
Novel Tricarbonylrhenium-Anthrapyrazole Complexes with DNA-Binding and Antitumor Properties: In Vitro and In Vivo Pharmacokinetic Studies with 99mTc-Analogue

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
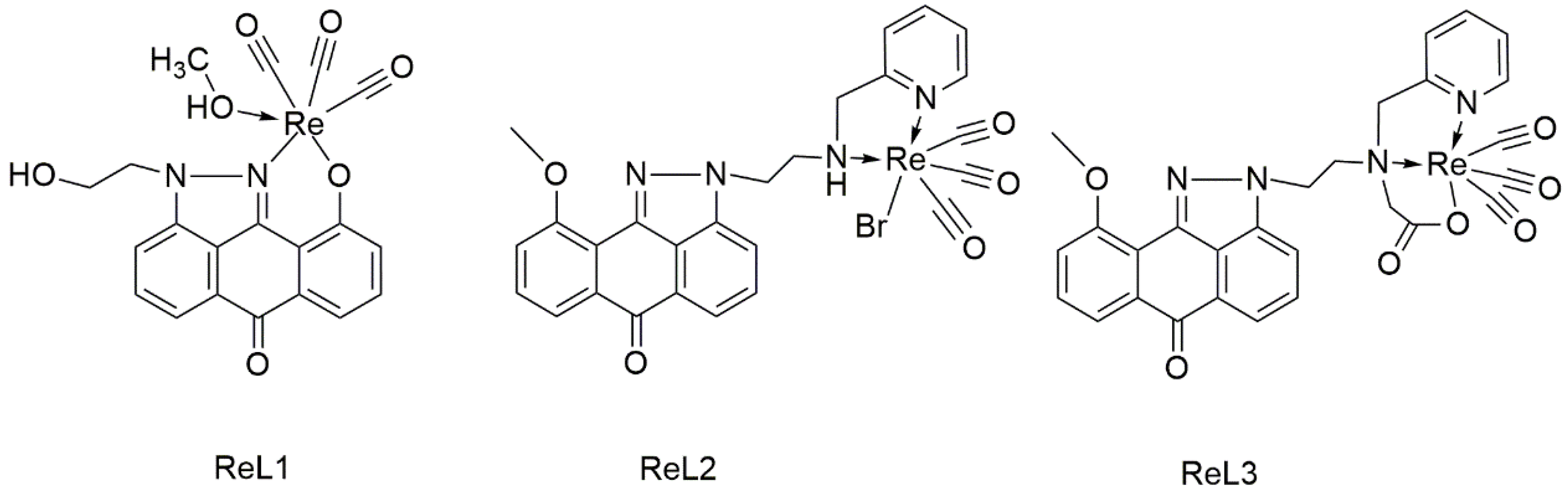
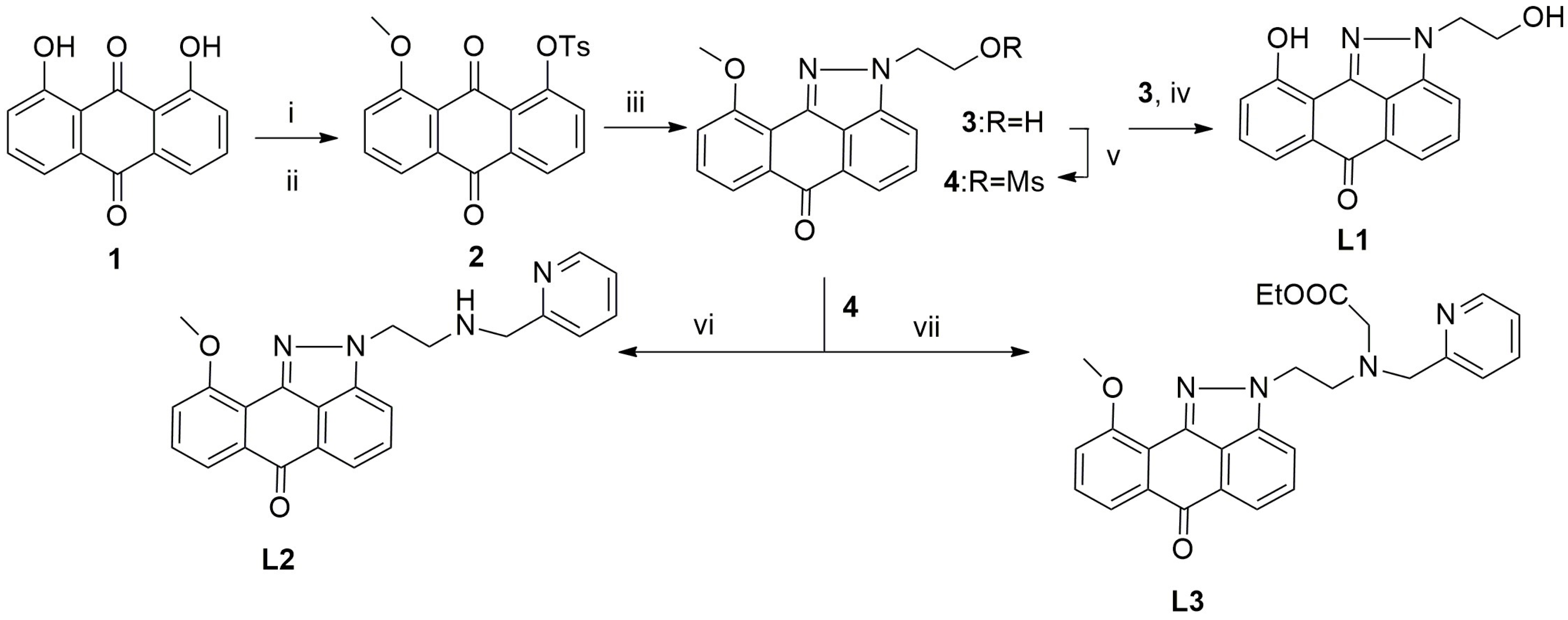
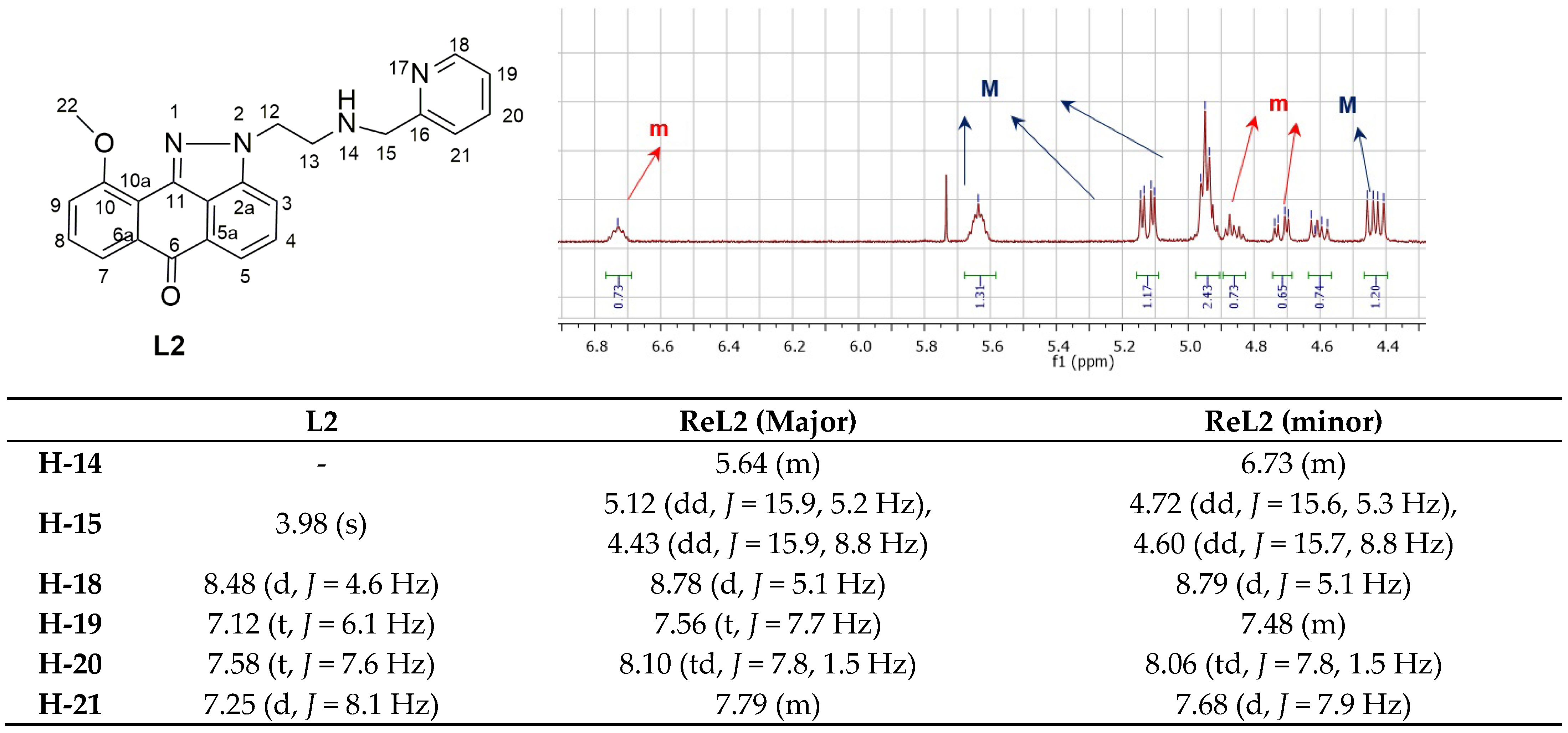
2.1. Synthesis and Spectroscopic Characterization of Rhenium Complexes
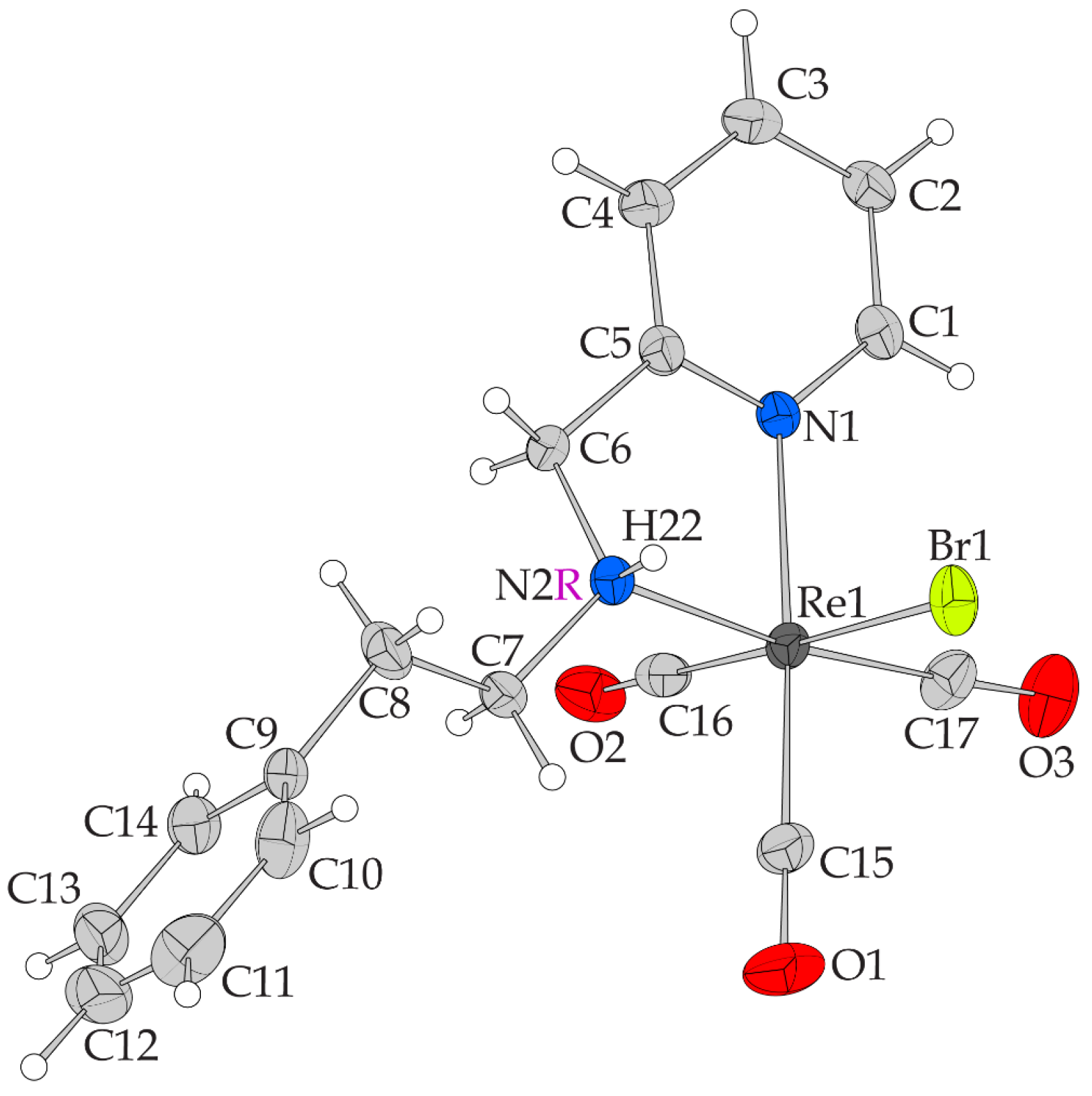
2.2. X-ray-Structure
2.3. DNA-Binding Studies
2.4. In Vitro Cell Studies
2.5. Radiotracer Studies
3. Materials and Methods
3.1. General
3.2. Syntheses
3.3. X-ray Crystallography of fac-[Re(CO)3Br(L4)]
3.4. DNA-Binding Studies
3.4.1. Study by UV–Vis Spectroscopy
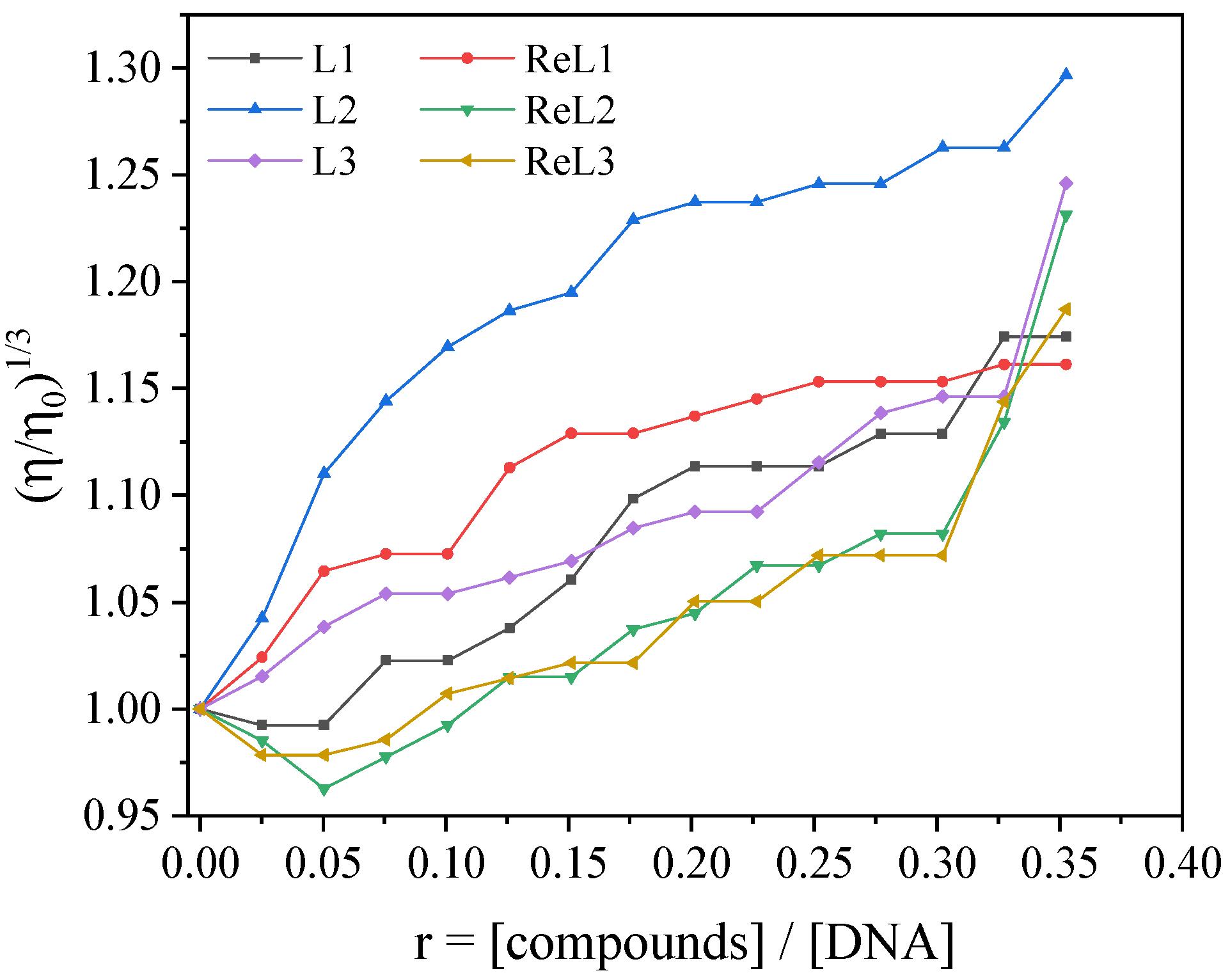
3.4.2. Viscometry
3.4.3. Competition Studies with Ethidium Bromide (EB) via Fluorescence Spectroscopy
3.5. In Vitro Cell Studies
3.5.1. Cell Cultures
3.5.2. Assessment of Proliferation Capacity of CT26 Cells Exposed to Anthrapyrazole Derivatives
3.6. 99mTc Radiochemistry and In Vitro Radiotracer Studies
3.7. Biodistribution Studies of 99mTcL2 in Mice
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muhammad, N.; Guo, Z. Metal-Based Anticancer Chemotherapeutic Agents. Curr. Opin. Chem. Biol. 2014, 19, 144–153. [Google Scholar] [CrossRef]
- Ye, R.; Tan, C.; Chen, B.; Li, R.; Mao, Z. Zinc-Containing Metalloenzymes: Inhibition by Metal-Based Anticancer Agents. Front. Chem. 2020, 8, 402. [Google Scholar] [CrossRef]
- Simpson, P.V.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-Based Antitumor Compounds: Beyond Cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-Class Ruthenium Anticancer Drug (KP1339/IT-139) Induces an Immunogenic Cell Death Signature in Colorectal Spheroids in Vitro†. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef]
- King, A.P.; Wilson, J.J. Endoplasmic Reticulum Stress: An Arising Target for Metal-Based Anticancer Agents. Chem. Soc. Rev. 2020, 49, 8113–8136. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Ortega, E.; Vigueras, G.; Ballester, F.J.; Ruiz, J. Targeting Translation: A Promising Strategy for Anticancer Metallodrugs. Coord. Chem. Rev. 2021, 446, 214129. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Clède, S.; Lambert, F.; Saint-Fort, R.; Plamont, M.A.; Bertrand, H.; Vessières, A.; Policar, C. Influence of the Side-Chain Length on the Cellular Uptake and the Cytotoxicity of Rhenium Triscarbonyl Derivatives: A Bimodal Infrared and Luminescence Quantitative Study. Chem. A Eur. J. 2014, 20, 8714–8722. [Google Scholar] [CrossRef] [PubMed]
- Leonidova, A.; Pierroz, V.; Adams, L.A.; Barlow, N.; Ferrari, S.; Gra, B. Enhanced Cytotoxicity through “Click” Conjugation of a Luminescent Re (I) Complex to a Cell-Penetrating Lipopeptide. ACS Med. Chem. Lett. 2014, 5, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Leonidova, A.; Pierroz, V.; Rubbiani, R.; Heier, J.; Ferrari, S.; Gasser, G. Towards Cancer Cell-Specific Phototoxic Organometallic Rhenium (I) Complexes. Dalton Trans. 2014, 43, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Knopf, K.M.; Murphy, B.L.; Macmillan, S.N.; Baskin, J.M.; Barr, M.P.; Boros, E.; Wilson, J.J. In Vitro Anticancer Activity and in Vivo Biodistribution of Rhenium (I) Tricarbonyl Aqua Complexes. J. Am. Chem. Soc. 2017, 139, 14302–14314. [Google Scholar] [CrossRef] [PubMed]
- Schindler, K.; Zobi, F. Anticancer and Antibiotic Rhenium Tri- and Dicarbonyl Complexes: Current Research and Future Perspectives. Molecules 2022, 27, 539. [Google Scholar] [CrossRef] [PubMed]
- Rescifina, A.; Zagni, C.; Varrica, M.G.; Pistarà, V.; Corsaro, A. Recent Advances in Small Organic Molecules as DNA Intercalating Agents: Synthesis, Activity, and Modeling. Eur. J. Med. Chem. 2014, 74, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-K.; Sadler, P.J. Metal Complexes as DNA Intercalators. Acc. Chem. Res. 2011, 44, 349–359. [Google Scholar] [CrossRef]
- Lee, L.C.-C.; Leung, K.-K.; Lo, K.K.-W. Recent Development of Luminescent Rhenium (i) Tricarbonyl Polypyridine Complexes as Cellular Imaging Reagents, Anticancer Drugs, and Antibacterial Agents. Dalton Trans. 2017, 46, 16357–16380. [Google Scholar] [CrossRef]
- Pagoni, C.C.; Xylouri, V.S.; Kaiafas, G.C.; Lazou, M.; Bompola, G.; Tsoukas, E.; Papadopoulou, L.C.; Psomas, G.; Papagiannopoulou, D. Organometallic Rhenium Tricarbonyl—Enrofloxacin and—Levofloxacin Complexes: Synthesis, Albumin—Binding, DNAi—Interaction and Cell Viability Studies. JBIC J. Biol. Inorg. Chem. 2019, 24, 609–619. [Google Scholar] [CrossRef]
- Nasiri Sovari, S.; Kolly, I.; Schindler, K.; Djuric, A.; Srdic-Rajic, T.; Crochet, A.; Pavic, A.; Zobi, F. Synthesis, Characterization, and in Vivo Evaluation of the Anticancer Activity of a Series of 5- and 6-(Halomethyl)-2,2′-Bipyridine Rhenium Tricarbonyl Complexes. Dalton Trans. 2023, 52, 6934–6944. [Google Scholar] [CrossRef]
- Frederick, C.A.; Williams, L.D.; Ughetto, G.; Van der Marel, G.A.; Van Boom, J.H.; Rich, A.; Wang, A.H.J. Structural Comparison of Anticancer Drug-DNA Complexes: Adriamycin and Daunomycin. Biochemistry 1990, 29, 2538–2549. [Google Scholar] [CrossRef]
- Mattioli, R.; Ilari, A.; Colotti, B.; Mosca, L.; Fazi, F.; Colotti, G. Doxorubicin and Other Anthracyclines in Cancers: Activity, Chemoresistance and Its Overcoming. Mol. Asp. Med. 2023, 93, 101205. [Google Scholar] [CrossRef]
- Imstepf, S.; Pierroz, V.; Rubbiani, R.; Felber, M.; Fox, T.; Gasser, G.; Alberto, R. Organometallic Rhenium Complexes Divert Doxorubicin to the Mitochondria. Angew. Chem. Int. Ed. 2016, 55, 2792–2795. [Google Scholar] [CrossRef]
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26. [Google Scholar] [CrossRef]
- Gogas, H.; Mansi, J.L. New Drugs. The Anthrapyrazoles. Cancer Treat. Rev. 1996, 21, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.H.; Zhang, Q.X.; Huang, Z.S.; Chen, Y.; Wang, X.D.; Gu, L.Q.; Wu, J.Y. Synthesis, DNA Binding and Cytotoxicity of New Pyrazole Emodin Derivatives. Eur. J. Med. Chem. 2006, 41, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Haiyang, H.; Morley, J.E.; Twamley, B.; Groeneman, R.H.; Bucar, D.K.; MacGillivray, L.R.; Benny, P.D. Investigation of the Coordination Interactions of S-(Pyridin-2-Ylmethyl)-L-Cysteine Ligands with M(CO)3+ (M = Re, 99mTc). Inorg. Chem. 2009, 48, 10625–10634. [Google Scholar]
- Makris, G.; Radford, L.; Gallazzi, F.; Jurisson, S.; Hennkens, H.; Papagiannopoulou, D. Synthesis and Evaluation of fac-[99mTc/Re(CO)3]+ Complexes with a New (N,S,N) Bifunctional Chelating Agent: The First Example of a fac-[Re(CO)3(N,S,N-Sst2-ANT)] Complex Bearing a Somatostatin Receptor Antagonist Peptide. J. Organomet. Chem. 2016, 805, 100–107. [Google Scholar] [CrossRef]
- He, H.; Lipowska, M.; Xu, X.; Taylor, A.T.; Marzilli, L.G. Rhenium Analogues of Promising Renal Imaging Agents with a {99mTc(CO)3}+ Core Bound to Cysteine-Derived Dipeptides, Including Lanthionine. Inorg. Chem. 2007, 46, 590–596. [Google Scholar] [CrossRef]
- Suárez-Ortiz, G.A.; Hernández-Correa, R.; Morales-Moreno, M.D.; Toscano, R.A.; Ramirez-Apan, M.T.; Hernandez-Garcia, A.; Amézquita-Valencia, M.; Araiza-Olivera, D. Diastereomeric Separation of Chiral Fac-Tricarbonyl (Iminopyridine) Rhenium (I) Complexes and Their Cytotoxicity Studies: Approach toward an Action Mechanism against Glioblastoma. J. Med. Chem. 2022, 65, 9281–9294. [Google Scholar] [CrossRef]
- Song, X.; Lim, M.H.; Mohamed, D.K.B.; Wong, S.M.; Zhao, J.; Hor, T.S.A. Re (I) Carbonyl Complexes Containing Pyridyl-Imine and Amine Ligands: Synthesis, Characterization and Their Catalytic Olefin Epoxidation Activities. J. Organomet. Chem. 2016, 814, 1–7. [Google Scholar] [CrossRef]
- Saund, S.S.; Siegler, M.A.; Thoi, V.S. Electrochemical Degradation of a Dicationic Rhenium Complex via Hoffman-Type Elimination. Inorg. Chem. 2021, 60, 13011–13020. [Google Scholar] [CrossRef]
- Liu, W.; Heinze, K. Rhenium (I) and Platinum (II) Complexes with Diimine Ligands Bearing Acidic Phenol Substituents: Hydrogen-Bonding, Acid–Base Chemistry and Optical Properties. Dalton Trans. 2010, 39, 9554–9564. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Spingler, B.; Alberto, R. Reactivity of 2-Pyridine–Aldehyde and 2-Acetyl–Pyridine Coordinated to [Re(CO)3]+ with Alcohols and Amines: Metal Mediated Schiff Base Formation and Dimerization. Inorganica Chim. Acta 2003, 355, 386–393. [Google Scholar] [CrossRef]
- Ketkaew, R.; Tantirungrotechai, Y.; Harding, P.; Chastanet, G.; Guionneau, P.; Marchivie, M.; Harding, D.J. OctaDist: A Tool for Calculating Distortion Parameters in Spin Crossover and Coordination Complexes. Dalton Trans. 2021, 50, 1086–1096. [Google Scholar] [CrossRef]
- Wolfe, A.; Shimer, G.; Meehan, T. Polycyclic Aromatic Hydrocarbons Physically Intercalate into Duplex Regions of Denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Lin, H.; Zhu, S.; Sun, H.; Chen, Y. Dinuclear Palladium (II) Complexes Containing Two Monofunctional [Pd(En)(Pyridine)Cl]+ Units Bridged by Se or S. Synthesis, Characterization, Cytotoxicity and Kinetic Studies of DNA-Binding. J. Inorg. Biochem. 1998, 70, 219–226. [Google Scholar] [CrossRef]
- Luis García-Giménez, J.; González-Álvarez, M.; Liu-González, M.; Macías, B.; Borrás, J.; Alzuet, G. Toward the Development of Metal-Based Synthetic Nucleases: DNA Binding and Oxidative DNA Cleavage of a Mixed Copper (II) Complex with N-(9H-Purin-6-Yl) Benzenesulfonamide and 1,10-Phenantroline. Antitumor Activity in Human Caco-2 Cells and Jurkat T Lymphocy. J. Inorg. Biochem. 2009, 103, 923–934. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Plenum Press: New York, NY, USA, 2006. [Google Scholar]
- Wilson, W.D.; Ratmeyer, L.; Zhao, M.; Strekowski, L.; Boykin, D. The Search for Structure-Specific Nucleic Acid-Interactive Drugs: Effects of Compound Structure on RNA versus DNA Interaction Strength. Biochemistry 1993, 32, 4098–4104. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.P.; Greenstock, C.L. Fluorescence Lifetime Analysis of DNA Intercalated Ethidium Bromide and Quenching by Free Dye. Biophys. Chem. 1994, 50, 305–312. [Google Scholar] [CrossRef]
- Sharma, A.; Özayral, S.; Caserto, J.S.; ten Cate, R.; Anders, N.M.; Barnett, J.D.; Kandala, S.K.; Henderson, E.; Stewart, J.; Liapi, E.; et al. Increased Uptake of Doxorubicin by Cells Undergoing Heat Stress Does Not Explain Its Synergistic Cytotoxicity with Hyperthermia. Int. J. Hyperth. 2019, 36, 712–720. [Google Scholar] [CrossRef]
- Shimolina, L.E.; Gulin, A.A.; Paez-Perez, M.; López-Duarte, I.; Druzhkova, I.N.; Lukina, M.M.; Gubina, M.V.; Brooks, N.J.; Zagaynova, E.V.; Kuimova, M.K.; et al. Mapping Cisplatin-Induced Viscosity Alterations in Cancer Cells Using Molecular Rotor and Fluorescence Lifetime Imaging Microscopy. J. Biomed. Opt. 2020, 25, 126004. [Google Scholar] [CrossRef]
- Showalter, H.D.; Johnson, J.L.; Hoftiezer, J.M.; Turner, W.R.; Werbel, L.M.; Leopold, W.R.; Shillis, J.L.; Jackson, R.C.; Elslager, E.F. Anthrapyrazole Anticancer Agents. Synthesis and Structure-Activity Relationships against Murine Leukemias. J. Med. Chem. 1987, 30, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Kostakis, I.K.; Magiatis, P.; Pouli, N.; Marakos, P.; Skaltsounis, A.L.; Pratsinis, H.; Léonce, S.; Pierré, A. Design, Synthesis, and Antiproliferative Activity of Some New Pyrazole-Fused Amino Derivatives of the Pyranoxanthenone, Pyranothioxanthenone, and Pyranoacridone Ring Systems: A New Class of Cytotoxic Agents. J. Med. Chem. 2002, 45, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Zee-Cheng, R.K.Y.; Podrebarac, E.G.; Menon, C.S.; Cheng, C.C. Structural Modification Study of Bis (Substituted Aminoalkylamino) Anthraquinones. An Evaluation of the Relationship of the [2-[(2-Hydroxyethyl)Amino]Ethyl] Amino Side Chain with Antineoplastic Activity. J. Med. Chem. 1979, 22, 501–505. [Google Scholar] [CrossRef]
- Papagiannopoulou, D. Technetium-99m Radiochemistry for Pharmaceutical Applications. J. Label. Comp. Radiopharm. 2017, 60, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Trogler, W.; Basolo, F. Pentacarbonyl Rhenium Halides. Inorg. Synth. 1985, 23, 41. [Google Scholar]
- Nitschke, J.; Schmidt, S.P.; Trogler, W.C. Properties of (Trifluoromethanesulfonato) Pentacarbonylmanganese (I) and Rhenium (I). Reactions in Superacid Solvents. Inorg. Chem. 1985, 24, 1972–1978. [Google Scholar] [CrossRef]
- Alberto, R.; Schibli, R.; Egli, A.; Schubiger, A.P.; Abram, U.; Kaden, T.A. A Novel Organometallic Aqua Complex of Technetium for the Labeling of Biomolecules: Synthesis of [99mTc(OH2)3(CO)3]+ from [99mTcO4]− in Aqueous Solution and Its Reaction with a Bifunctional Ligand. J. Am. Chem. Soc. 1998, 120, 7987–7988. [Google Scholar] [CrossRef]
- Marmur, J. A Procedure for the Isolation of Deoxyribonucleic Acid from Micro-Organisms. J. Mol. Biol. 1961, 3, 208-IN1. [Google Scholar] [CrossRef]
- Reichmann, M.E.; Rice, S.; Thomas, C.; Doty, P. A Further Examination of the Molecular Weight and Size of Desoxypentose Nucleic Acid. J. Am. Chem. Soc. 1954, 76, 3047–3053. [Google Scholar] [CrossRef]
- Policar, C.; Lambert, F.; Cesario, M.; Morgenstern-Badarau, I. An Inorganic Helix [Mn(IPG)(MeOH)]n[PF6]n[‡]: Structural and Magnetic Properties of a Syn-Anti Carboxylate-Bridged Manganese (II) Chain Involving a Tetradentate Ligand. Eur. J. Inorg. Chem. 1999, 1999, 2201–2207. [Google Scholar] [CrossRef]
- McInally, C. Apex2; Bruker Analytical X-Ray Systems, Inc.: Madison, WI, USA, 2006; p. M86-E01078. [Google Scholar]
- Sheldrick, G.M. SADABS: Area-Detector Absorption Correction; Siemens Industrial Automation, Inc.: Madison, WI, USA, 1996. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Watkin, D. The Control of Difficult Refinements. Acta Crystallogr. Sect. A 1994, 50, 411–437. [Google Scholar] [CrossRef]
- Prince, E. Mathematical Techniques in Crystallography and Materials Science; Springer: Berlin/Heidelberg, Germany, 1982. [Google Scholar]
- Watkin, D.J.; Prout, C.K.; Pearce, L.J. CAMERON, Chemical Crystallography Laboratory; University of Oxford: Oxford, UK, 1996. [Google Scholar]
- De Meulenaer, J.; Tompa, H. The Absorption Correction in Crystal Structure Analysis. Acta Crystallogr. 1965, 19, 1014–1018. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Re1—Br1 | 2.6219 (7) |
| Re1—N1 | 2.171 (4) |
| Re1—N2 | 2.223 (4) |
| Re1—C15 | 1.919 (6) |
| Re1—C16 | 1.894 (7) |
| Re1—C17 | 1.897 (6) |
| O1—C15 | 1.140 (8) |
| O2—C16 | 1.127 (7) |
| O3—C17 | 1.156 (7) |
| N1—C1 | 1.342 (7) |
| N1—C5 | 1.346 (7) |
| N2—C6 | 1.469 (7) |
| N2—C7 | 1.505 (7) |
| Br1—Re1—N1 | 85.82 (12) |
| Br1—Re1—N2 | 84.56 (12) |
| N1—Re1—N2 | 75.39 (16) |
| Br1—Re1—C15 | 92.5 (2) |
| N1—Re1—C15 | 174.8 (2) |
| N2—Re1—C15 | 99.5 (2) |
| Br1—Re1—C16 | 178.08 (18) |
| N1—Re1—C16 | 93.1 (2) |
| N2—Re1—C16 | 93.6 (2) |
| C15—Re1—C16 | 88.5 (3) |
| Br1—Re1—C17 | 91.2 (2) |
| N1—Re1—C17 | 98.5 (2) |
| N2—Re1—C17 | 172.8 (2) |
| C15—Re1—C17 | 86.5 (3) |
| C16—Re1—C17 | 90.5 (3) |
| Compound | λmax (nm) (ΔA/A0 (%) a, Δλmax (nm) b) | Kb (M−1) |
|---|---|---|
| L1 | 276 (−6 a, 0 b); 301 (sh) c (−5, 0); 323 (sh) (−7, 0); 439 (−7, 0) | 1.33 (±0.01) × 105 |
| L2 | 278 (−10, 0); 326 (sh) (−3.5, 0); 441 (−4, +2) | 7.74 (±0.03) × 104 |
| L3 | 277 (−7, +2); 325 (sh) (−7, 0); 441 (−6, 0) | 7.01 (±0.01) × 104 |
| ReL1 | 276 (−14, +3); 304 (−12, +2); 395 (−6.5, 0); 437 (sh) (−9, 0); 530 (−6, 0) | 1.24 (±0.04) × 105 |
| ReL2 | 278 (−20, +1); 300 (sh) (−10, 0); 435 (−5, +2) | 6.87 (±0.09) × 104 |
| ReL3 | 278 (−12, +2); 300 (sh) (−10, 0); 322 (sh) (−10, +2); 436 (−10, 0) | 1.09 (±0.15) × 106 |
| Compound | ΔI/Ι0 (%) | Ksv (M−1) | Kq (M−1s−1) |
|---|---|---|---|
| L1 | 53.9 | 5.65 (±0.09) × 105 | 2.46 (±0.04) × 1013 |
| L2 | 54.0 | 5.27 (±0.08) × 105 | 2.29 (±0.03) × 1013 |
| L3 | 56.5 | 5.93 (±0.04) × 105 | 2.58 (±0.02) × 1013 |
| ReL1 | 48.5 | 4.47 (±0.05) × 105 | 1.94 (±0.02) × 1013 |
| ReL2 | 51.4 | 4.52 (±0.07) × 105 | 1.96 (±0.03) × 1013 |
| ReL3 | 53.9 | 4.60 (±0.07) × 105 | 2.00 (±0.03) × 1013 |
| 15 min | 60 min | 120 min | 240 min | |
|---|---|---|---|---|
| 99mTcL2 | 1.29 ± 0.09% | 2.48 ± 0.25% | 3.94 ± 0.68% | 5.42 ± 0.19% |
| 99mTcL3 | 0.49 ± 0.06% | 0.53 ± 0.04% | 0.55 ± 0.08% | 0.75 ± 0.11% |
| Organ | %ID/g | |
|---|---|---|
| 30 min | 120 min | |
| Blood | 6.48 ± 2.02 | 4.16 ± 1.31 |
| Tumor (CT26) | 2.42 ± 0.04 | 3.10 ± 1.21 |
| Heart | 8.91 ± 3.77 | 5.90 ± 1.16 |
| Liver | 29.49 ± 9.43 | 24.51 ± 7.12 |
| Lungs | 15.36 ± 6.43 | 7.73 ± 2.29 |
| Muscle | 2.17 ± 0.25 | 2.20 ± 0.51 |
| Kidneys | 22.09 ± 8.91 | 17.04 ± 3.53 |
| Spleen | 6.78 ± 1.33 | 3.87 ± 0.87 |
| Intestine | 11.38 ± 3.43 | 19.84 ± 8.10 |
| Stomach | 8.58 ± 6.60 | 14.26 ± 7.53 |
| Crystal Data | |
|---|---|
| Chemical formula | C17H16BrN2O3Re |
| Mr | 562.43 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 295 |
| a, b, c (Å) | 11.5730 (8), 20.8663 (11), 7.7637 (5) |
| β (°) | 99.867 (2) |
| V (Å3) | 1847.1 (2) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 8.76 |
| Crystal size (mm) | 0.14 × 0.09 × 0.07 |
| Data collection | |
| Diffractometer | Bruker Kappa Apex2 |
| Absorption correction | Numerical Analytical Absorption [56] |
| Tmin, Tmax | 0.45, 0.54 |
| No. of measured, independent and observed [I > 2.0σ(I)] reflections | 17060, 3533, 3106 |
| Rint | 0.029 |
| (sin θ/λ)max (Å−1) | 0.613 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.036, 0.057, 1.00 |
| No. of reflections | 3106 |
| No. of parameters | 217 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 1.54, −1.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paparidis, G.; Akrivou, M.; Psomas, G.; Vizirianakis, I.S.; Hatzidimitriou, A.; Gabriel, C.; Sarigiannis, D.; Papagiannopoulou, D. Novel Tricarbonylrhenium-Anthrapyrazole Complexes with DNA-Binding and Antitumor Properties: In Vitro and In Vivo Pharmacokinetic Studies with 99mTc-Analogue. Inorganics 2024, 12, 254. https://doi.org/10.3390/inorganics12090254
Paparidis G, Akrivou M, Psomas G, Vizirianakis IS, Hatzidimitriou A, Gabriel C, Sarigiannis D, Papagiannopoulou D. Novel Tricarbonylrhenium-Anthrapyrazole Complexes with DNA-Binding and Antitumor Properties: In Vitro and In Vivo Pharmacokinetic Studies with 99mTc-Analogue. Inorganics. 2024; 12(9):254. https://doi.org/10.3390/inorganics12090254
Chicago/Turabian StylePaparidis, Georgios, Melpomeni Akrivou, George Psomas, Ioannis S. Vizirianakis, Antonios Hatzidimitriou, Catherine Gabriel, Dimosthenis Sarigiannis, and Dionysia Papagiannopoulou. 2024. "Novel Tricarbonylrhenium-Anthrapyrazole Complexes with DNA-Binding and Antitumor Properties: In Vitro and In Vivo Pharmacokinetic Studies with 99mTc-Analogue" Inorganics 12, no. 9: 254. https://doi.org/10.3390/inorganics12090254