4[Ag18Te11]. Z. Anorg. Allg. Chem. 2014, 640, 2939–2944. [Google Scholar] [CrossRef]
- Buxi, K.; Kuila, S.K.; Roy, A.; Jana, P.P. Atomic distributions of Ag and In in the γ-brass type Ag9In4. J. Solid State Chem. 2023, 327, 124247. [Google Scholar] [CrossRef]
- Assoud, A.; Guo, Q.; Sankar, C.R.; Kleinke, H. Crystal structure, electronic structure and physical properties of the new quaternary chalcogenides Tl2NdAg3Se4 and Tl2NdAg3Te4. Inorg. Chem. Front. 2017, 4, 315–323. [Google Scholar] [CrossRef]
- Buxi, K.; Mondal, A.; Wang, F.; Jana, P.P. Structural and theoretical investigations on the “coloring” scheme of γ-brass type phase Ag5Cd8. J. Solid State Chem. 2023, 323, 124019. [Google Scholar] [CrossRef]
- Stoyko, S.S.; Blanchard, P.E.R.; Mar, A. Crystal structure, electrical resistivity, and X-ray photoelectron spectroscopy of BaAg2As2. J. Solid State Chem. 2012, 194, 113–118. [Google Scholar] [CrossRef]
- Wang, F.; Pearson, K.N.; Miller, G.J. EuAgxAl11-x with the BaCd11-Type Structure: Phase Width, Coloring, and Electronic Structure. Chem. Mater. 2009, 21, 230–236. [Google Scholar] [CrossRef]
- Stoyko, S.S.; Khatun, M.; Mullen, C.S.; Mar, A. Ternary CaCu4P2-type pnictides AAg4Pn2 (A = Sr, Eu; Pn = As, Sb). J. Solid State Chem. 2012, 192, 325–330. [Google Scholar] [CrossRef]
- Assoud, A.; Soheilnia, N.; Kleinke, H. New Quaternary Barium Copper/Silver Selenostannates: Different Coordination Spheres, Metal-Metal Interactions, and Physical Properties. Chem. Mater. 2005, 17, 2255–2261. [Google Scholar] [CrossRef]
- Steinberg, S.; Brgoch, J.; Miller, G.J.; Meyer, G. Identifying a Structural Preference in Reduced Rare-Earth Metal Halides by Combining Experimental and Computational Techniques. Inorg. Chem. 2012, 51, 11356–11364. [Google Scholar] [CrossRef]
- Hempelmann, J.; Müller, P.C.; Ertural, C.; Dronskowski, R. The Orbital Origins of Chemical Bonding in Ge−Sb−Te Phase-Change Materials. Angew. Chem. Int. Ed. 2022, 61, e202115778. [Google Scholar] [CrossRef] [PubMed]
- Reitz, L.S.; Hempelmann, J.; Müller, P.C.; Dronskowski, R.; Steinberg, S. Bonding Analyses in the Broad Realm of Intermetallics: Understanding the Role of Chemical Bonding in the Design of Novel Materials. Chem. Mater. 2024, accepted. [Google Scholar] [CrossRef]
- McKinney, R.; Gorai, P.; Toberer, E.S.; Stevanovic, V. Rapid Prediction of Anisotropic Lattice Thermal Conductivity: Application to Layered Materials. Chem. Mater. 2019, 31, 2048–2057. [Google Scholar] [CrossRef]
- Shi, Y.; Sturm, C.; Kleinke, H. Chalcogenides as thermoelectric materials. J. Solid State Chem. 2019, 270, 273–279. [Google Scholar] [CrossRef]
- Yu, H.; Li, W.; Pei, Y.; Chen, Y. Pressure and doping effects on the structural stability of thermoelectric BaAg2Te2. J. Phys. Condens. Matter 2022, 34, 065401. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Guo, K.; Yang, X.; Xing, J.; Wang, K.; Luo, J.; Zhao, J.-T. Realizing High Thermoelectric Performance in BaCu2–xAgxTe2 through Enhanced Carrier Effective Mass and Point-Defect Scattering. ACS Appl. Energy Mater. 2019, 2, 889–895. [Google Scholar] [CrossRef]
- Tang, J.; Qin, C.; Yu, H.; Zeng, Z.; Cheng, L.; Ge, B.; Chen, Y.; Li, W.; Pei, Y. Ultralow lattice thermal conductivity enables high thermoelectric performance in BaAg2Te2 alloys. Mater. Today Phys. 2022, 22, 100591. [Google Scholar] [CrossRef]
- Seddik, T.; Rezini, B.; Djelid, K.; Haq, B.U.; Kim, S.-H.; Batouche, M.; Fahad, S.; Djelloul, A.; Yumnam, G. Electronic, optical, and thermoelectric properties of multifunctional zintl compound BaAg2Te2 for energy conversion. Phys. B Condens. Matter 2023, 668, 415209. [Google Scholar] [CrossRef]
- Yu, Y.; Cagnoni, M.; Cojocaru-Mirédin, O.; Wuttig, M. Chalcogenide Thermoelectrics Empowered by an Unconventional Bonding Mechanism. Adv. Funct. Mater. 2020, 30, 1904862. [Google Scholar] [CrossRef]
- Maier, S.; Steinberg, S.; Cheng, Y.; Schön, C.-F.; Schumacher, M.; Mazzarello, R.; Golub, P.; Nelson, R.; Cojocaru-Mirédin, O.; Raty, J.-Y.; et al. Discovering Electron-Transfer-Driven Changes in Chemical Bonding in Lead Chalcogenides (PbX, where X = Te, Se, S, O). Adv. Mater. 2020, 32, 2005533. [Google Scholar] [CrossRef]
- Zeier, W.G.; Zevalkink, A.; Gibbs, Z.M.; Hautier, G.; Kanatzides, M.G.; Snyder, G.J. Thinking Like a Chemist: Intuition in Thermoelectric Materials. Angew. Chem. Int. Ed. 2016, 55, 6826–6841. [Google Scholar] [CrossRef]
- Hempelmann, J.; Müller, P.C.; Konze, P.M.; Stoffel, R.P.; Steinberg, S.; Dronskowski, R. Long-Range Forces in Rock-Salt-Type Tellurides and How they Mirror the Underlying Chemical Bonding. Adv. Mater. 2021, 33, 2100163. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Gorai, P.; Ortiz, B.; Miller, S.; Barnett, S.A.; Mason, T.; Stevanovic, V.; Toberer, E.S. Material descriptors for predicting thermoelectric performance. Energy Environ. Sci. 2015, 8, 983–994. [Google Scholar] [CrossRef]
- Byström, A.; Evers, L. The Crystal Structures of Ag2PbO2 and Ag5Pb2O6. Acta Chem. Scand. 1950, 4, 613–627. [Google Scholar] [CrossRef]
- Jansen, M. Darstellung und Kristallstruktur von LiAg3O2/Preparation and Crystal Structure of LiAg3O2. Z. Naturforsch. B 1975, 30, 854–858. [Google Scholar] [CrossRef]
- Nuss, J.; Wedig, U.; Jansen, M. Synergistic Interaction between Attractive d10 Bonding and Localized Excess Electrons, the Cases of Subvalent Ag5SiO4 and Ag5GeO4. Z. Anorg. Allg. Chem. 2022, 648, e202200269. [Google Scholar] [CrossRef]
- Lofti, S.; Brgoch, J. Discovering Intermetallics Through Synthesis, Computation, and Data-Driven Analysis. Chem. Eur. J. 2020, 26, 8689–8697. [Google Scholar]
- Gautier, R.; Zhang, X.; Hu, L.; Yu, L.; Lin, Y.; Sunde, T.O.L.; Chon, D.; Poeppelmeier, K.R.; Zunger, A. Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds. Nat. Chem. 2015, 7, 308–316. [Google Scholar] [CrossRef]
- Pell, M.A.; Ibers, J.A. Layered Ternary and Quaternary Metal Chalcogenides. Chem. Ber. 1997, 130, 1–8. [Google Scholar] [CrossRef]
- Mitchell, K.; Ibers, J.A. Rare-earth transition-metal chalcogenides. Chem. Rev. 2002, 102, 1929–1952. [Google Scholar] [CrossRef] [PubMed]
- SAINT+, version 7.68; Bruker AXS Inc.: Madison, WI, USA, 2009.
- SADABS, version 2004/1; Bruker AXS Inc.: Madison, WI, USA, 2004.
- XPREP, Version 6.03; Bruker AXS Inc.: Madison, WI, USA, 2014.
- APEX2, v2014.11-0; Bruker Nonius. Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- WinXPow, 2.23 ed.; STOE & Cie GmbH: Darmstadt, Germany, 2005.
- Putz, H. Match! version 3.6.1.115; Crystal Impact GbR: Bonn, Germany, 2018.
- Domange, L.; Flahaut, J.; Pardo, M.-P.; Chirazi, A.N.; Guittard, M. Sur les tellurures de cérium, CeTe2 et Ce3Te4. C. R. Hebd. Séances Acad. Sci. 1960, 250, 857–858. [Google Scholar]
- Stoffel, R.P.; Wessel, C.; Lumey, M. Ab Initio Thermochemistry of Solid-State Materials. Angew. Chem. Int. Ed. 2010, 49, 5242–5266. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, R.P.; Dronskowski, R. Lattice Dynamics and Thermochemistry of Solid-State Materials from First-Principles Quantum-Chemical Calculations. In Handbook of Solid State Chemistry; Dronskowski, R., Kikkawa, S., Stein, A., Eds.; Wiley-VCH: Weinheim, Germany, 2017. [Google Scholar]
- Provino, A.; Steinberg, S.; Smetana, V.; Kulkarni, R.; Dhar, S.K.; Manfrinetti, P.; Mudring, A.-V. Gold-rich R3Au7Sn3: Establishing the interdependence between electronic features and physical properties. J. Mater. Chem. C 2015, 3, 8311–8321. [Google Scholar] [CrossRef]
- Smetana, V.; Steinberg, S.; Mudryk, Y.; Pecharsky, V.; Miller, G.J.; Mudring, A.-V. Cation-Poor Complex Metallic Alloys in Ba(Eu)−Au−Al(Ga) Systems: Identifying the Keys that Control Structural Arrangements and Atom Distributions at the Atomic Level. Inorg. Chem. 2015, 54, 10296–10308. [Google Scholar] [CrossRef] [PubMed]
- Bigun, I.; Steinberg, S.; Smetana, V.; Mudryk, Y.; Kalychak, Y.; Havela, L.; Pecharsky, V.; Mudring, A.-V. Magnetocaloric Behavior in Ternary Europium Indides EuT5In: Probing the Design Capability of First-Principles-Based Methods on the Multifaceted Magnetic Materials. Chem. Mater. 2017, 29, 2599–2614. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented wave-method. Phys. Rev. B Condens. Matter Mater. Phys. 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total energy calculations using a plane-wave basis set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Marsman, M.; Furthmüller, J. Vienna Ab-Initio Simulation Package VASP: The Guide; Computational Materials Physics, Faculty of Physics, Universität Wien: Vienna, Austria, 2014. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Ertural, C.; Steinberg, S.; Dronskowski, R. Development of a robust tool to extract Mulliken and Löwdin charges from plane waves and its applications to solid-state materials. RSC Adv. 2019, 9, 29821–29830. [Google Scholar] [CrossRef] [PubMed]
- Dronskowski, R.; Blöchl, P.E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets. J. Phys. Chem. A 2011, 115, 5461–5466. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic Projection From Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréff, A.L.; Dronskowski, R. LOBSTER: A tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef]
- Eck, B. wxDragon 2.2.3; RWTH Aachen University: Aachen, Germany, 2020. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RbCe2Ag3Te5 |
|---|---|
| fw | 1327.32 |
| space group | Cmcm |
| a (Å) | 4.665(2) |
| b (Å) | 16.142(6) |
| c (Å) | 18.874(7) |
| volume (Å3) | 1421.4(9) |
| Z | 4 |
| density (calc., g/cm3) | 6.203 |
| μ (mm−1) | 23.746 |
| F (000) | 2216 |
| θ ranges | 2.158–28.822 |
| index ranges | −5 ≤ h ≤ 6 −21 ≤ k ≤ 21 −24 ≤ l ≤ 25 |
| reflections collected | 5332 |
| independent reflections | 1072 |
| refinement method | full-matrix least squares on F2 |
| data/restraints/parameter | 1072/0/37 |
| goodness-of-fit on F2 | 0.995 |
| final R indices [I > 2σ(I)] | R1 = 0.0386; wR2 = 0.0762 |
| R indices (all data) | R1 = 0.0584; wR2 = 0.0832 |
| Rint | 0.0816 |
| largest diff. peak and hole (e−/Å3) | −2.008 and 2.387 |
| Atom | Wyckoff Position | x | y | z | Ueq, Å2 |
|---|---|---|---|---|---|
| Ce1 | 8f | 0 | 0.1904 (1) | 0.4055 (1) | 0.0141 (2) |
| Te2 | 8f | 0 | 0.5573 (1) | 0.1223 (1) | 0.0155 (2) |
| Te3 | 8f | 0 | 0.1700 (1) | 0.5740 (1) | 0.0140 (2) |
| Te4 | 4c | 0 | 0.2612 (1) | ¼ | 0.0157 (3) |
| Ag5 | 8f | 0 | 0.4134 (1) | 0.4732 (1) | 0.0233 (3) |
| Ag6 | 4c | ½ | 0.1647 (1) | ¼ | 0.0262 (4) |
| Rb7 | 4c | 0 | 0.0587 (1) | ¾ | 0.0270 (5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinelt, L.; Steinberg, S. Exploring the Nature of Ag–Ag Interactions in Different Tellurides by Means of the Crystal Orbital Bond Index (COBI). Inorganics 2024, 12, 192. https://doi.org/10.3390/inorganics12070192
Weinelt L, Steinberg S. Exploring the Nature of Ag–Ag Interactions in Different Tellurides by Means of the Crystal Orbital Bond Index (COBI). Inorganics. 2024; 12(7):192. https://doi.org/10.3390/inorganics12070192
Chicago/Turabian StyleWeinelt, Leander, and Simon Steinberg. 2024. "Exploring the Nature of Ag–Ag Interactions in Different Tellurides by Means of the Crystal Orbital Bond Index (COBI)" Inorganics 12, no. 7: 192. https://doi.org/10.3390/inorganics12070192
APA StyleWeinelt, L., & Steinberg, S. (2024). Exploring the Nature of Ag–Ag Interactions in Different Tellurides by Means of the Crystal Orbital Bond Index (COBI). Inorganics, 12(7), 192. https://doi.org/10.3390/inorganics12070192