Abstract
AAZTA (6-amino-6-methylperhydro-1,4-diazepinetetraacetic acid) is a mesocyclic chelating agent forming stable complexes with several metal ions. Over the past 20 years since its inception, AAZTA and its bifunctional derivatives have gained a growing role in several applications ranging from MRI contrast agents to diagnostics and nuclear medicine. The recent market restrictions applied to nitroethane preclude the easy preparation of AAZTA, prompting the search for a suitable alternative. In this work, we report the synthesis of two structural analogs (AAZTA-Bn and AAZTA-Et) from commercially available chemicals and the thermodynamic and kinetic study of their complexing ability towards selected metal ions. A comparison of the complexing properties of AAZTA-Bn and AAZTA-Et with the former AAZTA allows us to identify the possible heir of this efficient chelating agent.
1. Introduction
Metal complexes have been widely employed for decades in medicine for diagnostic, therapeutic, and theranostic purposes [1,2,3]. Since most metal ions are toxic to humans, they must be strongly coordinated with a suitable chelating agent before being administered to patients. Polyaminopolycarboxylic acids (PACs) are the most popular family of chelating agents (CAs), easily forming stable complexes with a large variety of metal ions. This property is exploited in a large number of applications ranging from the paper industry to metal separation, water softening, food, and cosmetic additives, and not least in medicine [4,5]. For example, Gd3+ complexes of PACs have been on the market for more than 35 years as magnetic resonance imaging (MRI) contrast agents [6,7] while several radiometal ions (e.g., 111In3+, 90Y3+, 68Ga3+, and 177Lu3+) are strongly coordinated by suitably designed PACs, providing radiopharmaceuticals exploited for PET/SPECT imaging and cancer therapy [8,9,10].
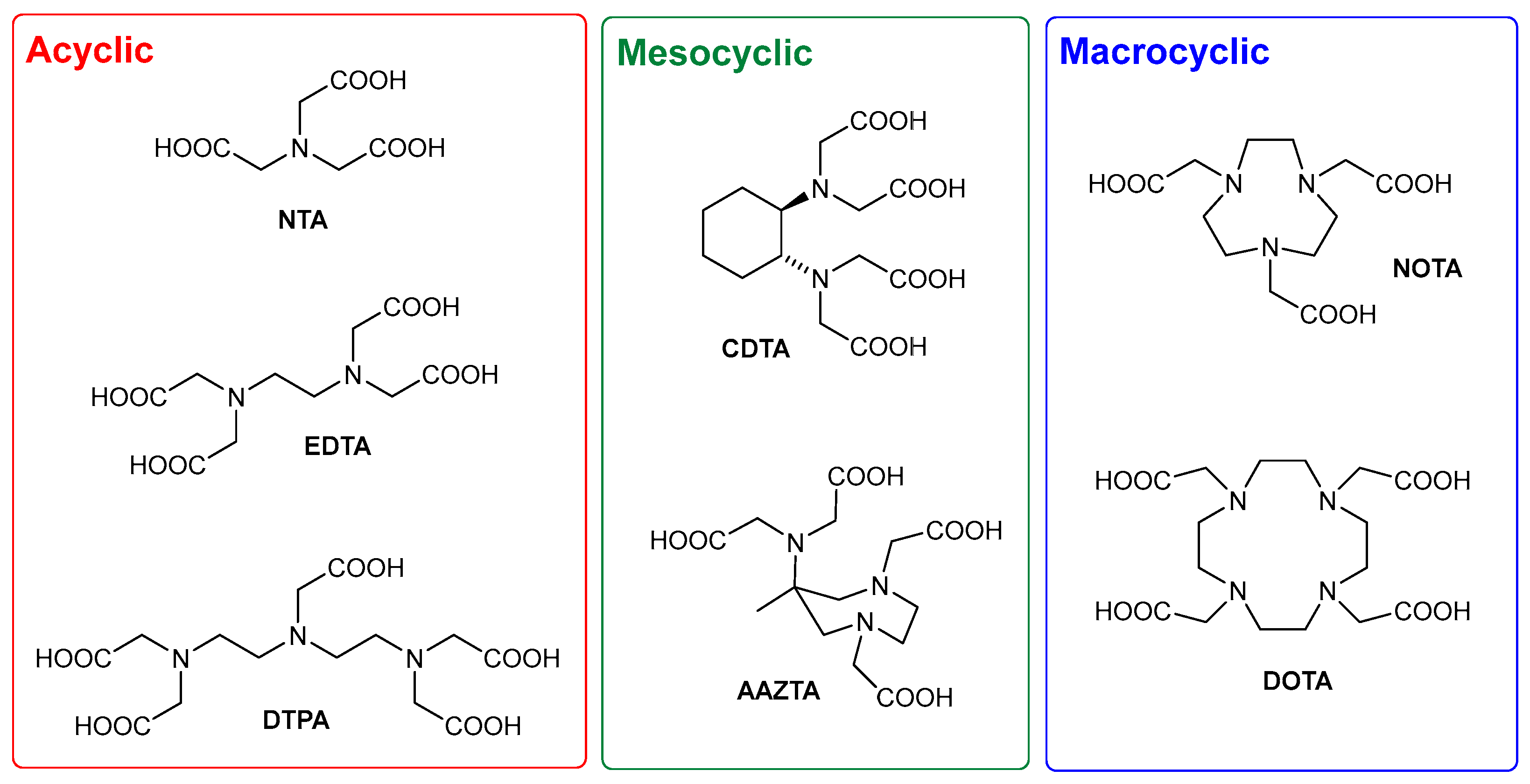
The teeming world of PACs has always been dominated by two main subfamilies (Figure 1): acyclic PACs such as NTA (nitrilotriacetic acid), EDTA (ethylenediaminotetraacetic acid), and DTPA (diethylenetriaminopentaacetic acid), and macrocyclic molecules such as NOTA (1,4,7-triazacyclononane-N,N′,N″-triacetic acid) and DOTA (1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid). Acyclic PACs are cheaper and share a generally simpler preparation with respect to macrocyclic ones, but the complexes of the latter are usually endowed with better thermodynamic and kinetic stability profiles. Mesocyclic CAs such as CDTA (trans-1,2-cyclohexanediamine-N,N,N′,N′-tetraacetic acid) and AAZTA (6-amino-6-methyl-perhydro-1,4-diazepine-N,N′,N″,N″-tetraacetic acid), based on medium-sized rings (5–8 atoms), have been developed in an attempt to combine the structural flexibility and the synthetic accessibility of acyclic CAs and the favorable structural preorganization of macrocyclic CAs, avoiding low-efficiency macrocyclization steps.
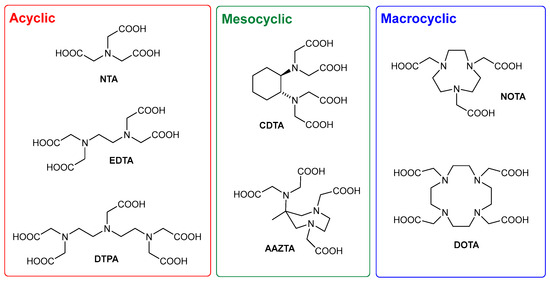
Figure 1.
Polyaminopolycarboxylic chelating agents (PACs).
Among mesocyclic CAs, AAZTA carved out a significant role since its first appearance in 2004 [11]. Originally designed to form an inert Gd3+ complex as an improved MRI contrast agent, AAZTA demonstrated to be a highly efficient chelating agent, forming stable complexes with other lanthanoid metal ions [12]. The good performance of the dihydrated [Gd(AAZTA)(H2O)2]− as an MRI contrast agent prompted the preparation of several derivatives, either modified to include specific structural moieties (e.g., lipophilic appendages to promote the formation of lipid-based nanoparticles [13]) or bearing additional functional groups leading to AAZTA-based bifunctional chelating agents [14,15], which are strategic starting materials for the preparation of paramagnetic conjugates of [Gd(AAZTA)]− by reaction with (bio)molecular vectors [16].
In addition, AAZTA proved to be a highly promising coordinating platform for metal ions of interest in nuclear medicine (NM). The mesocyclic diazepane ring of AAZTA allows for a combination of a high affinity for different metal ions with a fast kinetics of formation and a slow kinetics of dissociation, which is crucial for the prompt coordination of short-lived radioactive isotopes relevant for clinical NM applications [17]. Among the medical radionuclides, AAZTA was demonstrated to rapidly form stable chelates with the positron emitters 64Cu2+ [18], 68Ga3+ [18], and 44Sc3+ (PET) [19], and the γ-emitter 111In3+ (SPECT) [18], the β-emitter 177Lu3+ (radiotherapy) [20], and the α-emitter 213Bi3+ (targeted α-therapy, TAT) [21]. The generalized coordination ability of AAZTA is proposed to capitalize the theranostic pair 68Ga-177Lu. These excellent coordination properties have been exploited in a variety of radioconjugates [22], in which AAZTA complexes are covalently linked to small molecules such as RGD-peptidomimetics [23] and PSMA-ligands, peptides as octreotide analogs [24,25] and minigastrin, and even monoclonal antibodies [26]. Most of these probes have been tested in vivo, with two AAZTA-derived 68Ga3+ radioconjugates, ([68Ga]Ga-DATA5m.SA.FAPi [27] and [68Ga]Ga-DATA5m-LM4), and one 177Lu3+ radioconjugate ([177Lu]Lu-AAZTA5m-LM4) that was also evaluated in human oncologic patients, showing extremely promising results [28].
AAZTA has possibly approached its early demise owing to market restrictions recently applied to nitroethane, one of the starting materials for the preparation of AAZTA. As the end of nitroethane must not translate into the end of AAZTA, we took advantage of the relatively simple and short synthesis of AAZTA to find a possible heir of this useful and efficient mesocyclic PAC.
In this manuscript, we report the preparation of two closely related structural AAZTA analogs and the determination of the thermodynamic and kinetic properties of a selection of their complexes. The comparison of these results with those obtained in previous years for AAZTA complexes demonstrates the possibility of structural variations in the AAZTA molecules with no detrimental effects on its coordination behavior, rather with possible scope for improvement in the complexation ability.
2. Results and Discussion
2.1. Design and Synthesis of AAZTA Derivatives
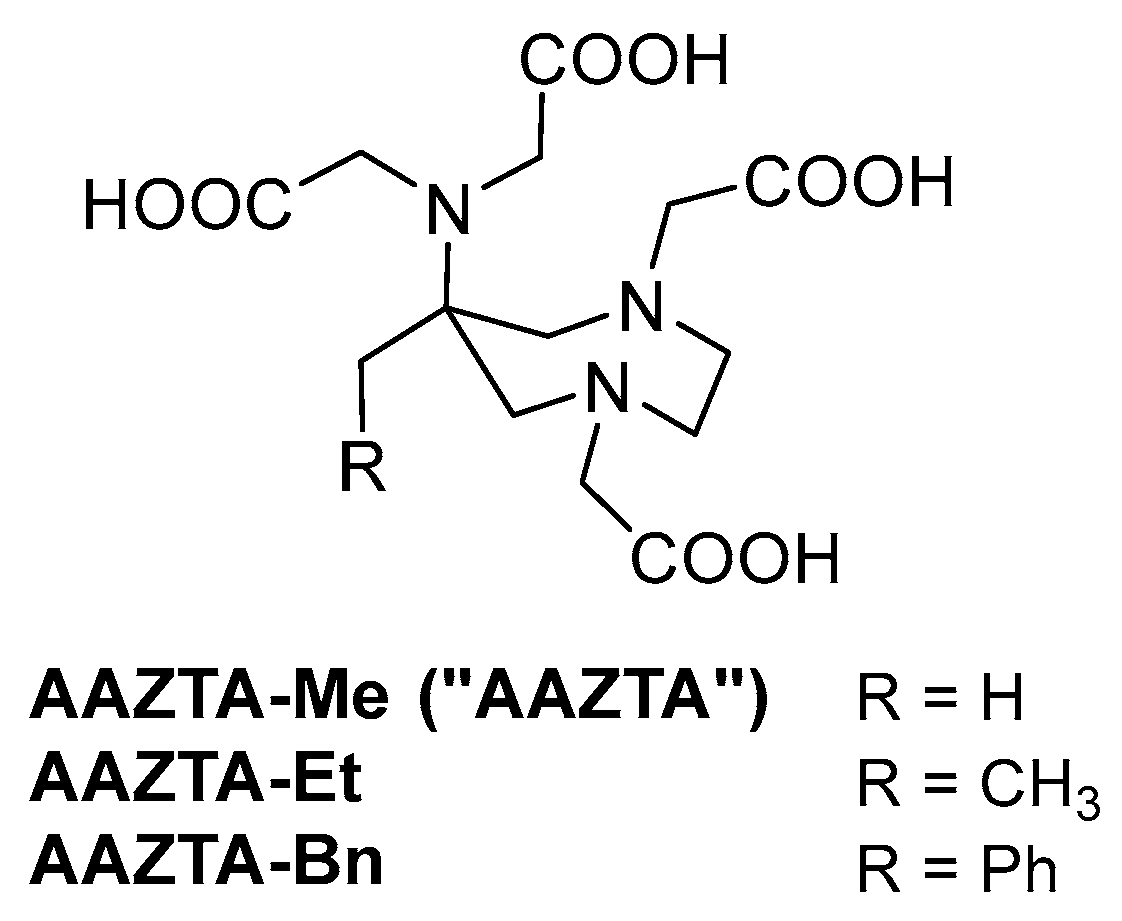
The molecular backbone of AAZTA is based on a seven-membered ring (1,4-diazepane) containing two endocyclic donor N-atoms and an additional exocyclic N-atom in position 6. The N-atoms are decorated with coordinating carboxymethyl residues, completing the heptadentate PAC. The exocyclic N-atom is located at position 6, allowing the correct connectivity for stable coordination. In the same position, AAZTA bears a methyl group, functional to the first step of the synthesis but relatively uninfluential to the coordination sphere of the corresponding complexes. Several AAZTA derivatives were prepared by replacing this methyl group, especially with a functional group containing aliphatic moieties, providing the bifunctional PACs used for the synthesis of metal complex–(bio)vector conjugates. Unfunctionalized AAZTA analogs, representing useful models for studying the coordination ability of this mesocyclic PAC towards different metal ions, have not been investigated in depth. In this work, we undertook the preparation of two AAZTA analogs and the preliminary investigation of their coordination properties in comparison with the parent AAZTA.
The AAZTA analogs discussed in this work are depicted in Figure 2. The parent AAZTA (R = H) is for clarity indicated hereafter as AAZTA-Me to highlight the substitution. AAZTA-Et represents the simplest homolog, with a single additional methylene group, and could be prepared by 1-nitropropane, a cheap starting material devoid of market restrictions. AAZTA-Bn (R = Ph), previously reported by us in a patent [29], bears a 6-benzyl substituent, in which the aromatic ring gives the possibility of further functionalization and is a chromophore suitable for the development of luminescent metal complexes and for tracing this PAC and its complexes by UV-Vis-based analytical methods.
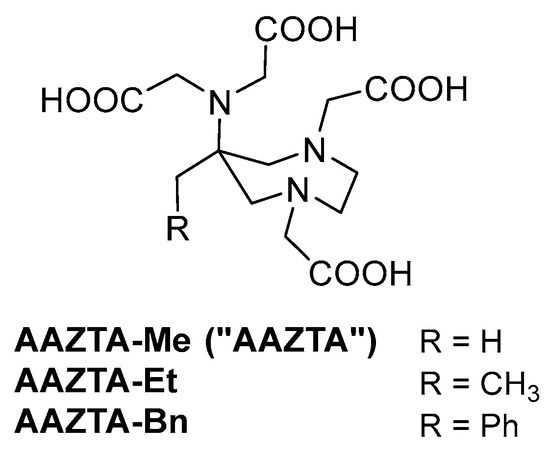
Figure 2.
AAZTA and structural analogs.
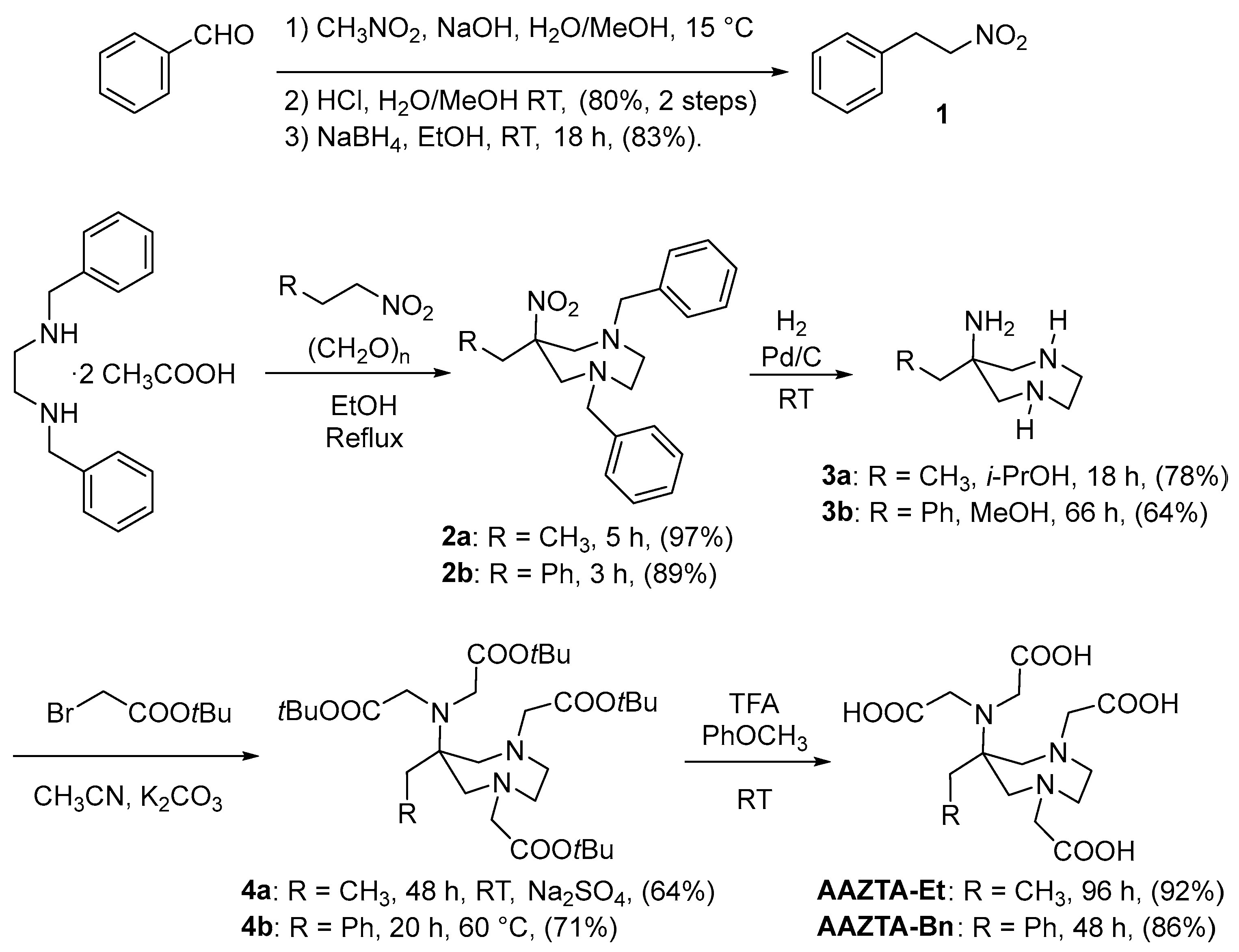
The preparation of AAZTA-Et and AAZTA-Bn is directly mutated by the original synthetic access to AAZTA-Me, with an initial double nitro-Mannich reaction of the suitable primary nitroalkane with N,N′-dibenzylethylenediamine (benzathine) and formaldehyde, leading to the one-pot formation of the mesocyclic diazepane ring. The primary nitroalkane is represented by 1-nitropropane for the synthesis of AAZTA-Et and 2-nitroethylbenzene for AAZTA-Bn.
AAZTA-Et is prepared through a four-step synthesis, starting from benzathine diacetate, refluxed with nitropropane and paraformaldehyde in ethanol for 5 h, resulting in the nitrodiamine 2a in 97% yield. Compound 2a is hydrogenated (H2 10 bar, Pd/C 10%) at room temperature for 18 h in isopropanol providing compound 3a in 78% yield. The latter is alkylated with tert-butyl bromoacetate in acetonitrile at room temperature for 48 h in the presence of potassium carbonate and sodium sulfate resulting in compound 4a in 64% yield. The addition of sodium sulfate proved beneficial in this reaction to avoid the formation of byproducts. Finally, 4a is treated with TFA and anisole at room temperature for 96 h resulting in a 92% yield of the ligand AAZTA-Et (Scheme 1) and completing its preparation in an overall 45% yield from 1-nitropropane (compared with AAZTA-Me, 48% overall yield from nitroethane [11]).
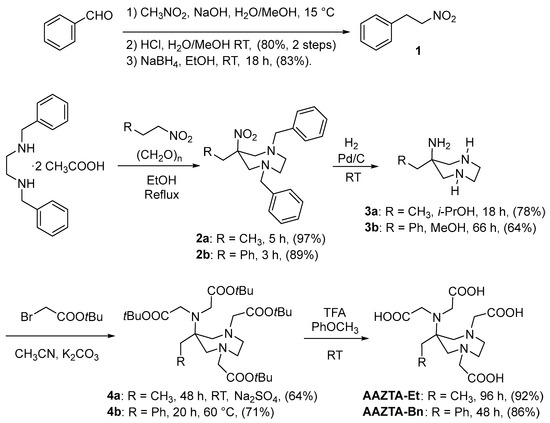
Scheme 1.
Preparation of AAZTA structural analogs.
While 1-nitropropane is commercialized in bulk, 2-nitroethylbenzene 1 is currently available only in limited quantities and must be prepared by base-promoted Henry condensation of nitromethane with benzaldehyde followed by acid-catalyzed dehydration to nitrostyrene, later selectively reduced to 2-nitroethylbenzene 1 (Scheme 1). To this purpose, benzaldehyde was reacted with nitromethane and NaOH in water/methanol. The heterogeneous reaction mixture was then treated with a dilute aqueous HCl solution leading to the precipitation of β-nitrostyrene, recovered in 80% yield. The latter is selectively reduced with sodium borohydride in ethanol at room temperature, obtaining compound 1 in 83% yield. 2-Nitroethylbenzene 1 is reacted with benzathine diacetate and paraformaldehyde in ethanol at reflux temperature for 3 h to give the mesocyclic nitrodiamine 2b in 89% yield. Compound 2b is hydrogenated (H2 10 bar, Pd/C 10%) at room temperature for 66 h in methanol with a yield of 64%. The triamine 3b is exhaustively alkylated with tert-butyl bromoacetate in acetonitrile at 60 °C for 20 h in the presence of potassium carbonate, resulting in compound 4b in 71% yield. The longer reaction time and heating required for the alkylation step may be ascribed to an increased steric hindrance in the triamine 3b compared to the congener 3a. Finally, 4b is deprotected by treatment with TFA and anisole at room temperature for 48 h (86% yield), resulting in the desired ligand AAZTA-Bn in an overall 23% yield from benzaldehyde.
2.2. Acid–Base Properties of AAZTA-Et and AAZTA-Bn Ligands
The protonation constants of H4AAZTA-Et and H4AAZTA-Bn, defined by Equation (1), where i = 1, 2, …, 7, were determined by pH potentiometry and the logK1H values are listed in Table 1 (standard deviations are shown in parentheses).

Table 1.
Protonation constants of the AAZTA-Et, AAZTA-Bn, AAZTA-C2-COOH, AAZTA-C4-COOH, and AAZTA-Me ligands (25 °C).
The stepwise protonation of the parent AAZTA-Me ligand was already fully characterized by both spectroscopic and potentiometric methods [12]. Based on the results of 1H NMR measurements, the first protonation takes place at the endo- and exocyclic nitrogen atoms of the ligand (partial protonation of all nitrogen atoms). The second protonation occurs on the endocyclic nitrogen, whereas the first proton is shifted to the exocyclic nitrogen due to the electrostatic repulsion and the better charge separation. Further protonation processes belong to the ring–carboxylate groups attached to the non-protonated endocyclic nitrogen and/or the carboxylate pendant arms, respectively. Based on the similarities, we can safely assume that the protonations of the AAZTA-Et, AAZTA-Bn, and AAZTA-Me ligands take place in an analog manner. A comparison of the protonation constants determined in the 0.15 M NaCl solution (Table 1) indicates that the logK1H values of the AAZTA-Et and AAZTA-Bn ligands are slightly higher than the corresponding protonation constants of the parent AAZTA-Me and similar to those of the AAZTA-C2-COOH and AAZTA-C4-COOH ligands. The second, third, and fourth protonation constants of AAZTA-Et and AAZTA-Bn are very similar to the corresponding logKiH values of AAZTA-Me, AAZTA-C2-COOH, and AAZTA-C4-COOH. Surprisingly, the pH potentiometric studies of AAZTA-Bn allow the determination of four protonation constants only, which might be explained by the very low basicity of the carboxylate group. It should be highlighted that the third protonation of AAZTA-C2-COOH and AAZTA-C4-COOH occurs at the carboxylate group of the ω-carboxyalkyl side chains [30].
The somewhat higher logK1H value of AAZTA-Et might be interpreted by the electron-donating properties of the ethyl group, leading to a small increase in the basicity of the exo- and endocyclic nitrogen atoms. Similarly, the electron donation of the ω-carboxyalkyl side chains improves the basicity of the nitrogen donor atoms in the backbone of the AAZTA-C2-COOH and AAZTA-C4-COOH ligands. Obviously, the slightly higher logK1H value of AAZTA-Bn cannot be interpreted by the inductive effect of the benzyl substituent. Several studies highlighted that the logK1H value of AAZTA-Me and its derivative ligands obtained in 0.15 M NaCl solution is significantly lower than the value determined in 0.1 M KCl or 0.1 M Me4NCl solutions [12,18,32,33], due to the competition between the first protonation process and the formation of Na+ complexes. A slightly higher first protonation constant of AAZTA-Bn might be interpreted by the lower stability of the Na+ complex.
2.3. Complexation Properties of AAZTA-Et and AAZTA-Bn Ligands with M2+/3+ Cations
The stability and protonation constants of the Ca2+, Mn2+, Zn2+, Cu2+, and Ln3+ complexes formed with the AAZTA-Et and AAZTA-Bn ligands are defined by Equations (2) and (3):
where i = 1, 2, …, 4. The protonation and stability constants of the Ca2+, Mn2+, Zn2+, Cu2+, and Ln3+ complexes formed with the AAZTA-Et and AAZTA-Bn ligands were calculated from the pH potentiometric titration curves obtained at 1:1 metal/ligand concentration ratios. The best fitting of the pH potentiometric data was obtained by assuming the formation of ML, MHL, and MH2L species in equilibrium. The formation of the deprotonated [M(L)H−1]n− complexes takes place at pH > 10.0, as indicated by an extra base-consuming process in the titration curves (M = Zn2+ and Cu2+, n = 2). These processes can be interpreted by the coordination of an OH− ion with the formation of [M(L)H−1]n− species. The protonation constant of the [M(L)H−1]n− species is defined by Equation (4):
The stability and protonation constants of the Cu2+ complex with AAZTA-Et and AAZTA-Bn were determined by spectrophotometry. The equilibrium reaction (Equation (5)) has been studied in the [H+] range of 0.01–1.0 M (the ionic strength was constant I = [Na+] + [H+] = 0.15 in the samples [H+] ≤ 0.15 M), where the formation of Cu2+, CuH2L, and HxL species was assumed (x = 4 and 5; y = 2 and 3). Some characteristic absorption spectra are shown in Figures S29 and S30.
Cu2+ + HxL ⇌ [Cu(Hx–yL)] + yH+
The stability constants of the Gd3+ and Lu3+ complexes with AAZTA-Et and AAZTA-Bn were determined by the measurement of the proton relaxation rates (r1) to investigate the competition reaction of the AAZTA-Et and AAZTA-Bn ligands with DTPA for the Gd3+ ion (Equation (6)) and by following the integrals of the 1H NMR signal of the -CH2-CH3 (δ = 1.40 ppm) and benzyl-CH2 (δ = 2.72 ppm) groups in the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes formed in the competition reaction of the AAZTA-Et and AAZTA-Bn ligands with DTPA for the Lu3+ ion (Equation (6)) at pH = 3.6 and 3.8 in 0.15 M NaCl solution:
where M3+ = Gd3+ or Lu3+, L = AAZTA-Et or AAZTA-Bn, x = 2 and 3, and y = 2 and 3. The relaxivity values of the Gd3+-AAZTA-Et-DTPA and Gd3+-AAZTA-Bn-DTPA systems are shown in Figures S31 and S32. The relaxivity values (r1) of the Gd3+-AAZTA-Et-DTPA and Gd3+-AAZTA-Bn-DTPA systems at pH = 3.6 and 21 MHz can be expressed by Equation (7):
where r1Gd(DTPA), r1GdL, and r1w are the relaxivity values of the [Gd(DTPA)]2−, [Gd(AAZTA-Et)]−, and [Gd(AAZTA-Bn)]− complexes and the aqueous solution in the absence of paramagnetic species (r1Gd(DTPA) = 4.8 mM−1s−1, r1Gd(AAZTA−Et)=7.4 mM−1s−1, r1Gd(AAZTA−Bn) = 9.2 mM−1s−1, and r1w = 0.38 s−1, pH = 3.6, 21 MHz, 298K, 0.15 M NaCl). Considering the protonation constants of the DTPA (DTPA: logK1H = 9.93, logK2H = 8.37, logK3H = 4.18, logK4H = 2.71, logK5H = 2.00, 0.15 M NaCl, 25 °C), AAZTA-Et, and AAZTA-Bn ligands (Table 1), the relaxivity values of the [Gd(DTPA)]2−, [Gd(AAZTA-Et)]−, and [Gd(AAZTA-Bn)]− complexes and the stability constant of the [Gd(DTPA)]2− ([Gd(DTPA)]2−: logKGdL = 22.03, 0.15 M NaCl, 25 °C) [34], the stability constant (logKGdL) of the [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− complexes could be calculated by fitting the relaxivity values in Figures S31 and S32 to Equation (7). The logKGdL values of the [Gd(AAZTA-Et)]− complex obtained from the 1H NMR relaxometry studies agree well with the stability constants determined by pH potentiometry.
[M(L)]− + HxDTPA ⇌ [M(DTPA)]2− + HyL
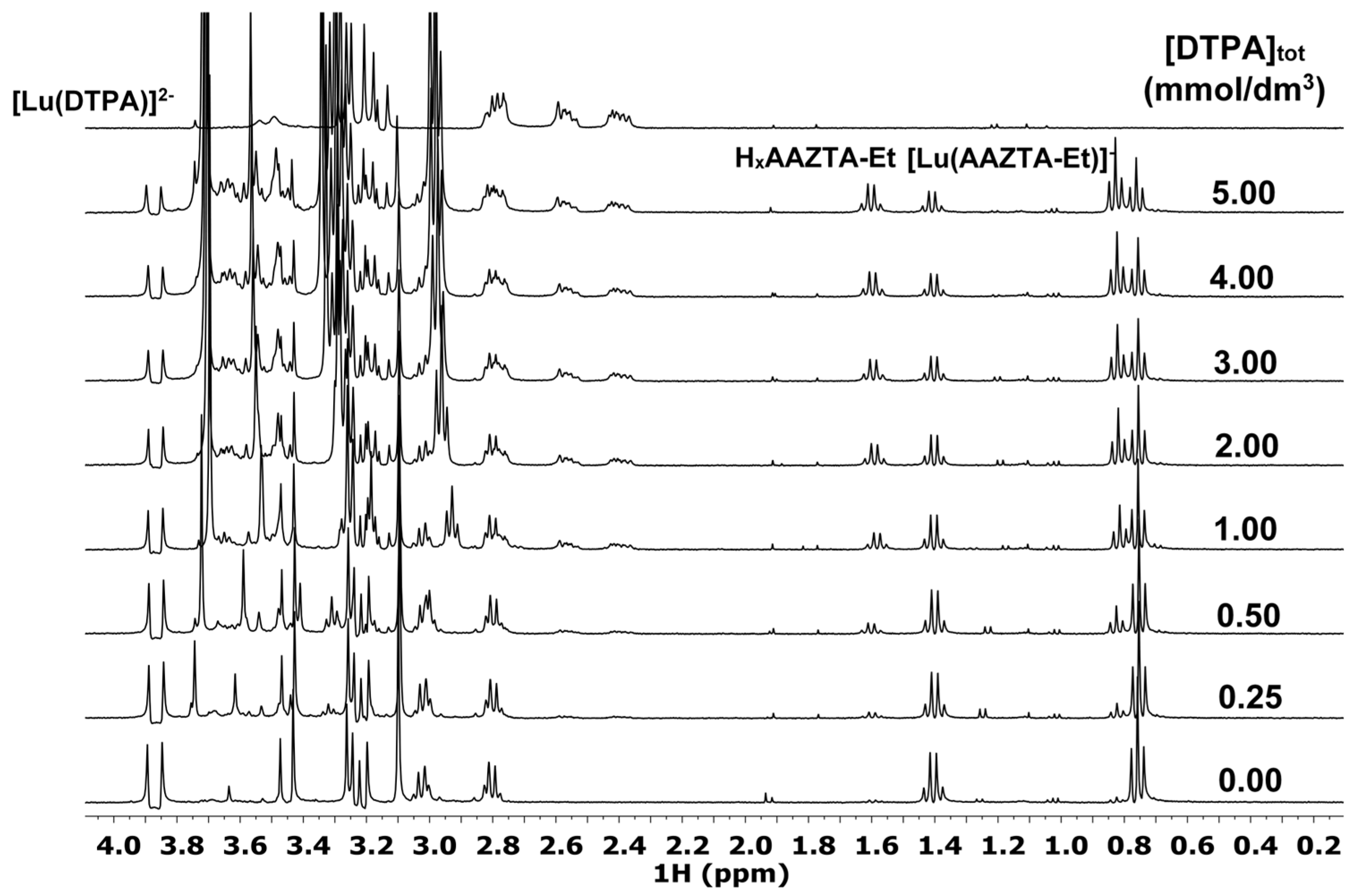
The 1H NMR spectra, the species distribution diagram, and the integral values of the 1H NMR signals of -CH2-CH3 and benzyl-CH2 groups in the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes of the Lu3+-AAZTA-Et-DTPA and Lu3+-AAZTA-Bn-DTPA systems are shown in Figure 3, Figure 4 and Figures S33 and S34, respectively.
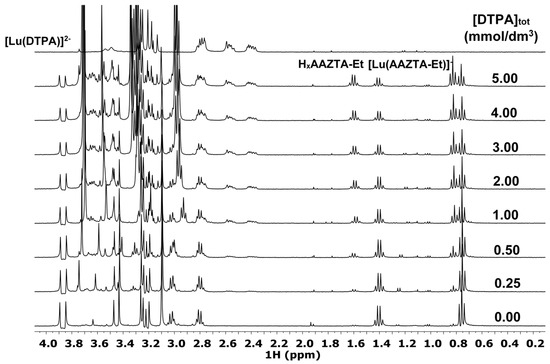
Figure 3.
1H NMR spectra of the Lu3+-AAZTA-Et-DTPA systems at 400 MHz and pH = 3.8 ([Lu3+] = [AAZTA-Et] = 1.0 mM, 25 °C, 0.15 M NaCl).
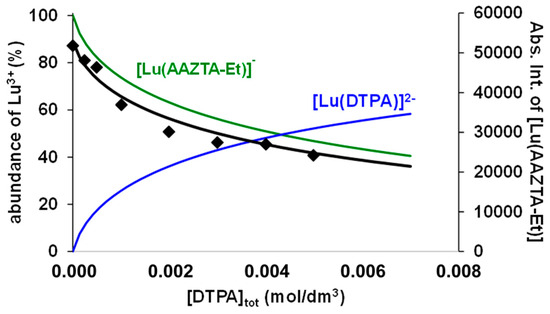
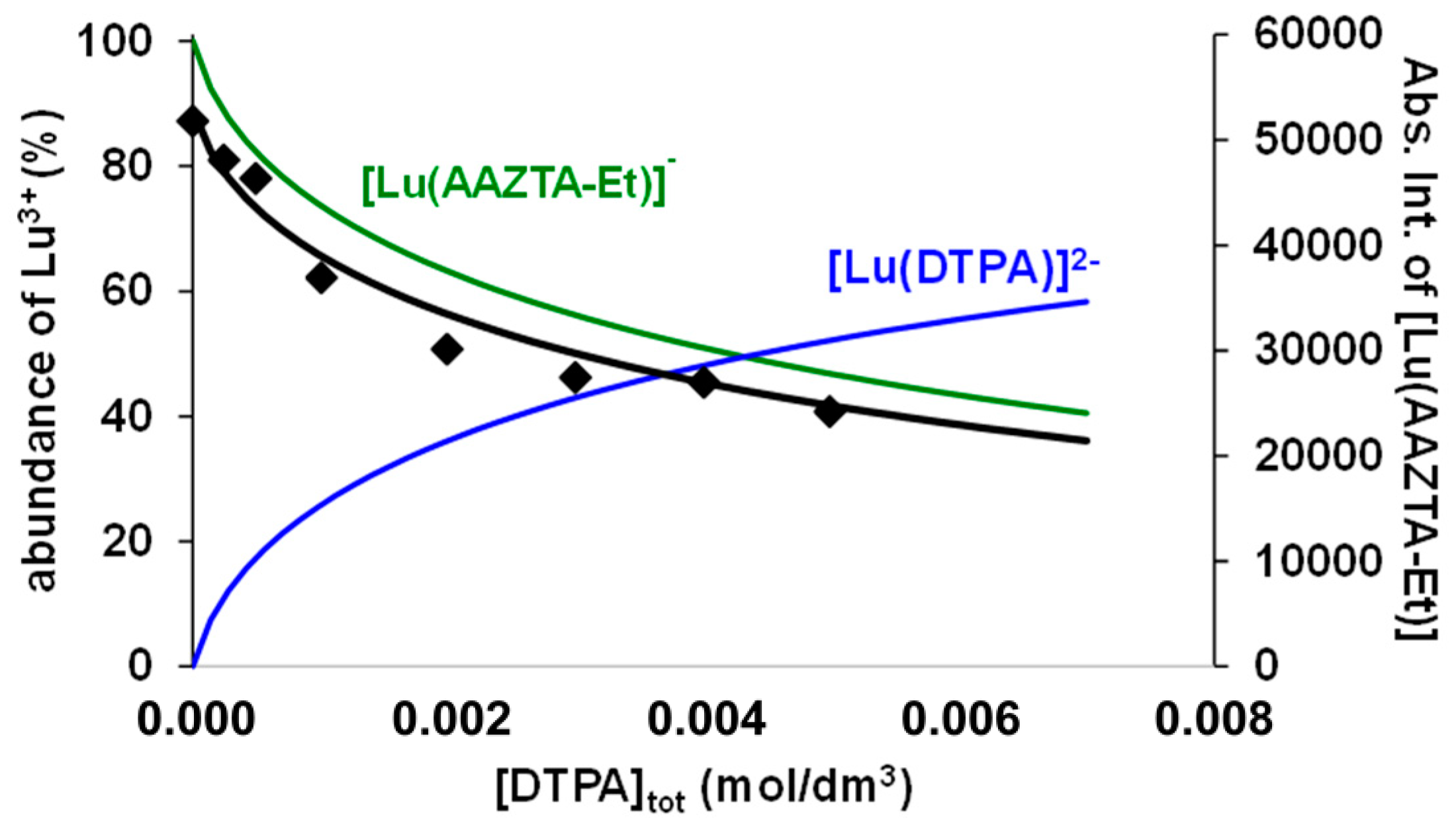
Figure 4.
Abs. Int. values of the [Lu(AAZTA-Et)]− in the Lu3+-AAZTA-Et-DTPA systems at 400 MHz and pH = 3.8 ([Lu3+] = [AAZTA-Et] = 1.0 mM, 25 °C, 0.15 M NaCl).
Considering the protonation constants of the DTPA (DTPA: logK1H = 9.93, logK2H = 8.37, logK3H = 4.18, logK4H = 2.71, and logK5H = 2.00, 0.15 M NaCl, 25 °C), AAZTA-Et, and AAZTA-Bn ligands (Table 1), the molar integral values of the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes ([Lu(AAZTA-Et)]−: Mol. Int. = (5.5 ± 0.1) × 107 dm3/mol; [Lu(AAZTA-Bn)]−: Mol. Int. = (4.9 ± 0.1) × 107 dm3/mol; pH = 3.8, 0.1 M NaCl, 25 °C) and the stability constant of the [Lu(DTPA)]2− obtained by pH potentiometric studies ([Lu(DTPA)]2−: logKLuL = 22.19 (1), 0.15 M NaCl, 25 °C), the stability constant (logKLuL) of the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes could be calculated from the integral values of the 1H NMR signals of -CH2-CH3 and benzyl-CH2 groups in the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes in Figure 4 and Figure S34.
The stability and protonation constants of the Ca2+, Mn2+, Zn2+, Cu2+, and Ln3+ complexes formed with the AAZTA-Et and AAZTA-Bn ligands obtained by using pH potentiometric titration, spectrophotometry, 1H NMR relaxometry, and 1H NMR spectroscopy techniques are presented and compared with those of the corresponding AAZTA-C2-COOH, AAZTA-C4-COOH, and AAZTA-Me complexes in Table 2.
Table 2.
Stability and protonation constants of the AAZTA-Et, AAZTA-Bn, AAZTA-C2-COOH, AAZTA-C4-COOH, and AAZTA-Me complexes formed with Ca2+, Mn2+, Zn2+, Cu2+, and Ln3+ ions (25 °C).
The comparison of the stability constants summarized in Table 2 indicates that the Ca2+, Mn2+, Zn2+, and Ln3+ complexes with the AAZTA-Et and AAZTA-Bn ligands are comparable and very similar to those of the corresponding complexes of AAZTA-C2-COOH and AAZTA-C4-COOH. Surprisingly, the stability constants for the Ca2+, Mn2+, Zn2+, and Ln3+ complexes of AAZTA-Et and AAZTA-Bn are slightly higher than those of the parent AAZTA-Me complexes. In order to compare the affinity of the different ligands to the Gd3+ ion, the pGd values were calculated at pH = 7.4, 25 °C, [Gd3+]tot = 1.0 × 10−6 M, and [L]tot = 1.0 × 10−6 M (Table 2). Based on the pGd values, the conditional stability of [Gd(AAZTA-Bn)]− is comparable with that of the parent [Gd(AAZTA-Me)]− and slightly higher than that of [Gd(AAZTA-Et)]− due to the higher total basicity of the AAZTA-Et ligand (ΣlogKiH, Table 1). Among the AAZTA derivatives, [Gd(AAZTA-C2-COO−)]− and [Gd(AAZTA-C2-COO−)]− are characterized by the highest conditional stability.
Interestingly, the stability constants for the [Cu(AAZTA-Bn)]2− are lower by 1.2 and 2.8 logK units than that of the [Cu(AAZTA-Et)]2− and the parent [Cu(AAZTA-Me)]2−, respectively.
Metal complexes of AAZTA-Et and AAZTA-Bn can be protonated at low pH values. The logKMHL values of the Ca2+, Mn2+, Zn2+, Cu2+, and Ln3+ complexes with AAZTA-Et and AAZTA-Bn are very similar to those of the corresponding AAZTA-Me complexes (Table 2). According to the logKMHL values of these complexes with AAZTA-Et and AAZTA-Bn, the protonation takes place on the weakly coordinated carboxylate oxygen donor atom [30]. The first protonation of the Ca2+, Zn2+, Cu2+, and Ln3+ complexes with the AAZTA-C2-COOH and AAZTA-C4-COOH ligands takes place on the ω-carboxyalkyl side chains of the ligands. At lower pH values, a second protonation process could be identified for the Zn2+ and Cu2+ complexes of AAZTA-Et and AAZTA-Bn, which might be explained by the presence of another weakly coordinated donor atom (carboxylate oxygen) protonated in the pH range of 2 to 4.
2.4. Kinetic Inertness of [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− Complexes
In order to investigate the effect of ethyl and benzyl groups on the kinetic inertness of the Gd3+ complexes, the rates of the transmetallation reactions between the [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− complexes and Cu2+ were studied by spectrophotometry in the presence of excess Cu2+. The mechanisms proposed for the transmetallation reactions of the [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− complexes with Cu2+ are shown in Scheme 2. Experimental details and equations used to evaluate the kinetic parameters are summarized in ESI. The rate and equilibrium constants that characterize the transmetallation reaction of [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− are shown and compared with those of [Gd(AAZTA-Me)]−, [Gd(AAZTA-C2-COO−)], and [Gd(AAZTA-C4-COO−)] in Table 3.
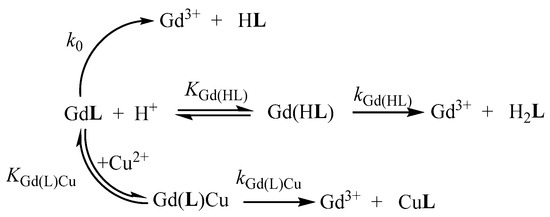
Scheme 2.
Proposed mechanism of the transmetallation reactions for [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]−.
Table 3.
Rate (ki) and equilibrium (Ki) constants and half-life (t1/2 = ln2/kd) values characterizing the transmetallation reactions of the Gd3+ complexes with AAZTA-Et, AAZTA-Bn, AAZTA-C2-COOH, AAZTA-C4-COOH, and AAZTA-Me (25 °C).
The value of k0 rate constants arising from the fitting procedure is very low and its error is very high, indicating that the spontaneous dissociation has no relevant contribution to the overall dissociation rate of [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− in our experimental condition (pH = 2.8–4.5). The comparison of the k1 rate constants reveals that the rate of the proton-assisted dissociation of [Gd(AAZTA-Et)]− and [Gd(AAZTA-Me)]− are comparable and about three times faster than that of [Gd(AAZTA-Bn)]−. On the other hand, the rate constant k3Cu, characterizing the Cu2+-assisted dissociation of [Gd(AAZTA-Bn)]−, cannot be evaluated from the kinetic data due to its relatively small contribution to the dissociation rate of [Gd(AAZTA-Bn)]−. Based on this evidence, it might be assumed that the heterodinuclear [Gd(AAZTA-Bn)Cu]+ intermediate has no substantial role in the dissociation of the [Gd(AAZTA-Bn)]− in our experimental conditions ([GdL] = 1.0 mM, [Cu2+] = 10–40 mM, pH = 2.8–4.5). However, the rate of the Cu2+-assisted dissociation of [Gd(AAZTA-Et)]− is similar to that of the parent [Gd(AAZTA-Me)]−. Interestingly, the stability constants of the heterodinuclear [Gd(AAZTA-Et)Cu]+and [Gd(AAZTA-Bn)Cu]+ intermediates (KGd(L)Cu, Table 3) are comparable and somewhat higher than that of [Gd(AAZTA-Me)Cu]+.
The kinetic studies reveal that the most important pathway of the transmetallation reactions is the proton-assisted dissociation of the [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− complexes. The proton-assisted dissociation of [Gd(AAZTA-Me)]− and its derivates takes place by the formation of protonated Gd(HL) intermediate. The dissociation of the Gd(HL) intermediate presumably takes place by the proton transfer from the carboxylate group to the nitrogen atom, resulting in the release of the Gd3+ ion from the coordination cage. According to the kinetic data, it can be assumed that this proton transfer in [Gd(HAAZTA-Bn)] is less probable due to a stronger interaction between the Gd3+ ion and the donor atoms of the AAZTA-Bn ligand. Moreover, the higher affinity of the Gd3+ ion to AAZTA-Bn (pGd value in Table 2) may determine the lower transfer rate of the functional groups from the Gd3+ to the attacking Cu2+ ion in the heterodinuclear [Gd(AAZTA-Bn)Cu]+ intermediate. This hypothesis is further supported by the lack of the Cu2+-assisted dissociation of the Gd(AAZTA-C4-COOH) complex due to the stronger coordination of the Gd3+ ion with the more basic nitrogen atoms of the ligand. The higher kinetic inertness of [Gd(AAZTA-Bn]− is also confirmed by the calculated dissociation rate (kd) and half-life (t1/2) values at pH = 7.4 (Table 3). The comparison of the kd and t1/2 values also reveals that the dissociation rate of [Gd(AAZTA-Bn)]− and [Gd(AAZTA-C4-COO−)]− are comparable and about 2–2.5 times lower than that of [Gd(AAZTA-Et)]− and the parent [Gd(AAZTA-Me)]−.
3. Materials and Methods
3.1. Synthesis—General Information
Reagents and solvents were obtained from Merck KGaA (Darmstadt, Germany) or TCI Europe N.V. (Zwijndrecht, Belgium) and used without further purification. All aqueous solutions were prepared from ultrapure laboratory-grade water (18 MΩ·cm) obtained from the Millipore/MilliQ purification system (Merck KGaA, Darmstadt, Germany). 1H and 13C NMR spectra were recorded at 400 MHz on a Bruker Avance Neo 400 spectrometer (Bruker Italia srl, Milan, Italy). Chemical shifts are reported in ppm with the protic impurities of the deuterated solvent as the internal reference. Mass spectra were obtained with a Thermo Finnigan LCQ-Deca XP-PLUS ion trap spectrometer equipped with an electrospray source (Waltham, MA, USA). High-resolution mass spectra were registered on a ThermoScientific Q-Exactive Plus spectrometer (Waltham, MA, USA). TLC was performed with silica gel (MN Kieselgel 60F254, Carlo Erba Reagents, Cornaredo (MI), Italy)) and visualized by UV or sprayed with Dragendorff reagent or alkaline KMnO4. Column chromatography was carried out on Macherey–Nagel Silica gel 60 (0.063–0.200 mm) (Carlo Erba Reagents, Cornaredo (MI), Italy).
3.1.1. 1,4-Dibenzyl-6-ethyl-6-nitro-1,4-diazepane (2a)
Benzathine diacetate (10.2 g, 28.4 mmol) was dissolved in EtOH (70 mL) and 1-nitropropane (2.7 mL, 29.8 mmol), and paraformaldehyde (3.41, 114 mmol) was added. Then, the suspension was refluxed for 5 h, during which it became homogeneous. The reaction mixture was evaporated under a vacuum, the residue was taken up with petroleum ether and shaken with a sat. aq. Na2CO3 solution, and then the organic phase was separated. The aqueous layer was extracted two more times with petroleum ether, and then pooled organic phases were dried over Na2CO3 and Na2SO4, filtered, and evaporated under vacuum. The crude product was purified by adsorption on silica gel (61.32 g), followed by sequential washings with 2 × 200 mL of petroleum ether, 2 × 200 mL of petroleum ether/ethyl acetate 95:5, and 3 × 200 mL of petroleum ether/ethyl acetate 9:1 (1 volume = 200 mL), and each digestion lasted 15 min. The eluted washings were checked by TLC, pooled, and evaporated under vacuum to give pure 2a (9.71 g, 97%) as a thick yellow oil. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 7.37–7.26 (m, 10H), 3.78 (d, J = 13.1 Hz, 2H), 3.64 (d, J = 13.1 Hz, 2H), 3.58 (d, J = 14.2 Hz, 2H), 3.02 (d, J = 14.2 Hz, 2H), 2.68–2.56 (m, 4H), 1.72 (q, J = 7.4 Hz, 2H), 0.55 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 139.3 (C), 129.1 (CH), 128.4 (CH), 127.3 (CH), 95.6 (C), 64.1 (CH2), 61.7 (CH2), 58.6 (CH2), 30.1 (CH2), 7.4 (CH3) ppm. HRMS (ESI+): m/z = 354.21727 (100%, [M + H]+). Calc. for C21H27O2N3 + H+: 354.21760.
3.1.2. 6-Ethyl-1,4-diazepan-6-amine (3a)
Compound 2a (9.42 g, 26.7 mmol) was dissolved in i-PrOH (100 mL) and then Pd/C (10% wetted with 50% water, 2.10 g) was added. The suspension was stirred at RT for 18 h under a hydrogen atmosphere (10 bar). The mixture was vacuum-filtered through Celite®, evaporated under vacuum, and then separated by column chromatography (CH2Cl2/CH3OH/aq. NH3 7:3:0.5) to give 3a (2.98 g, 78%) as a yellow oil. 1H NMR (400.2 MHz, D2O, 298 K) δ 3.44–3.36 (m, 2H), 3.31–3.23 (m, 4H), 3.17 (d, J = 14.2 Hz, 2H), 1.82 (q, J = 7.6 Hz, 2H), 1.18 (t, J = 7.6 Hz, 3H) ppm. 13C NMR (100.6 MHz, D2O, 298 K) δ 57.0 (C), 55.4 (CH2), 48.3 (CH2), 30.0 (CH2), 6.4 (CH3) ppm. HRMS (ESI+): m/z = 144.14950 (100%, [M + H]+). Calc. for C7H17N3 + H+: 144.14952.
3.1.3. Di-tert-butyl 2,2′-((1,4-Bis(2-(tert-butoxy)-2-oxoethyl)-6-ethyl-1,4-diazepan-6-yl)azanediyl)diacetate (4a)
Compound 3a (1.50 g, 10.5 mmol) was dissolved in acetonitrile (50 mL), and potassium carbonate (8.68 g, 62.8 mmol) and sodium sulfate (8.92 g, 62.8 mmol) were added. tert-Butyl bromoacetate (7.7 mL, 52.4 mmol) was added dropwise for 1 h at 0 °C, and then the reaction was stirred at RT for 48 h. The mixture was filtered to remove the inorganic salts and evaporated under vacuum. The residue was taken up with ether and washed with deionized water. The aqueous phase was extracted twice with ether and the pooled organic phases were dried over Na2CO3 and Na2SO4, filtered, and then the solvent was removed under vacuum. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 95:5–9:1) to yield compound 4a (4.00 g, 64%) as a yellow oil. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 3.62 (s, 4H), 3.22 (br s, 4H), 2.97 (d, J = 14.1 Hz, 2H), 2.78–2.72 (m, 2H), 2.68–2.61 (m, 4H), 1.59 (q, J = 7.3 Hz, 2H), 1.43 (s, 18H), 1.42 (s, 18H), 0.85 (t, J = 7.3 Hz, 3H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 173.0 (C), 170.9 (C), 80.8 (C), 80.3 (C), 65.2 (CH2), 63.1 (C), 62.6 (CH2), 59.4 (CH2), 52.0 (CH2), 29.9 (CH2), 28.3 (CH3), 28.2 (CH3), 7.0 (CH3) ppm. HRMS (ESI+): m/z = 600.42134 (100%, [M + H]+). Calc. for C31H57O8N3 + H+: 600.42184.
3.1.4. 2,2′-((1,4-Bis(carboxymethyl)-6-ethyl-1,4-diazepan-6-yl)azanediyl)diacetic Acid (AAZTA-Et)
Compound 4a (2.20 g, 3.67 mmol) was mixed with anisole (2.4 mL, 22.0 mmol) and TFA (25 mL). The mixture was left at RT for 4 d, and then evaporated under vacuum. The residue was stirred with diethyl ether, centrifugated, and then the supernatant was discarded. The procedure was repeated three times with ether and then three times with acetone to give pure AAZTA-Et (1.27 g, 92%) as an amorphous solid. 1H NMR (400.2 MHz, D2O, 298 K) δ 3.92–3.69 (m, 10H), 3.60–3.52 (m, 6H), 1.64 (q, J = 7.5 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (100.6 MHz, D2O, 298 K) δ 175.1 (C), 170.6 (C), 64.2 (C), 58.9 (CH2), 58.5 (CH2), 53.6 (CH2), 51.9 (CH2), 26.3 (CH2), 7.2 (CH3) ppm. HRMS (ESI+): m/z = 376.17107 (100%, [M + H]+). Calc. for C15H25O8N3 + H+: 376.17144.
3.1.5. 2-Nitroethylbenzene (1) [29]
Benzaldehyde (50.3 mL, 493 mmol) and nitromethane (26.4 mL, 493 mmol) were dissolved in methanol (100 mL) in a two-necked flask equipped with a thermometer and a pressure-equalizing dropping funnel. A solution of NaOH (25.6 g, 641 mmol) in water (50 mL) was added dropwise to the solution, keeping the temperature below 15° C. A fine slurry formed during the addition and methanol was added to allow stirring. After the addition, the slurry was diluted with ice/water, and then the resulting clear solution was added dropwise to a solution of conc. HCl (60 mL) in H2O (200 mL); a yellow precipitate was formed during the addition. The precipitate was filtered on a Buchner funnel, washed with water, and recrystallized from ethanol to yield the intermediate β-nitrostyrene (73.5 g, 80%) as yellow needles. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 8.00 (d, J = 13.7 Hz, 1H), 7.59 (d, J = 13.7 Hz, 1H), 7.56–7.43 (m, 5H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 139.2 (CH), 137.2 (CH), 132.3 (CH), 130.2 (C), 129.5 (CH), 129.3 (CH) ppm.
In a 500 mL two-necked flask equipped with a thermometer and a pressure-equalizing dropping funnel, NaBH4 (5.33 g, 0.140 mol) was suspended in 100 mL of THF and 30 mL of ethanol. A solution of β-nitrostyrene (10.0 g, 67.0 mmol) in 100 mL of THF was added dropwise to the solution over 45 min, keeping the temperature below 30° C. A fine slurry was formed during the addition. After the addition, the slurry was stirred for 18 h. The mixture was then concentrated under vacuum and the residue was dissolved in water and extracted twice with dichloromethane. The organic phase was washed with water (2 × 30 mL), brine (1 × 30 mL), dried (Na2SO4), filtered, and then evaporated under vacuum, obtaining 2-nitroethylbenzene 1 (8.40 g, 83%) as a yellow oil. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 7.46–7.18 (m, 5H), 4.64 (t, J = 7.4 Hz, 2H), 3.35 (t, J = 7.4 Hz, 2H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 135.8 (C), 129.1 (CH), 128.7 (CH), 127.5 (CH), 76.4 (CH2), 33.5 (CH2) ppm.
3.1.6. 1,4-Dibenzyl-6-benzyl-6-nitro-1,4-diazepane (2b) [29]
Benzathine diacetate (59.6 g, 165 mmol) and 2-nitroethylbenzene (1, 25.0 g, 165 mmol) were dissolved in ethanol (200 mL). Paraformaldehyde (17.4 g, 579 mmol) was added portion-wise to the solution and the resulting suspension was heated to reflux. The mixture became progressively homogeneous and evaporated after 3 h under vacuum. The residue was recrystallized from methanol to give (2b) (61.3 g, 89%) as a yellow solid. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 7.38–7.22 (m, 13H), 6.86–6.83 (m, 2H), 3.78 (d, J = 13.3 Hz, 2H), 3.67 (d, J = 13.2 Hz, 2H), 3.50 (d, J = 14.2 Hz, 2H), 3.14 (d, J = 14.3 Hz, 2H), 3.03 (s, 2H), 2.60 (br s, 4H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 139.3 (C), 134.3 (C), 129.8 (CH), 129.1 (CH), 128.6 (CH), 128.5 (CH), 127.6 (CH), 127.4 (CH), 95.6 (C), 64.1 (CH2), 62.4 (CH2), 58.5 (CH2), 43.1 (CH2) ppm. HRMS (ESI+): m/z = 416.23271 (100%, [M + H]+). Calc. for C26H29O2N3 + H+: 416.23325.
3.1.7. 6-Benzyl-1,4-diazepan-6-ylamine (3b) [29]
The nitrodiamine 2b (4.10 g, 9.87 mmol) was dissolved in methanol (40 mL) and then Pd/C (10% wetted with 50% water, 2.05 g) was added. The suspension was stirred at RT for 66 h under a hydrogen atmosphere (10 bar) and periodically checked by TLC (ethyl acetate or CH2Cl2/CH3OH/aq. NH3 7:3:0.5). The mixture was filtered through Celite® and evaporated under vacuum. The residue was redissolved in methanol (10 mL) and 3 mL of 37% aqueous HCl was added in 5 min at 0 °C. The mixture was refluxed for 5 min to obtain a clear solution and kept overnight at 4 °C. Colorless-to-white crystals of 3b hydrochloride form were isolated by suction filtration, washed with cold water, ethanol, and ether, and then dried under vacuum. The mother liquor was reduced to half volume under vacuum and left overnight at 4 °C, resulting in an additional crop of 3b hydrochloride. The crops were pooled and dissolved in 30% aq. NaOH and extracted with CH2Cl2. The aqueous layer was extracted three times with CH2Cl2. The pooled organic extracts were dried over Na2CO3 and Na2SO4, filtered, and then the solvent was removed under vacuum to give the triamine-free base 3b (1.30 g, 64%) as a yellow oil. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 7.32–7.20 (m, 5H), 2.98–2.81 (m, 6H), 2.68 (s, 2H), 2.61 (d, J = 13.6 Hz, 2H), 1.86 (s, 4H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 137.5 (C), 130.7 (CH), 128.3 (CH), 126.5 (CH), 61.0 (CH2), 56.8 (C), 52.3 (CH2), 45.8 (CH2) ppm. HRMS (ESI+): m/z = 206.16500 (100%, [M + H]+). Calc. for C12H19N3 + H+: 206.16517.
3.1.8. Di-tert-butyl 2,2′-((6-Benzyl-1,4-bis(2-(tert-butoxy)-2-oxoethyl)-1,4-diazepan-6-yl)azanediyl)diacetate (4b) [29]
Compound 3b (1.01 g, 4.92 mmol) was dissolved in acetonitrile (10 mL) and potassium carbonate (4.08 g, 29.5 mmol) was added, and then the solution was cooled to 0–5 °C. tert-Butyl bromoacetate (3.6 mL, 24.6 mmol) was added dropwise for 15 min, and then the reaction mixture was heated at 60 °C for 20 h. Then, the mixture was filtered to remove the inorganic salts and evaporated under vacuum. Dichloromethane and an aqueous saturated solution of sodium carbonate were added to the residue. The organic phase was separated, and then the aqueous phase was extracted three times with CH2Cl2. The pooled organic phases were dried over Na2CO3 and Na2SO4, filtered, and then the solvent was removed under vacuum. The crude product was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 9:1–8:2) to give the ester 4b (2.30 g, 71%) as a yellow oil. 1H NMR (400.2 MHz, CDCl3, 298 K) δ 7.55 (d, J = 6.9 Hz, 2H), 7.23 (t, J = 7.6 Hz, 2H), 7.15 (tt, J = 7.3/1.7 Hz, 1H), 3.80 (s, 4H), 3.23 (d, J = 16.7 Hz, 2H), 3.18 (d, J = 16.6 Hz, 2H), 3.00 (d, J = 14.2 Hz, 2H), 2.95 (s, 2H), 2.81–2.62 (m, 6H), 1.44 (s, 18H), 1.39 (s, 18H) ppm. 13C NMR (100.6 MHz, CDCl3, 298 K) δ 172.6 (C), 170.9 (C), 138.2 (C), 130.7 (CH), 128.0 (CH), 125.9 (CH), 80.9 (C), 80.5 (C), 65.1 (CH2), 64.3 (CH2), 62.8 (CH2), 59.4 (CH2), 52.3 (CH2), 41.1 (CH2), 28.3 (CH3) ppm. HRMS (ESI+): m/z = 662.43660 (100%, [M + H]+). Calc. for C36H59O8N3 + H+: 662.43749.
3.1.9. [6-Bis(carboxymethylamino)-4-carboxymethyl-6-benzyl-1,4-diazepan-1-yl]acetic Acid (AAZTA-Bn) [29]
Compound 9 (2.20 g, 3.32 mmol) was mixed with anisole (3 mL) and TFA (30 mL). The reaction was heated to reflux for 5 min, then left at RT for 48 h. The mixture was evaporated under vacuum and the residue was stirred with diethyl ether, centrifugated, and the supernatant discarded. This procedure was repeated three times. The product was dried under vacuum and recrystallized from water. After 18h at 4 °C, the crystalline product was recovered by vacuum filtration and sequentially washed with cold water, isopropanol, and diethyl ether. An additional crop was obtained by concentration of the mother liquor, and overnight standing at 4 °C. The pooled crops were dried under vacuum to obtain AAZTA-Bn (1.25 g, 86%, 2 crops) as white crystals. 1H NMR (400.2 MHz, D2O, 298 K) δ 7.43–7.35 (m, 3H), 7.26 (dd, J = 7.9/1.6 Hz, 2H), 3.85–3.70 (m, 10H), 3.72 (d, J = 14.8 Hz, 2H), 3.49–3.43 (m, 4H), 2.95 (s, 2H) ppm. 13C NMR (100.6 MHz, D2O, 298 K) δ 176.1 (C), 170.4 (C), 133.9 (C), 130.7 (CH), 129.0 (CH), 127.7 (CH), 63.0 (C), 59.3 (CH2), 59.1 (CH2), 53.8 (CH2), 51.6 (CH2), 39.4 (CH2) ppm. HRMS (ESI+): m/z = 436.17274 (100%, [M + H]+). Calc. for C20H27O8N3 + H+: 436.17254.
3.2. Equilibrium Properties—General Information
3.2.1. Materials
The chemicals used for the experiments were of the highest analytical grade. The concentration of the CaCl2, MnCl2, ZnCl2, CuCl2, and LnCl3 solutions was determined by complexometric titration with standardized Na2H2EDTA and xylenol orange (ZnCl2 and LnCl3), murexide (CuCl2), eriochrome black t (MnCl2), and Patton and Reeder (CaCl2) as indicators. The concentration of the H4AAZTA-Et, H4AAZTA-Bn, and H5DTPA stock solutions was determined by pH potentiometric titration in the presence and absence of a large (40-fold) excess of CaCl2. The pH potentiometric titrations were made with standardized 0.2 M NaOH.
3.2.2. pH Potentiometry
The protonation constants of ligands, the stability, and the protonation constants of the Ca2+, Mn2+, La3+, and Gd3+ complexes were determined by pH potentiometric titrations. The metal-to-ligand concentration ratio was 1:1 (the concentration of the ligand was generally 0.002 M). The protonation constants of the Cu2+ and Gd3+ complexes were determined using pH potentiometry by titrating the pre-prepared complexes from pH = 2.0 to pH = 12.0 with 0.2 M NaOH. For the pH measurements and titrations, a Metrohm 888 Titrando automatic titration workstation with a Metrohm-6.0234.110 combined electrode were used (Metrohm Italiana Srl, Origgio (VA), Italy). Equilibrium measurements were carried out at a constant ionic strength (0.15 M NaCl) in 6 mL samples at 25 °C under magnetic stirring and N2 bubbling. The titrations were made in the pH range of 1.7–12.0. KH-phthalate (pH = 4.005) and borax (pH = 9.177) buffers were used to calibrate the pH meter. For the calculation of [H+] from the measured pH values, the method proposed by Irving et al. was used [36]. A 0.01M HCl solution was titrated with the standardized NaOH solution in the presence of 0.15 M NaCl ionic strength. The differences (A) between the measured (pHread) and calculated pH (−log[H+]) values were used to obtain the equilibrium H+ concentration from the pH values measured in the titration experiments (A = 0.019). For the equilibrium calculations, the stoichiometric water ionic product (pKw) was also needed to calculate [OH−] values under basic conditions. The VNaOH–pHread data pairs of the HCl-NaOH titration obtained in the pH range 10.5–12.0 were used to calculate the pKw value (pKw = 13.787).
3.2.3. Spectrophotometry
The stability constants of Cu2+ complexes formed with AAZTA-Et and AAZTA-Bn were determined by spectrophotometry studying the Cu2+-AAZTA-Et and Cu2+-AAZTA-Bn systems at the absorption band of the Cu2+ complexes at [H+] = 0.01–1.0 M in the wavelength range of 400–800 nm. The concentrations of Cu2+, H4AAZTA-Et, and H4AAZTA-Bn were 0.002 M. The H+ concentration in the samples was adjusted with the addition of calculated amounts of 3 M HCl, and the ionic strength was also maintained constant to 0.15 M by the addition of appropriate amounts of NaCl so that I = [Na+] + [H+] = 0.15 M, where [H+] ≤ 0.15 M. The samples were kept at 25 °C for a week. The absorbance values of the samples were determined at 11 wavelengths (575, 595, 615, 635, 655, 675, 695, 715, 735, 755, and 775 nm). For the calculations of the stability and protonation constants of [Cu(AAZTA-Et)]2− and [Cu(AAZTA-Bn)]2−, the molar absorptivities of CuCl2, [Cu(HL)]−, and [Cu(H2L)] species were determined by recording the spectra of 1.0 × 10−3, 1.5 × 10−3, 2.0 × 10−3, and 2.5 × 10−3 M solutions of CuCl2, [Cu(AAZTA-Et)]2−, and [Cu(AAZTA-Bn)]2− in the pH range 1.7–7.5. The pH was adjusted by stepwise addition of concentrated NaOH or HCl solutions. The spectrophotometric measurements were made on a PerkinElmer Lambda 365 UV-Vis spectrophotometer (Perkin Elmer Italia SpA, Milan, Italy) at 25 °C, using 1.0 cm cells. The protonation and stability constants were calculated with the PSEQUAD program [37].
3.2.4. 1H NMR Relaxometry
The relaxivity values were calculated from the longitudinal relaxation time of H2O protons (T1) measured at 21 MHz on a Stelar relaxometer connected to a Bruker WP80 NMR electromagnet adapted to variable-field measurements (15−80 MHz proton Larmor frequency) (Bruker Italia srl, Milan, Italy). The temperature of the sample holder was controlled with a thermostated air stream.
The stability constant of the Gd3+ complexes with AAZTA-Et and AAZTA-Bn ligands was determined by the measurement of the proton relaxation rates (r1) studying the competition reaction of AAZTA-Et and AAZTA-Bn with DTPA for the Gd3+ ion at pH = 3.6 in 0.15 M NaCl solution. In these experiments, the concentration of Gd3+, AAZTA-Et, and AAZTA-Bn was 1.0 mM, while that of the DTPA was varied between 1.0 and 5.0 mM (8 × 1.5 mL samples). The pH was adjusted to pH = 3.6 by stepwise addition of concentrated NaOH or HCl solutions. The samples were kept at 25 °C for two weeks to attain the equilibrium (the time needed to reach the equilibria was determined by 1H-NMR relaxometry). The longitudinal relaxation time was measured with the “inversion recovery” method (180°–τ–90°) by using 12 different τ values with a typical 90° pulse width of 6.4 μs, 4 scans. The measurements were performed with a 1 mM solution of the Gd3+ complexes, so the relaxivity values were given as r1 = 1/T1p + 1/T1w, where T1p and T1w were the relaxation time of bulk water protons in the presence and absence of paramagnetic species, respectively. The stability constants of the [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− complexes were calculated by considering the protonation constants of the DTPA ligand and the stability constants of the [Gd(DTPA)]− complex (DTPA: logK1H = 9.93, logK2H = 8.37, logK3H = 4.18, logK4H = 2.71, logK5H = 2.00; [Gd(DTPA)]2−: logKGdL = 22.03, 0.15 M NaCl, 25 °C) [34]. The equilibrium constants were calculated with the program PSEQUAD [37].
3.2.5. NMR Experiments
1H NMR measurements were performed using a Bruker Avance III (9.4 T) spectrometer equipped with a Bruker Variable Temperature Unit (BVT), a Bruker Cooling Unit (BCU), and a BB inverse z gradient probe (5 mm) (Bruker Italia srl, Milan, Italy). The stability constants of the Lu3+ complexes with AAZTA-Et and AAZTA-Bn were determined by 1H NMR spectroscopy studying the competition reaction of AAZTA-Et and AAZTA-Bn with DTPA for the Lu3+ ion at pH = 3.8 in 0.15 M NaCl aqueous solution. In these experiments, the concentration of the Lu3+, AAZTA-Et, and AAZTA-Bn ligands was 1.0 mM, while that of DTPA was varied between 1.0 and 5.0 mM (8 × 1.5 mL samples). A capillary with D2O was used for locking. The pH was adjusted to 3.8 by stepwise addition of concentrated NaOH or HCl solutions. The samples were kept at 25 °C for two weeks to attain equilibrium (the time needed to reach equilibrium was determined by 1H-NMR spectroscopy). Since the ligand exchange reactions are slow on the actual NMR timescale, the amount of the Lu3+ complexes formed in the equilibrium was calculated by using the integrals of the 1H NMR signal of the -CH2-CH3 (δ = 1.40 ppm) and benzyl-CH2 (δ = 2.72 ppm) groups in the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes, respectively. The molar integral values of the 1H NMR signals of the -CH2-CH3 and benzyl-CH2 groups in the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes were determined by recording the 1H NMR spectra of 1.0, 1.5, 2.0, and 2.5 mM solutions of the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes ([Lu(AAZTA-Et)]−: Mol. Int. = (5.5 ± 0.1) × 107 dm3/mol; [Lu(AAZTA-Bn)]−: Mol. Int. = (4.9 ± 0.1) × 107 dm3/mol; pH = 3.8, 0.1 M NaCl, 25 °C). The stability constants of the [Lu(AAZTA-Et)]− and [Lu(AAZTA-Bn)]− complexes were calculated by considering the protonation constants of the DTPA ligand and the stability constant of the [Lu(DTPA)]2− complex obtained by pH potentiometric titration of the DTPA ligand and Lu3+-DTPA systems at a 1:1 metal-to-ligand concentration ratio ([Lu(DTPA)]2−: logKLuL = 22.19 (1), logKLu(HL) = 2.10 (1), 0.15 M NaCl, 25 °C). The equilibrium constants were calculated with the program PSEQUAD [37].
3.2.6. Kinetics—Transmetallation
The kinetic inertness of the [Gd(AAZTA-Et)]− and [Gd(AAZTA-Bn)]− complexes was characterized by the rates of the exchange reactions taking place between the Gd3+ complexes (GdL) and Cu2+. The transmetallation reactions with Cu2+ were studied by spectrophotometry by following the formation of the Cu2+ complexes at 320 nm with the use of 1.0 cm cells and a PerkinElmer Lambda 365 UV-Vis spectrophotometer (Perkin Elmer Italia SpA, Milan, Italy). The concentration of the GdL complexes was 0.0010 M, whereas the concentration of the Cu2+ was 10, 20, 30, or 40 times larger to ensure pseudo-first-order conditions. The temperature was maintained at 25 °C and the ionic strength of the solutions was kept constant, 0.15 M by NaCl. The exchange rates were studied in the pH range of about 2.8–4.5. To keep the pH values constant, monochloroacetic acid (pH range 2.8–3.1), N,N′-dimethylpiperazine (pH range 3.1–4.1), and N-methylpiperazine (pH range 4.1–5.2) buffers (0.01 M) were used. The pseudo-first-order rate constants (kd) were calculated by fitting the absorbance data to Equation (8):
where At, A0, and Ap are the absorbance values at time t, at the start of the reaction and at equilibrium, respectively. The calculation of the kinetic parameters was performed by fitting the absorbance–time, and relaxation rate–time data pairs with the Micromath Scientist computer program (version 2.0, Salt Lake City, UT, USA).
4. Conclusions
The unique combination of stability, fast kinetics of formation, and slow kinetics of dissociation of metal–AAZTA chelates make this mesocyclic chelating agent a reference point for the development of clinically relevant metal complexes. The preparation of the archetypal AAZTA ligand (AAZTA-Me) may be hampered by the global market restrictions recently applied to a key starting material (nitroethane), and the future unavailability of AAZTA-Me may be an obstacle to the steadily growing number of applications of this unique PAC. In this work, we reported the preparation of two closely related AAZTA analogs (AAZTA-Et and AAZTA-Bn) by short and efficient syntheses and a comprehensive investigation of their coordination chemistry with respect to a selection of metal ions. The results of this study unambiguously show that AAZTA-Et and AAZTA-Bn have a coordination behavior fully comparable to that of the parent AAZTA-Me, with stability constants of the corresponding complexes almost identical and even slightly higher. The methyl substituent on the diazepane ring of AAZTA-Me can be safely replaced with an ethyl or a benzyl group with no detrimental effects on the coordination chemistry of this important chelating platform.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/inorganics12090235/s1. Figure S1. 1H NMR spectrum of compound 2a. Figure S2. 13C APT NMR spectrum of compound 2a. Figure S3. 1H NMR spectrum of compound 3a. Figure S4. 13C APT NMR spectrum of compound 3a. Figure S5. 1H NMR spectrum of compound 4a. Figure S6. 13C APT NMR spectrum of compound 4a. Figure S7. 1H NMR spectrum of AAZTA-Et. Figure S8. 13C APT NMR spectrum of AAZTA-Et. Figure S9. 1H NMR spectrum of β-nitrostyrene. Figure S10. 13C NMR spectrum of β-nitrostyrene. Figure S11. 1H NMR spectrum of compound 1. Figure S12. 13C APT NMR spectrum of compound 1. Figure S13. 1H NMR spectrum of compound 2b. Figure S14. 13C APT NMR spectrum of compound 2b. Figure S15. 1H NMR spectrum of compound 3b. Figure S16. 13C APT NMR spectrum of compound 3b. Figure S17. 1H NMR spectrum of compound 4b. Figure S18. 13C APT NMR spectrum of compound 4b. Figure S19. 1H NMR spectrum of AAZTA-Bn. Figure S20. 13C APT NMR spectrum of AAZTA-Bn. Figure S21. HRMS of compound 2a. Figure S22. HRMS of compound 3a. Figure S23. HRMS of compound 4a. Figure S24. HRMS of AAZTA-Et. Figure S25. HRMS of compound 2b. Figure S26. HRMS of compound 3b. Figure S27. HRMS of compound 9. Figure S28. HRMS of AAZTA-Bn. Figure S29. Absorption spectra of Cu2+-AAZTA-Et systems. Figure S30. Absorption spectra of Cu2+-AAZTA-Bn systems. Figure S31. Relaxivity values of the Gd3+-AAZTA-Et-DTPA systems at 21 MHz and pH = 3.6. Figure S32. Relaxivity values of the Gd3+-AAZTA-Bn-DTPA systems at 21 MHz and pH = 3.6. Figure S33. 1H NMR spectra of the Lu3+-AAZTA-Bn-DTPA systems at 400 MHz and pH = 3.8. Figure S34. Abs. Int. values of the [Lu(AAZTA-Bn)]− in Lu3+-AAZTA-Bn-DTPA systems at 400 MHz and pH = 3.8. Figure S35. Pseudo-first order rate constants (kd) characterize the transmetalation reactions of [Gd(AAZTA-Et)]− with Cu2+ as a function of pH and [H+]. Figure S36. Pseudo-first order rate constants (kd) characterize the transmetalation reactions of [Gd(AAZTA-Bn)]− with Cu2+ as a function of pH and [H+]. References [12,30,38,39,40] are cited in the supplementary materials.
Author Contributions
Conceptualization, L.L., G.B.G. and Z.B.; formal analysis, Z.B.; investigation, F.F., F.T., M.R. and M.B.; writing—original draft preparation, F.F., F.T., M.R. and M.B.; writing—review and editing, L.L., G.B.G. and Z.B.; visualization, Z.B.; supervision, L.L. and G.B.G.; funding acquisition, L.L. and G.B.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data are contained within the article or Supplementary Materials.
Conflicts of Interest
Authors Federico Forgione and Giovanni B. Giovenzana have a collaboration with the company CAGE Chemicals srl. Authors Madalina Ranga, Mariangela Boccalon, Zsolt Baranyai and Luciano Lattuada were employed by the company Bracco Imaging SpA. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Mjos, K.D.; Orvig, C. Metallodrugs in Medicinal Inorganic Chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef]
- Gaynor, D.; Griffith, D.M. The Prevalence of Metal-Based Drugs as Therapeutic or Diagnostic Agents: Beyond Platinum. Dalton Trans. 2012, 41, 13239–13257. [Google Scholar] [CrossRef]
- Alessio, E. (Ed.) Bioinorganic Medicinal Chemistry, 1st ed.; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32631-0. [Google Scholar]
- Hart, J.R. Ethylenediaminetetraacetic Acid and Related Chelating Agents. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; ISBN 978-3-527-30673-2. [Google Scholar]
- Lattuada, L.; Barge, A.; Cravotto, G.; Giovenzana, G.B.; Tei, L. The Synthesis and Application of Polyamino Polycarboxylic Bifunctional Chelating Agents. Chem. Soc. Rev. 2011, 40, 3019–3049. [Google Scholar] [CrossRef] [PubMed]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2019, 119, 957–1057. [Google Scholar] [CrossRef]
- Merbach, A.S.; Helm, L.; Tóth, É. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed.; Wiley: Hoboken, NJ, USA, 2013; ISBN 978-1-119-99176-2. [Google Scholar]
- Herrero Álvarez, N.; Bauer, D.; Hernández-Gil, J.; Lewis, J.S. Recent Advances in Radiometals for Combined Imaging and Therapy in Cancer. ChemMedChem 2021, 16, 2909–2941. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. Matching Chelators to Radiometals for Radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Ramogida, C.F.; Orvig, C. Tumour Targeting with Radiometals for Diagnosis and Therapy. Chem. Commun. 2013, 49, 4720–4739. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Calabi, L.; Cavallotti, C.; Gianolio, E.; Giovenzana, G.B.; Losi, P.; Maiocchi, A.; Palmisano, G.; Sisti, M. [Gd-AAZTA]−: A New Structural Entry for an Improved Generation of MRI Contrast Agents. Inorg. Chem. 2004, 43, 7588–7590. [Google Scholar] [CrossRef]
- Baranyai, Z.; Uggeri, F.; Giovenzana, G.B.; Bényei, A.; Brücher, E.; Aime, S. Equilibrium and Kinetic Properties of the Lanthanoids(III) and Various Divalent Metal Complexes of the Heptadentate Ligand AAZTA. Chem. Eur. J. 2009, 15, 1696–1705. [Google Scholar] [CrossRef]
- Gianolio, E.; Giovenzana, G.B.; Longo, D.; Longo, I.; Menegotto, I.; Aime, S. Relaxometric and Modelling Studies of the Binding of a Lipophilic Gd-AAZTA Complex to Fatted and Defatted Human Serum Albumin. Chem. A Eur. J. 2007, 13, 5785–5797. [Google Scholar] [CrossRef]
- Artali, R.; Bombieri, G.; Giovenzana, G.B.; Galli, M.; Lattuada, L.; Meneghetti, F. Preparation, Crystallographic and Theoretical Study on a Bifunctional Gd-AAZTA Derivative as Potential MRI Contrast Agent Precursor. Inorganica Chim. Acta 2013, 407, 306–312. [Google Scholar] [CrossRef]
- Minazzi, P.; Lattuada, L.; Menegotto, I.G.; Giovenzana, G.B. An Enzymatic Approach to Bifunctional Chelating Agents. Org. Biomol. Chem. 2014, 12, 6915–6921. [Google Scholar] [CrossRef] [PubMed]
- Giovenzana, G.B.; Lattuada, L.; Negri, R. Recent Advances in Bifunctional Paramagnetic Chelates for MRI. Isr. J. Chem. 2017, 57, 825–832. [Google Scholar] [CrossRef]
- Travagin, F.; Lattuada, L.; Giovenzana, G.B. AAZTA: The Rise of Mesocyclic Chelating Agents for Metal Coordination in Medicine. Coord. Chem. Rev. 2021, 438, 213908–213931. [Google Scholar] [CrossRef]
- Baranyai, Z.; Uggeri, F.; Maiocchi, A.; Giovenzana, G.B.; Cavallotti, C.; Takács, A.; Tóth, I.; Bányai, I.; Bényei, A.; Brucher, E.; et al. Equilibrium, Kinetic and Structural Studies of AAZTA Complexes with Ga3+, In3+ and Cu2+. Eur. J. Inorg. Chem. 2013, 2013, 147–162. [Google Scholar] [CrossRef]
- Nagy, G.; Szikra, D.; Trencsényi, G.; Fekete, A.; Garai, I.; Giani, A.M.; Negri, R.; Masciocchi, N.; Maiocchi, A.; Uggeri, F.; et al. AAZTA: An Ideal Chelating Agent for the Development of 44Sc PET Imaging Agents. Angew. Chem. 2017, 56, 2118–2122. [Google Scholar] [CrossRef]
- Pfister, J.; Summer, D.; Rangger, C.; Petrik, M.; von Guggenberg, E.; Minazzi, P.; Giovenzana, G.B.; Aloj, L.; Decristoforo, C. Influence of a Novel, Versatile Bifunctional Chelator on Theranostic Properties of a Minigastrin Analogue. EJNMMI Res. 2015, 5, 74. [Google Scholar] [CrossRef]
- Horváth, D.; Vágner, A.; Szikra, D.; Trencsényi, G.; Demitri, N.; Guidolin, N.; Maiocchi, A.; Ghiani, S.; Travagin, F.; Giovenzana, G.B.; et al. Boosting Bismuth(III) Complexation for Targeted α-Therapy (TAT) Applications with the Mesocyclic Chelating Agent AAZTA. Angew. Chem. Int. Ed. 2022, 61, e202207120. [Google Scholar] [CrossRef]
- Fersing, C.; Masurier, N.; Rubira, L.; Deshayes, E.; Lisowski, V. AAZTA-Derived Chelators for the Design of Innovative Radiopharmaceuticals with Theranostic Applications. Pharmaceuticals 2022, 15, 234. [Google Scholar] [CrossRef]
- Manzoni, L.; Belvisi, L.; Arosio, D.; Bartolomeo, M.P.; Bianchi, A.; Brioschi, C.; Buonsanti, F.; Cabella, C.; Casagrande, C.; Civera, M.; et al. Synthesis of Gd and 68Ga Complexes in Conjugation with a Conformationally Optimized RGD Sequence as Potential MRI and PET Tumor-Imaging Probes. ChemMedChem 2012, 7, 1084–1093. [Google Scholar] [CrossRef]
- Nock, B.A.; Kaloudi, A.; Nagel, J.; Sinnes, J.-P.; Roesch, F.; Maina, T. Novel Bifunctional DATA Chelator for Quick Access to Site-Directed PET 68Ga-Radiotracers: Preclinical Proof-of-Principle with [Tyr3]Octreotide. Dalton Trans. 2017, 46, 14584–14590. [Google Scholar] [CrossRef] [PubMed]
- Sinnes, J.-P.; Nagel, J.; Waldron, B.P.; Maina, T.; Nock, B.A.; Bergmann, R.K.; Ullrich, M.; Pietzsch, J.; Bachmann, M.; Baum, R.P.; et al. Instant Kit Preparation of 68Ga-Radiopharmaceuticals via the Hybrid Chelator DATA: Clinical Translation of [68Ga]Ga-DATA-TOC. EJNMMI Res. 2019, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Klasen, B.; Moon, E.S.; Rösch, F. AAZTA5-Squaramide Ester Competing with DOTA-, DTPA- and CHX-A″-DTPA-Analogues: Promising Tool for 177Lu-Labeling of Monoclonal Antibodies under Mild Conditions. Nucl. Med. Biol. 2021, 96–97, 80–93. [Google Scholar] [CrossRef]
- Greifenstein, L.; Kramer, C.S.; Moon, E.S.; Rösch, F.; Klega, A.; Landvogt, C.; Müller, C.; Baum, R.P. From Automated Synthesis to In Vivo Application in Multiple Types of Cancer—Clinical Results with [68Ga]Ga-DATA5m.SA.FAPi. Pharmaceuticals 2022, 15, 1000. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kanellopoulos, P.; Moon, E.S.; Rouchota, M.; Loudos, G.; Ballal, S.; Yadav, M.P.; Bal, C.; Mishra, P.; Sheokand, P.; et al. [111In]In/[177Lu]Lu-AAZTA5-LM4 SST2R-Antagonists in Cancer Theranostics: From Preclinical Testing to First Patient Results. Pharmaceutics 2023, 15, 776. [Google Scholar] [CrossRef] [PubMed]
- Giovenzana, G.B.; Palmisano, G.; Sisti, M.; Cavallotti, C.; Aime, S.; Calabi, L.; Swenson, R.; Kondareddiar, R.; Lattuada, L.; Morosini, P. Multidentate Aza Ligands Able to Complex Metal Ions and the Use Thereof in Diagnostics and Therapy. U.S. Patent US20060034773A1, 16 February 2006. [Google Scholar]
- Kock, F.V.C.; Forgács, A.; Guidolin, N.; Stefania, R.; Vágner, A.; Gianolio, E.; Aime, S.; Baranyai, Z. [Gd(AAZTA)]− Derivatives with n-Alkyl Acid Side Chains Show Improved Properties for Their Application as MRI Contrast Agents. Chem. A Eur. J. 2021, 27, 1849–1859. [Google Scholar] [CrossRef]
- Farkas, E.; Nagel, J.; Waldron, B.P.; Parker, D.; Tóth, I.; Brücher, E.; Rösch, F.; Baranyai, Z. Equilibrium, Kinetic and Structural Properties of Gallium(III) and Some Divalent Metal Complexes Formed with the New DATAm and DATA5m Ligands. Chem. Eur. J. 2017, 23, 10358–10371. [Google Scholar] [CrossRef]
- Vágner, A.; D’Alessandria, C.; Gambino, G.; Schwaiger, M.; Aime, S.; Maiocchi, A.; Tóth, I.; Baranyai, Z.; Tei, L. A Rigidified AAZTA-like Ligand as Efficient Chelator for 68Ga Radiopharmaceuticals. ChemistrySelect 2016, 1, 163–171. [Google Scholar] [CrossRef]
- Vágner, A.; Gianolio, E.; Aime, S.; Maiocchi, A.; Tóth, I.; Baranyai, Z.; Tei, L. High Kinetic Inertness of a Bis-Hydrated Gd-Complex with a Constrained AAZTA-like Ligand. Chem. Commun. 2016, 52, 11235–11238. [Google Scholar] [CrossRef]
- Baranyai, Z.; Pálinkás, Z.; Uggeri, F.; Brücher, E. Equilibrium Studies on the Gd3+, Cu2+ and Zn2+ Complexes of BOPTA, DTPA and DTPA-BMA Ligands: Kinetics of Metal-Exchange Reactions of [Gd(BOPTA)]2−. Eur. J. Inorg. Chem. 2010, 2010, 1948–1956. [Google Scholar] [CrossRef]
- Tei, L.; Gugliotta, G.; Fekete, M.; Kálmán, F.K.; Botta, M. Mn(II) Complexes of Novel Hexadentate AAZTA-like Chelators: A Solution Thermodynamics and Relaxometric Study. Dalton Trans. 2011, 40, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Irving, H.M.; Miles, M.G.; Pettit, L.D. A Study of Some Problems in Determining the Stoicheiometric Proton Dissociation Constants of Complexes by Potentiometric Titrations Using a Glass Electrode. Anal. Chim. Acta 1967, 38, 475–488. [Google Scholar] [CrossRef]
- Zekany, L.; Nagypal, I. PSEQUAD. In Computational Methods for the Determination of Formation Constants; Leggett, D.J., Ed.; Modern Inorganic Chemistry; Springer: Boston, MA, USA, 1985; pp. 291–353. ISBN 978-1-4684-4934-1. [Google Scholar]
- Baranyai, Z.; Pálinkás, Z.; Uggeri, F.; Maiocchi, A.; Aime, S.; Brücher, E. Dissociation Kinetics of Open-Chain and Macrocyclic Gadolinium(III)-Aminopolycarboxylate Complexes Related to Magnetic Resonance Imaging: Catalytic Effect of Endogenous Ligands. Chem. Eur. J. 2012, 18, 16426–16435. [Google Scholar] [CrossRef] [PubMed]
- Sarka, L.; Burai, L.; Brücher, E. The Rates of the Exchange Reactions between [Gd(DTPA)]2− and the Endogenous Ions Cu2+ and Zn2+: A Kinetic Model for the Prediction of the In Vivo Stability of [Gd(DTPA)]2−, Used as a Contrast Agent in Magnetic Resonance Imaging. Chem. Eur. J. 2000, 6, 719–724. [Google Scholar] [CrossRef]
- Brücher, E.; Laurenczy, G. Aminopolycarboxylates of Rare Earths—VIII: Kinetic Study of Exchange Reactions between Eu3+ Ions and Lanthanide(III) Diethylenetriaminepentaacetate Complexes. J. Inorg. Nucl. Chem. 1981, 43, 2089–2096. [Google Scholar] [CrossRef]
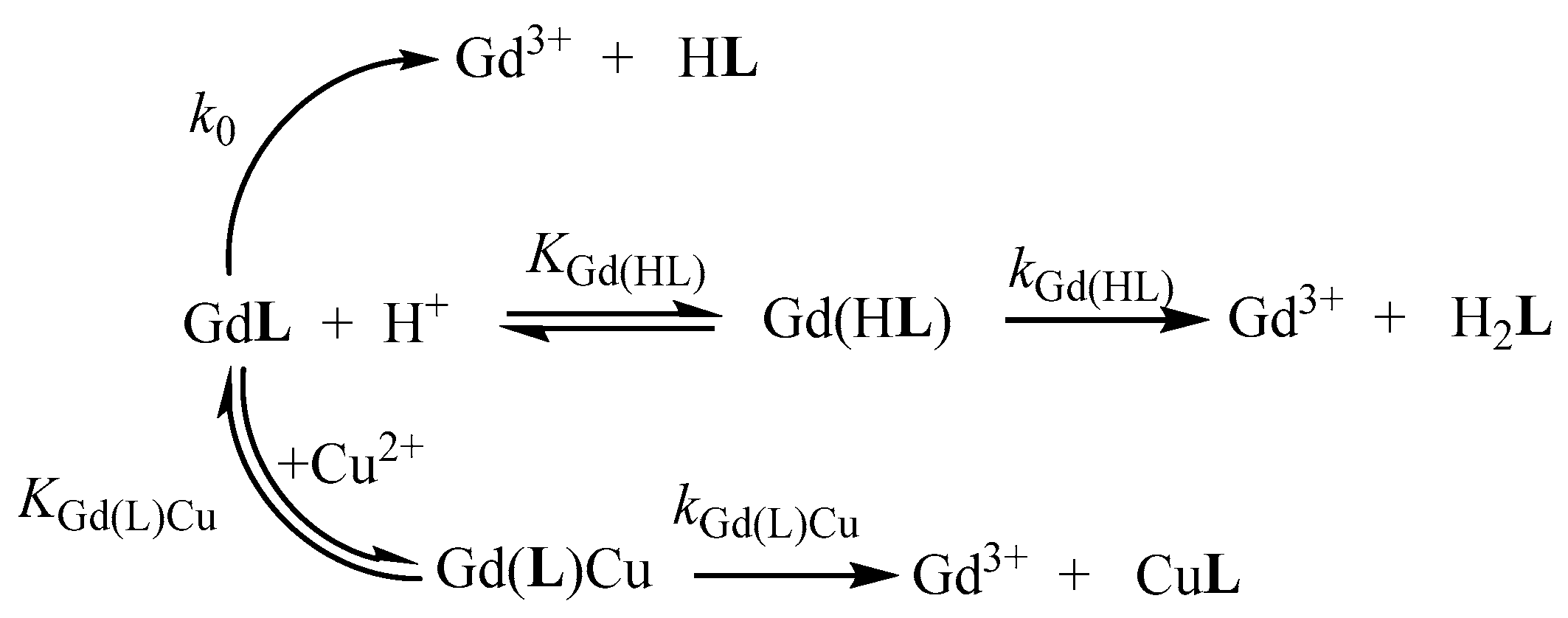
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).