
Zirconium-Catalyzed Alkene Hydrophosphination and Dehydrocoupling with an Air-Stable, Fluorescent Primary Phosphine



Abstract
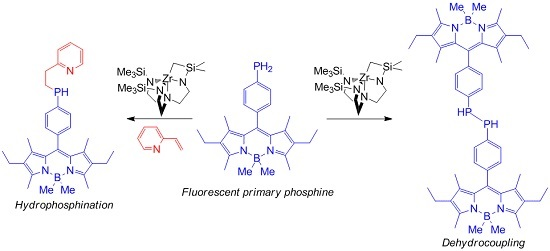
:
{kind=link}
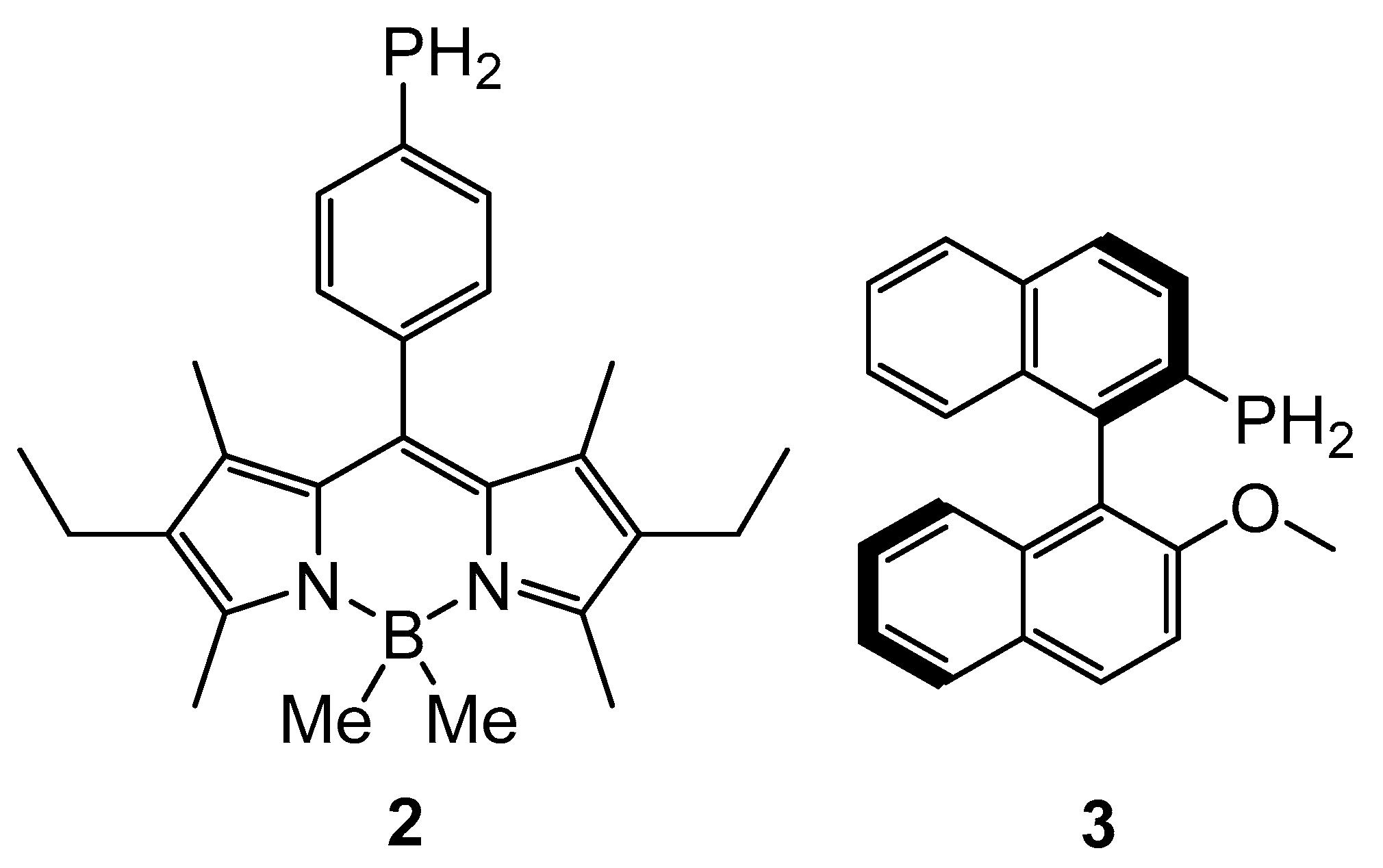{kind=link}
{kind=link}
{kind=link}
1. Introduction
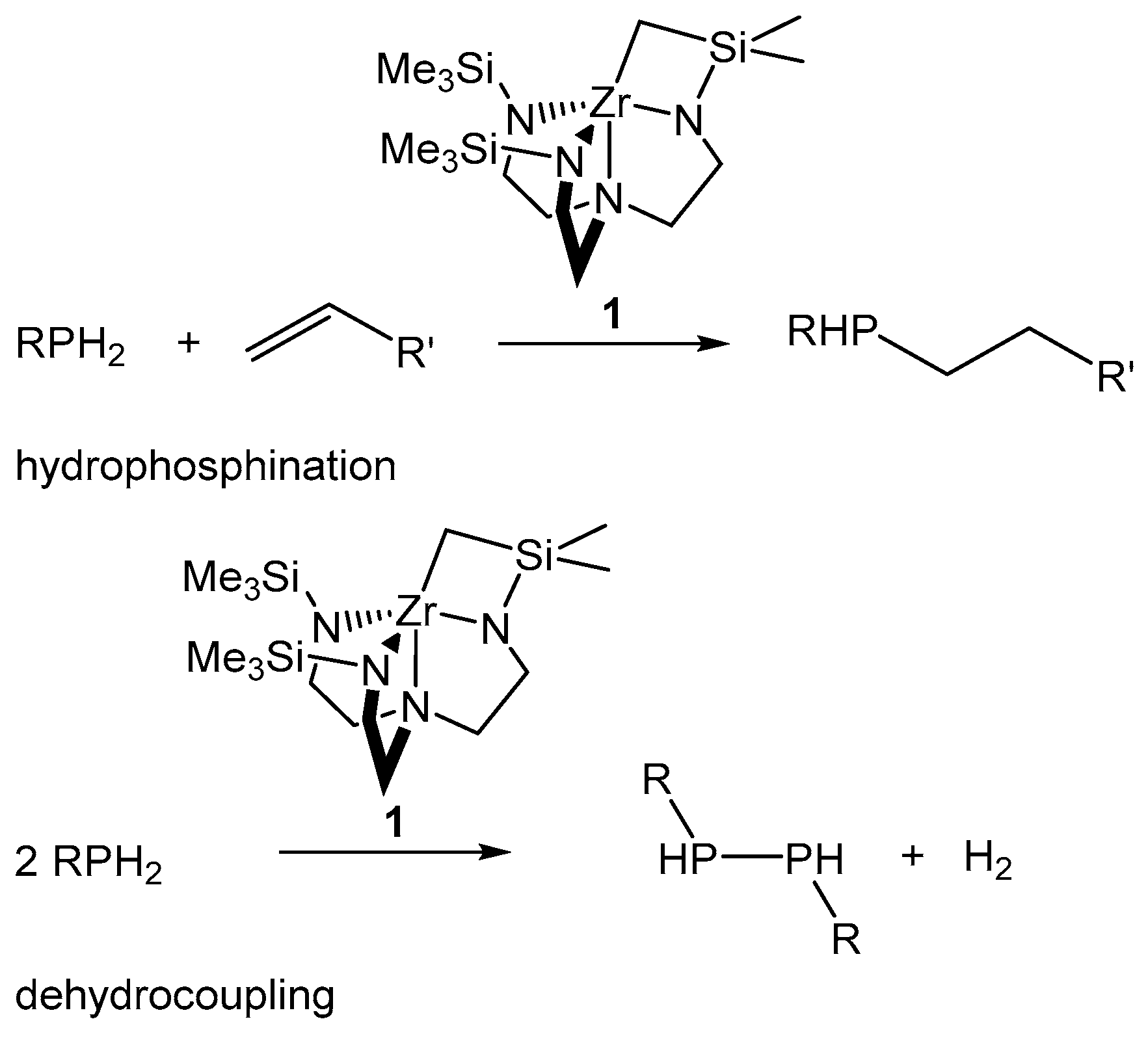
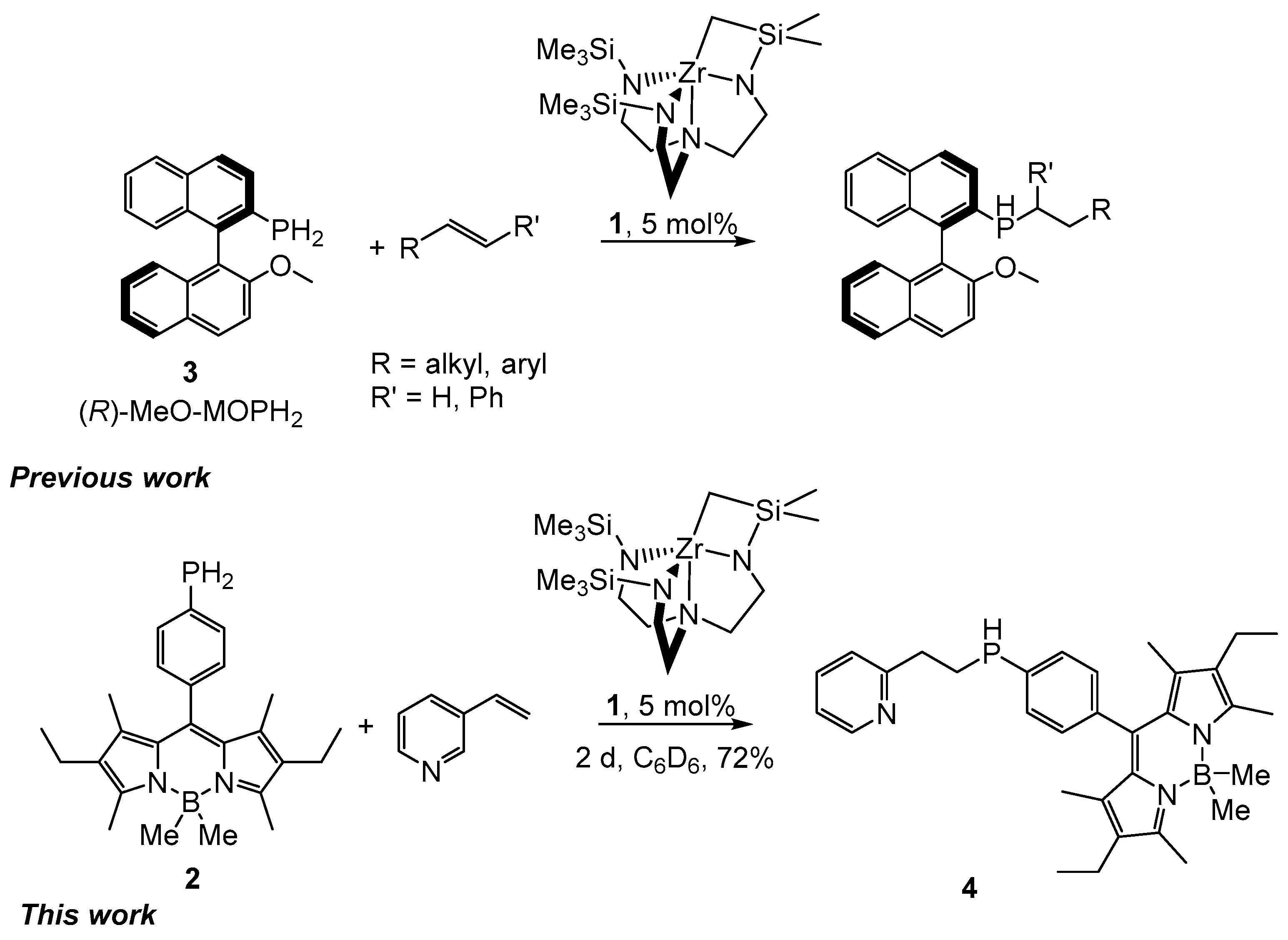
2. Results and Discussion


3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gladysz, J.A.; Bedford, R.B.; Fujita, M.; Gabbai, F.P.; Goldberg, K.I.; Holland, P.L.; Kiplinger, J.L.; Krische, M.J.; Louie, J.; Lu, C.C.; et al. Organometallics Roundtable 2013–2014. Organometallics 2014, 33, 1505–1527. [Google Scholar] [CrossRef]
- Bange, C.A.; Waterman, R. Challenges in Catalytic Hydrophosphination. Chem. Eur. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R. Dehydrogenative bond-forming catalysis involving phosphines. Curr. Org. Chem. 2008, 12, 1322–1339. [Google Scholar] [CrossRef]
- Ghebreab, M.B.; Bange, C.A.; Waterman, R. Intermolecular Zirconium-Catalyzed Hydrophosphination of Alkenes and Dienes with Primary Phosphines. J. Am. Chem. Soc. 2014, 136, 9240–9243. [Google Scholar] [CrossRef] [PubMed]
- Roering, A.J.; Leshinski, S.E.; Chan, S.M.; Shalumova, T.; MacMillan, S.N.; Tanski, J.M.; Waterman, R. Insertion Reactions and Catalytic Hydrophosphination by Triamidoamine-Supported Zirconium Complexes. Organometallics 2010, 29, 2557–2565. [Google Scholar] [CrossRef]
- Bange, C.A.; Ghebreab, M.B.; Ficks, A.; Mucha, N.T.; Higham, L.; Waterman, R. Zirconium-catalyzed intermolecular hydrophosphination using a chiral, air-stable primary phosphine. Dalton Trans. 2016, 45, 1863–1867. [Google Scholar] [CrossRef] [PubMed]
- Roering, A.J.; MacMillan, S.N.; Tanski, J.M.; Waterman, R. Zirconium-Catalyzed Heterodehydrocoupling of Primary Phosphines with Silanes and Germanes. Inorg. Chem. 2007, 46, 6855–6857. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R. Selective Dehydrocoupling of Phosphines by Triamidoamine Zirconium Catalysts. Organometallics 2007, 26, 2492–2494. [Google Scholar] [CrossRef]
- Ghebreab, M.B.; Shalumova, T.; Tanski, J.M.; Waterman, R. Triamidoamine-supported zirconium complexes in the catalytic dehydrocoupling of 1,2-bisphosphinobenzene and -ethane. Polyhedron 2010, 29, 42–45. [Google Scholar] [CrossRef]
- Rosenberg, L. Mechanisms of Metal-Catalyzed Hydrophosphination of Alkenes and Alkynes. ACS Catal. 2013, 3, 2845–2855. [Google Scholar] [CrossRef]
- Koshti, V.; Gaikwad, S.; Chikkali, S.H. Contemporary avenues in catalytic PH bond addition reaction: A case study of hydrophosphination. Coord. Chem. Rev. 2014, 265, 52–73. [Google Scholar] [CrossRef]
- Davies, L.H.; Stewart, B.; Higham, L.J. Air-stable, fluorescent primary phosphines. Organomet. Chem. 2014, 39, 51–71. [Google Scholar]
- Stewart, B.; Harriman, A.; Higham, L.J. Predicting the Air Stability of Phosphines. Organometallics 2011, 30, 5338–5343. [Google Scholar] [CrossRef]
- Ficks, A.; Martinez-Botella, I.; Stewart, B.; Harrington, R.W.; Clegg, W.; Higham, L.J. Taming functionality: Easy-to-handle chiral phosphiranes. Chem. Commun. 2011, 47, 8274–8276. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.T.; Higham, L.J. Primary phosphine chemistry. Coord. Chem. Rev. 2015, 297–298, 127–145. [Google Scholar] [CrossRef]
- Davies, L.H.; Stewart, B.; Harrington, R.W.; Clegg, W.; Higham, L.J. Air-Stable, Highly Fluorescent Primary Phosphanes. Angew. Chem. Int. Ed. 2012, 51, 4921–4924. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.H.; Wallis, J.F.; Probert, M.R.; Higham, L.J. Efficient multigram syntheses of air-stable, fluorescent primary phosphines via palladium-catalyzed phosphonylation of aryl bromides. Synthesis 2014, 46, 2622–2628. [Google Scholar]
- Davies, L.H.; Harrington, R.W.; Clegg, W.; Higham, L.J. BR2BodPR2: Highly fluorescent alternatives to PPh3 and PhPCy2. Dalton Trans. 2014, 43, 13485–13499. [Google Scholar] [CrossRef] [PubMed]
- Ficks, A.; Hiney, R.M.; Harrington, R.W.; Gilheany, D.G.; Higham, L.J. MOP-phosphonites: A novel ligand class for asymmetric catalysis. Dalton Trans. 2012, 41, 3515–3522. [Google Scholar] [CrossRef] [PubMed]
- Hiney, R.M.; Ficks, A.; Muller-Bunz, H.; Gilheany, D.G.; Higham, L.J. Air-stable chiral primary phosphines part (I) synthesis, stability and applications. In Organometallic Chemistry; Fairlamb, I.J.S., Lynam, J.M., Eds.; The Royal Society of Chemistry: London, UK, 2011; Volume 37, pp. 27–45. [Google Scholar]
- Davies, L.H.; Kasten, B.B.; Benny, P.D.; Arrowsmith, R.L.; Ge, H.; Pascu, S.I.; Botchway, S.W.; Clegg, W.; Harrington, R.W.; Higham, L.J. Re and 99mTc complexes of BodP3—Multi-modality imaging probes. Chem. Commun. 2014, 50, 15503–15505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, S.; Burke, B.P.; Davies, L.H.; Domarkas, J.; Wallis, J.F.; Waddell, P.G.; Waby, J.S.; Benoit, D.M.; Seymour, A.-M.; Cawthorne, C.; et al. Structurally optimized BODIPY derivatives for imaging of mitochondrial dysfunction in cancer and heart cells. Chem. Commun. 2016, 52, 7114–7117. [Google Scholar] [CrossRef] [PubMed]
- Blaurock, S.; Hey-Hawkins, E. Unexpected reduction of [Cp*TaCl4(PH2R)] (R = But, Cy, Ad, Ph, 2,4,6-Me3C6H2; Cp* = C5Me5) by reaction with DBU—Molecular structure of [(DBU)H][Cp*TaCl4] (DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene). Z. Anorg. Allg. Chem. 2002, 628, 37–40. [Google Scholar] [CrossRef]
- Baudler, M.; Gruner, C.; Tschaebunin, H.; Hahn, J. Contributions to the chemistry of phosphorus. 110. 1,2-Di-tert-butyldiphosphine and tert-butyldiphosphine. Chem. Ber. 1982, 115, 1739–1745. [Google Scholar] [CrossRef]
- Roering, A.J.; Maddox, A.F.; Elrod, L.T.; Chan, S.M.; Ghebreab, M.B.; Donovan, K.L.; Davidson, J.J.; Hughes, R.P.; Shalumova, T.; MacMillan, S.N.; et al. General Preparation of (N3N)ZrX (N3N = N(CH2CH2NSiMe3)33−) Complexes from a Hydride Surrogate. Organometallics 2009, 28, 573–581. [Google Scholar] [CrossRef]
- Leshinski, S.; Shalumova, T.; Tanski, J.M.; Waterman, R. Insertion reactions involving a triamidoamine-supported zirconium complex. Dalton Trans. 2010, 39, 9073–9078. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bange, C.A.; Mucha, N.T.; Cousins, M.E.; Gehsmann, A.C.; Singer, A.; Truax, T.; Higham, L.J.; Waterman, R. Zirconium-Catalyzed Alkene Hydrophosphination and Dehydrocoupling with an Air-Stable, Fluorescent Primary Phosphine. Inorganics 2016, 4, 26. https://doi.org/10.3390/inorganics4030026
Bange CA, Mucha NT, Cousins ME, Gehsmann AC, Singer A, Truax T, Higham LJ, Waterman R. Zirconium-Catalyzed Alkene Hydrophosphination and Dehydrocoupling with an Air-Stable, Fluorescent Primary Phosphine. Inorganics. 2016; 4(3):26. https://doi.org/10.3390/inorganics4030026
Chicago/Turabian StyleBange, Christine A., Neil T. Mucha, Morgan E. Cousins, Abigail C. Gehsmann, Anna Singer, Taylor Truax, Lee J. Higham, and Rory Waterman. 2016. "Zirconium-Catalyzed Alkene Hydrophosphination and Dehydrocoupling with an Air-Stable, Fluorescent Primary Phosphine" Inorganics 4, no. 3: 26. https://doi.org/10.3390/inorganics4030026
APA StyleBange, C. A., Mucha, N. T., Cousins, M. E., Gehsmann, A. C., Singer, A., Truax, T., Higham, L. J., & Waterman, R. (2016). Zirconium-Catalyzed Alkene Hydrophosphination and Dehydrocoupling with an Air-Stable, Fluorescent Primary Phosphine. Inorganics, 4(3), 26. https://doi.org/10.3390/inorganics4030026