
Synthesis and Characterization of a Sulfonyl- and Iminophosphoryl-Functionalized Methanide and Methandiide


Abstract
:
1. Background
2. Results
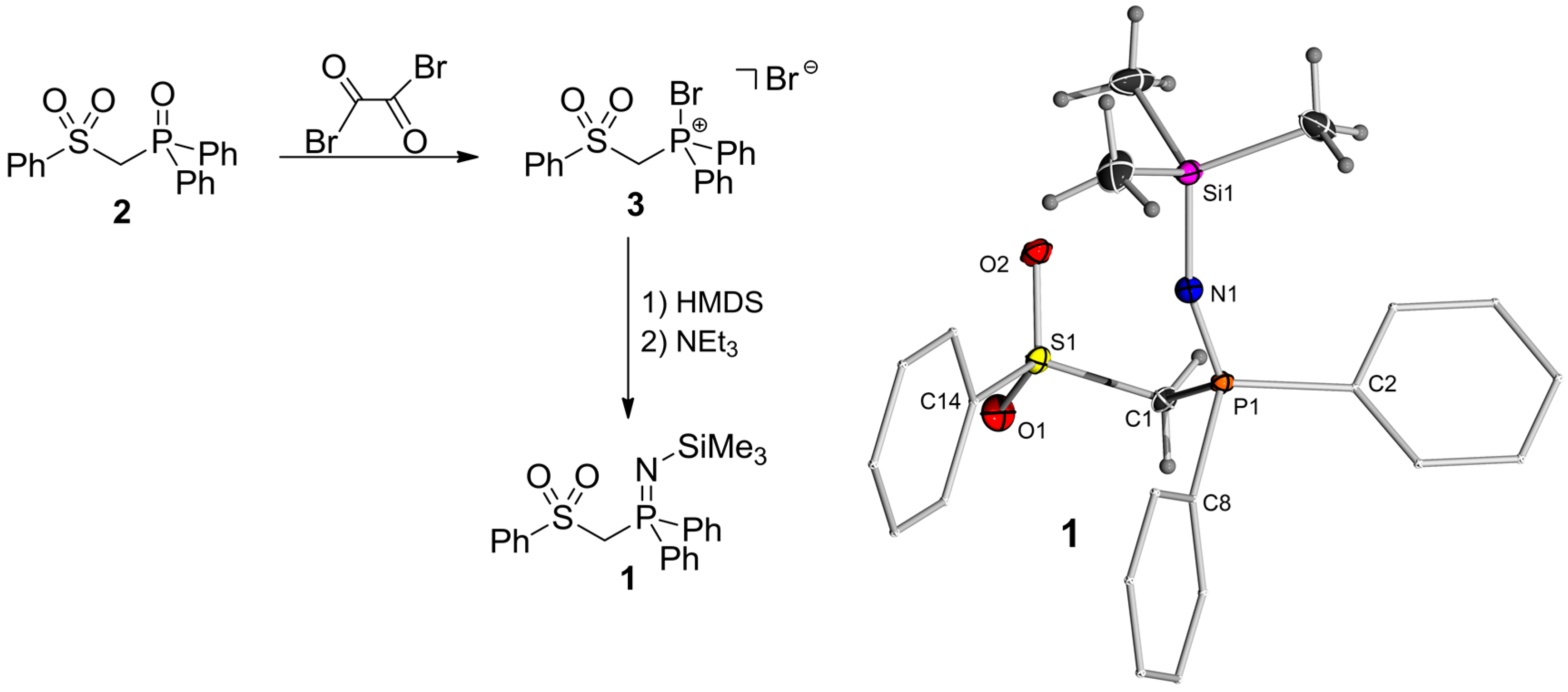
2.1. Synthesis of the Protonated Precursor 1
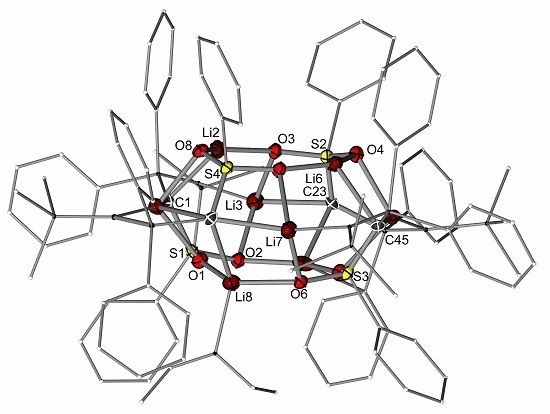
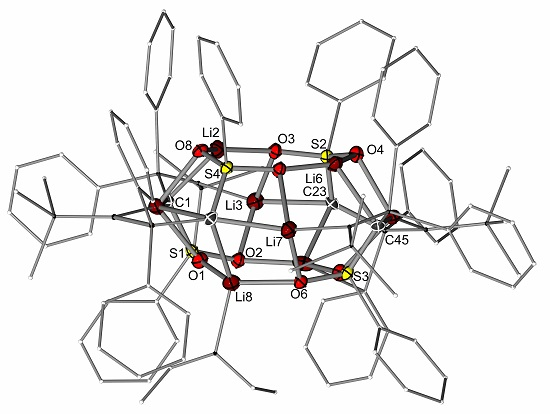
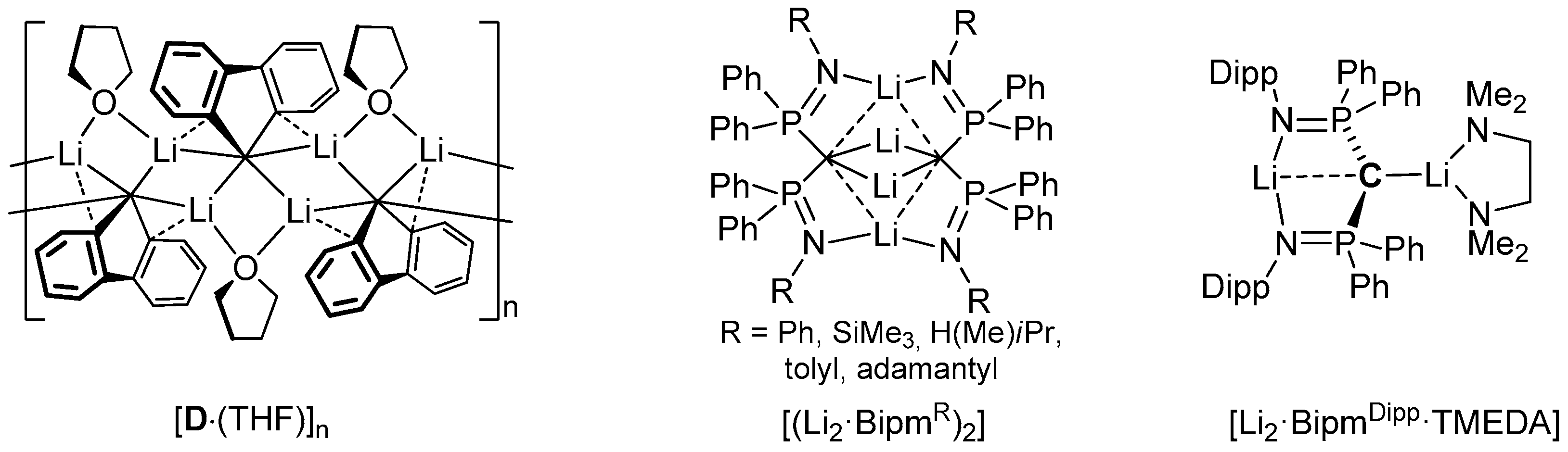
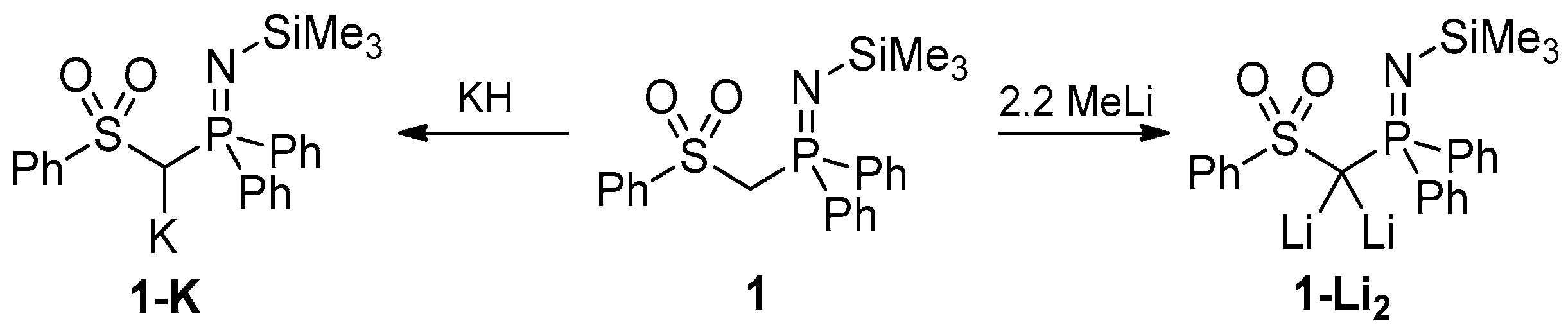
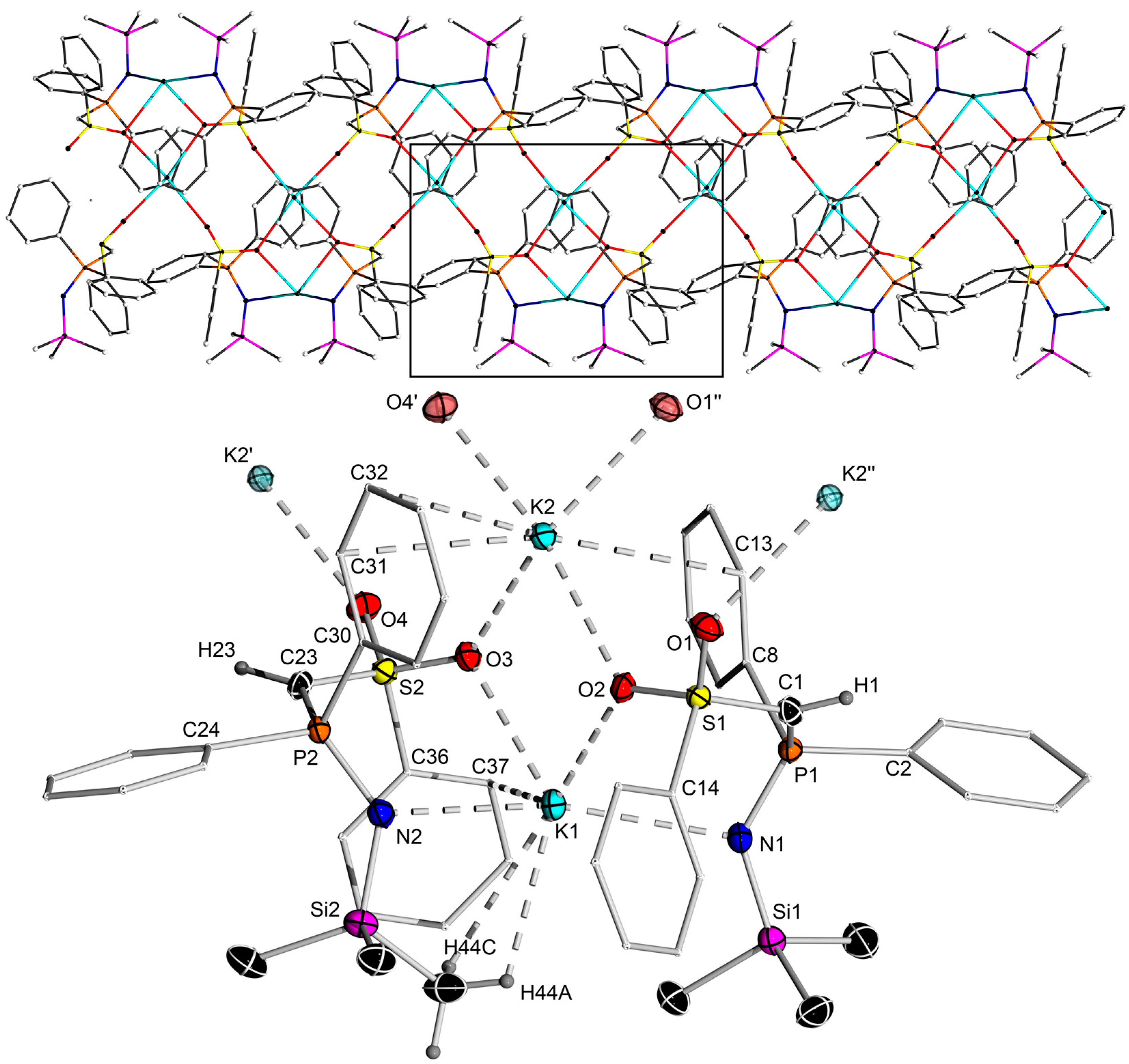
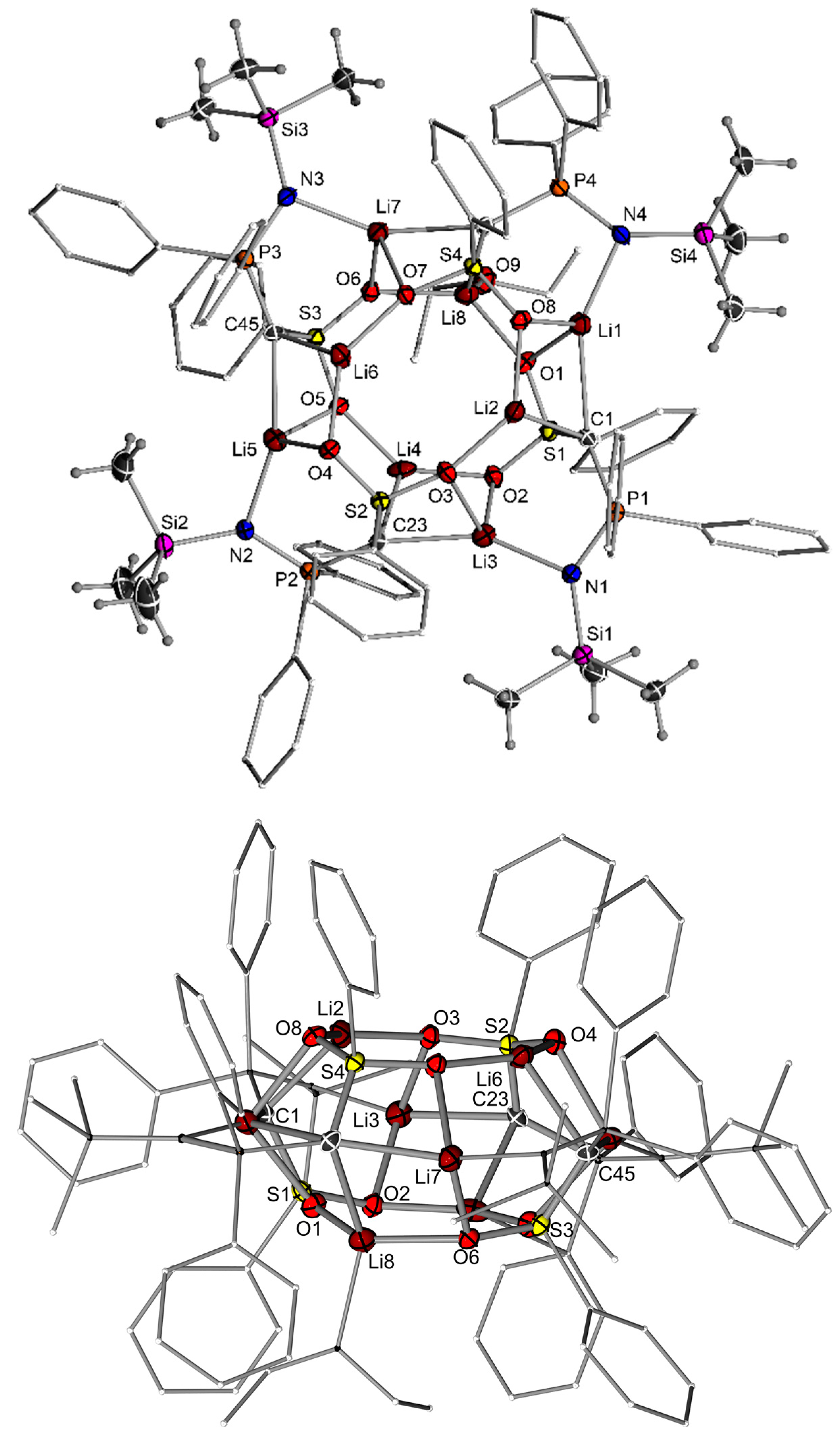
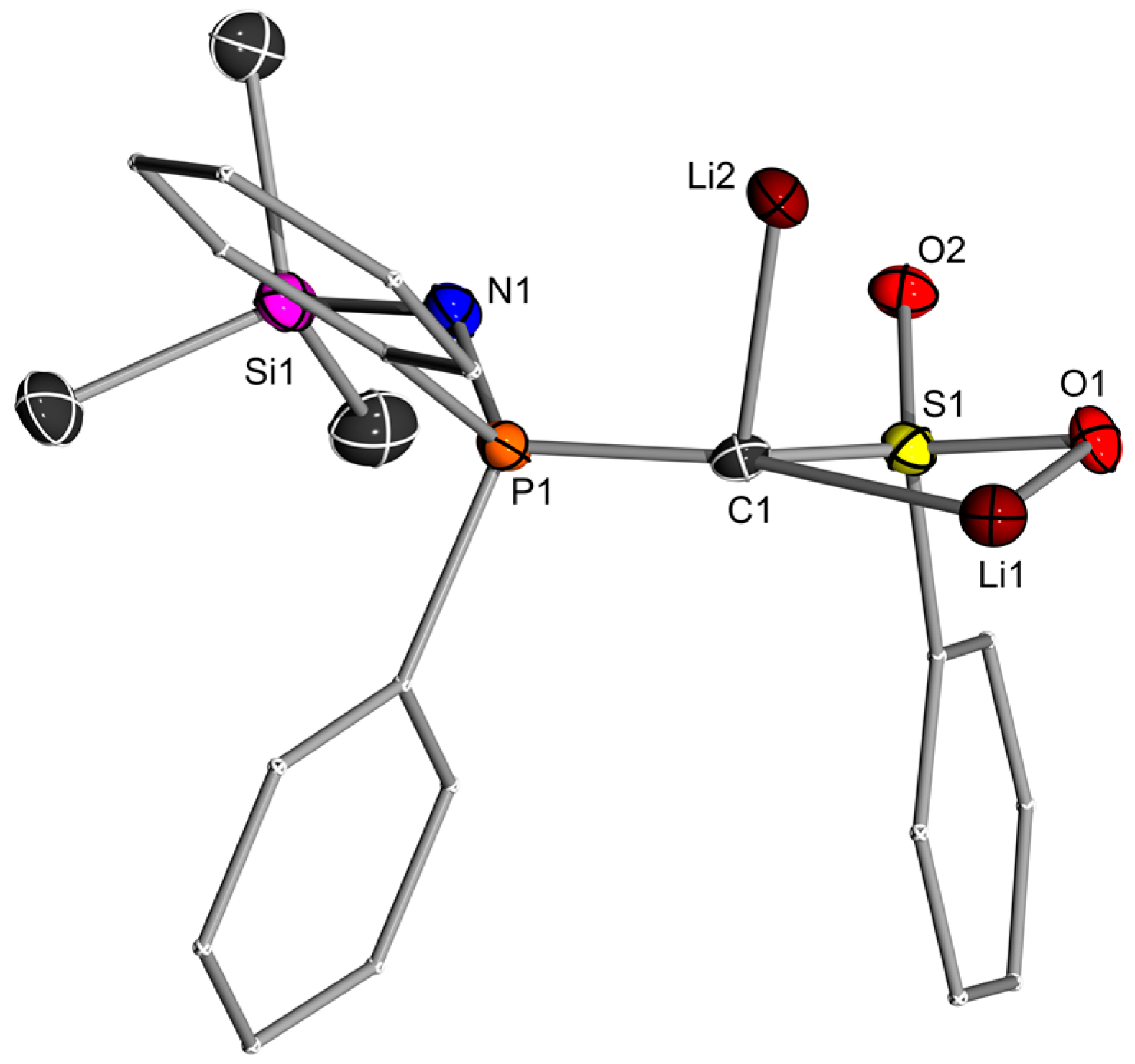
2.2. Preparation of Methanide 1-K and Methandiide 1-Li2
3. Experimental Section
3.1. General Procedures
3.2. Syntheses
3.3. X-ray Crystallography
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gessner, V.H.; Becker, J.; Feichtner, K.-S. Carbene Complexes Based on Dilithium Methandiides. Eur. J. Inorg. Chem. 2015, 2015, 1841–1859. [Google Scholar] [CrossRef]
- Harder, S. Geminal dianions stabilized by phosphonium substituents. Coord. Chem. Rev. 2011, 255, 1252–1267. [Google Scholar] [CrossRef]
- Liddle, S.T.; Mills, D.P.; Wooles, A.J. Early metal bis(phosphorus-stabilised)carbene chemistry. Chem. Soc. Rev. 2011, 40, 2164–2176. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, L.; Jansen, G.; Harder, S. Synthesis, Structure, and Reactivity of a Stabilized Calcium Carbene: R2CCa. J. Am. Chem. Soc. 2006, 128, 14676–14684. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, L.; Harder, S. Isolation of an Intermediate in the Catalytic Trimerization of Isocyanates by a Monomeric Calcium Carbene with Chelating Iminophosphorane Substituents. Organometallics 2007, 26, 2144–2148. [Google Scholar] [CrossRef]
- Thirumoorthi, R.; Chivers, T.; Vargas-Baca, I. S,C,S-Pnictogen bonding in pincer complexes of the methanediide [C(Ph2PS)2]2−. Dalton Trans. 2011, 40, 8086–8088. [Google Scholar] [CrossRef] [PubMed]
- Chivers, T.; Konu, J.; Thirumoorthi, R. PCP-bridged chalcogen-centred anions: Coordination chemistry and carbon-based reactivity. Dalton Trans. 2012, 41, 4283–4295. [Google Scholar] [CrossRef] [PubMed]
- Cantat, T.; Mézailles, N.; Ricard, L.; Jean, Y.; Le Floch, P. A Bis(thiophosphinoyl)methanediide Palladium Complex: Coordinated Dianion or Nucleophilic Carbene Complex? Angew. Chem. Int. Ed. 2004, 43, 6382–6385. [Google Scholar] [CrossRef] [PubMed]
- Gessner, V.H.; Meier, F.; Uhrich, D.; Kaupp, M. Synthesis and Bonding in Carbene Complexes of an Unsymmetrical Dilithio Methandiide: A Combined Experimental and Theoretical Study. Chem. Eur. J. 2013, 19, 16729–16739. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.P.K.; McDonald, R.; Cavell, R.G. The first hafnium methandiide complexes: The assembly of an entire triad of group 4 metal “pincer” bis(phosphinimine) complexes possessing the M=C carbene–ylide structure. Chem. Commun. 2000, 6, 481–482. [Google Scholar] [CrossRef]
- Leung, W.-P.; So, C.-W.; Wang, J.-Z.; Mak, T.C.W. A novel synthesis of metallogermacyclopropane and molybdenum bis(iminophosphorano)carbene complexes from bisgermavinylidene. Chem. Commun. 2003, 2, 248–249. [Google Scholar] [CrossRef]
- Cadierno, V.; Diez, J.; García-Álvarez, J.; Gimeno, J. (η6-Arene)−Ruthenium(II) Complexes Containing Methanide and Methandiide Anions of Ph2P(=S)CH2P(=NR)Ph2: Unprecedented Insertion of Isocyanide into a Ruthenium−Carbene Bond. Organometallics 2008, 27, 1809–1822. [Google Scholar] [CrossRef]
- Aparna, K.; Ferguson, M.; Cavell, R.G. A Monomeric Samarium Bis(Iminophosphorano) Chelate Complex with a S=C Bond. J. Am. Chem. Soc. 2000, 122, 726–727. [Google Scholar] [CrossRef]
- Cantat, T.; Arliguie, T.; Noël, A.; Thuéry, P.; Ephritikhine, M.; Le Floch, P.; Mézailles, N. The U=C Double Bond: Synthesis and Study of Uranium Nucleophilic Carbene Complexes. J. Am. Chem. Soc. 2009, 131, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.J.; Mills, D.P.; McMaster, J.; Moro, F.; Davies, E.S.; Lewis, W.; Blake, A.J.; Liddle, S.T. Uranium–Carbon Multiple Bonding: Facile Access to the Pentavalent Uranium Carbene [U{C(PPh2NSiMe3)2}(Cl)2(I)] and Comparison of UV=C and UIV=C Bonds. Angew. Chem. Int. Ed. 2011, 50, 2383–2386. [Google Scholar] [CrossRef] [PubMed]
- Mills, D.P.; Moro, F.; McMaster, J.; van Slageren, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A delocalized arene-bridged diuranium single-molecule magnet. Nat. Chem. 2011, 3, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Aparna, K.; Babu, R.P.K.; McDonald, R.; Cavell, R.G. [Ph2P(NSiMe3)]2CLi2: A Dilithium Dianionic Methanide Salt with an Unusual Li4C2 Cluster Structure. Angew. Chem. Int. Ed. 1999, 38, 1483–1484. [Google Scholar]
- Ong, C.M.; Stephan, D.W. Lithiations of Bis-diphenyl-N-trimethylsilylphosphiniminomethane: An X-ray Structure of a 1,1-Dilithiomethane Derivative. J. Am. Chem. Soc. 1999, 121, 2939–2940. [Google Scholar] [CrossRef]
- Gais, H.-J.; Vollhardt, J. Solid-state and solution structure of dilithium trimethyl[(phenylsulfonyl)methyl]silane, a true dilithiomethane derivative. J. Am. Chem. Soc. 1988, 110, 978–980. [Google Scholar] [CrossRef]
- Linti, G.; Rodig, A.; Pritzkow, H. 9,9-Dilithiofluorene: The First Crystal-Structure Analysis of an α,α-Dilithiated Hydrocarbon. Angew. Chem. Int. Ed. 2002, 41, 4503–4505. [Google Scholar] [CrossRef]
- Müller, J.F.K.; Neuburger, M.; Spingler, B. Structural Investigation of a Dilithiated Phosphonate in the Solid State. Angew. Chem. Int. Ed. 1999, 38, 92–94. [Google Scholar] [CrossRef]
- Cavell, R.G.; Kamalesh Babu, R.P.; Kasani, A.; McDonald, R. Novel Metal-Carbon Multiply Bonded Twelve-Electron Complexes of Ti and Zr Supported by a Bis(Phosphoranimine) Chelate. J. Am. Chem. Soc. 1999, 121, 5805–5806. [Google Scholar] [CrossRef]
- Cantat, T.; Ricard, L.; Le Floch, P.; Mézailles, N. Phosphorus-Stabilized Geminal Dianions. Organometallics 2006, 25, 4965–4976. [Google Scholar] [CrossRef]
- Chen, J.-H.; Guo, J.; Li, Y.; So, C.-W. Synthesis and Structure of [Li2C(PPh2=NSiMe3)(PPh2=S)]: A Geminal Dianionic Ligand. Organometallics 2009, 28, 4617–4620. [Google Scholar] [CrossRef]
- Heuclin, H.; Fustier-Boutignon, M.; Ho, S.Y.-F.; Le Goff, X.-F.; Carenco, S.; So, C.-W.; Mézailles, N. Synthesis of Phosphorus(V)-Stabilized Geminal Dianions. The Cases of Mixed P=X/P→BH3 (X = S, O) and P=S/SiMe3 Derivatives. Organometallics 2013, 32, 498–508. [Google Scholar] [CrossRef]
- Schröter, P.; Gessner, V.H. Tetrahedral versus Planar Four-Coordinate Carbon: A Sulfonyl-Substituted Methandiide. Chem. Eur. J. 2012, 18, 11223–11227. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Gessner, V.H. Synthesis and Electronic Structure of Carbene Complexes Based on a Sulfonyl-Substituted Dilithio Methandiide. Organometallics 2014, 33, 1310–1317. [Google Scholar] [CrossRef]
- Becker, J.; Modl, T.; Gessner, V.H. Methandiide as Non-Innocent Ligand in Carbene Complexes: From the Electronic Structure to Bond Activation Reactions and Cooperative Catalysis. Chem. Eur. J. 2014, 20, 11295–11299. [Google Scholar] [CrossRef] [PubMed]
- Weismann, J.; Waterman, R.; Gessner, V.H. Metal-Ligand Cooperativity in a Methandiide Derived Iridium Carbene Complex. Chem. Eur. J. 2016, 22, 3846–3855. [Google Scholar] [CrossRef] [PubMed]
- Gessner, V.H.; Däschlein, C.; Strohmann, C. Structure Formation Principles and Reactivity of Organolithium Compounds. Chem. Eur. J. 2009, 15, 3320–3335. [Google Scholar] [CrossRef] [PubMed]
- Harrison-Marchand, A.; Mongin, F. Mixed AggregAte (MAA): A Single Concept for All Dipolar Organometallic Aggregates. 1. Structural Data. Chem. Rev. 2013, 113, 7470–7562. [Google Scholar] [CrossRef] [PubMed]
- Stey, T.; Stalke, D. Lead structures in lithium organic chemistry. In The Chemistry of Organolithium Compounds; Rappoport, Z., Marek, I., Eds.; John Wiley & Sons: New York, NY, USA, 2004; pp. 47–120. [Google Scholar]
- Stucky, G.D.; Eddy, M.M.; Harrison, W.H.; Lagow, R.; Kawa, H.; Cox, D.E. Some Observations Concerning the Structure of Dilithiomethane. J. Am. Chem. Soc. 1990, 112, 2425–2427. [Google Scholar] [CrossRef]
- Feichtner, K.-S.; Gessner, V.H. Synthesis and stability of Li/Cl carbenoids based on bis(iminophosphoryl)methanes. Dalton Trans. 2014, 43, 14399–14408. [Google Scholar] [CrossRef] [PubMed]
- Demange, M.; Boubekeur, L.; Auffrant, A.; Mézailles, N.; Ricard, L.; Le Goff, R.; Le Floch, P. A new and convenient approach towards bis(iminophosphoranyl)methane ligands and their dicationic, cationic, anionic and dianionic derivatives. New J. Chem. 2006, 30, 1745–1754. [Google Scholar] [CrossRef]
- Cooper, O.J.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Synthesis and structure of [U{C(PPh2NMes)2}2] (Mes = 2,4,6-Me3C6H2): A homoleptic uranium bis(carbene) complex with two formal U=C double bonds. Dalton Trans. 2010, 39, 5074–5076. [Google Scholar] [CrossRef] [PubMed]
- Sindlinger, C.P.; Stasch, A. Syntheses, structures and flexible coordination of sterically demanding di- and “tri”-lithiated methandiides. Dalton Trans. 2014, 43, 14334–14345. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Noll, B.C.; Henderson, K.W. Structural Characterization and Dynamic Solution Behavior of the Disodio and Lithio−Sodio Geminal Organodimetallics [{{Ph2P(Me3Si)N}2CNa2}2] and [{{Ph2P(Me3Si)N}2CLiNa}2]. Organometallics 2006, 25, 4072–4074. [Google Scholar] [CrossRef]
- Orzechowski, L.; Jansen, G.; Harder, S. Methandiide Complexes (R2CM2) of the Heavier Alkali Metals (M = Potassium, Rubidium, Cesium): Reaching the Limit? Angew. Chem. Int. Ed. 2009, 48, 3825–3829. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Carmichael, I.; Noll, B.C.; Henderson, K.W. Homo- and Heterodimetallic Geminal Dianions Derived from the Bis(phosphinimine) {Ph2P(NSiMe3)}2CH2 and the Alkali Metals Li, Na, and K. Chem. Eur. J. 2008, 14, 3939–3953. [Google Scholar] [CrossRef] [PubMed]
- Cooper, O.J.; Wooles, A.J.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A Monomeric Dilithio Methandiide with a Distorted trans-Planar Four-Coordinate Carbon. Angew. Chem. Int. Ed. 2010, 49, 5570–5573. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; Schleyer, P.v.R.; Seeger, R.; Pople, J.A. Stabilization of Planar Tetracoordinate Carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Feichtner, K.-S.; Englert, S.; Gessner, V.H. Preparation and Isolation of a Chiral Methandiide and its Application as Cooperative Ligand in Bond Activation. Chem. Eur. J. 2016, 22, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, K.; Müller-Bunz, H.; Gilheany, D. Direct evidence of a multicentre halogen bond: Unexpected contraction of the P–XXX–P fragment in triphenylphosphine dihalides. Chem. Commun. 2013, 49, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Mardersteig, H.G.; Meinel, L.; Nöth, H. N-Diphenylphosphino-triphenylphosphazen. Z. Anorg. Allg. Chem. 1969, 368, 254–261. [Google Scholar] [CrossRef]
- Müller, A.; Möhlen, M.; Neumüller, B.; Faza, N.; Massa, W.; Dehnicke, K. Die Kristallstrukturen der silylierten Phosphanimine Me3SiNP(c-C6H11)3 und (Me3SiNPPh2)2CH2. Z. Anorg. Allg. Chem. 1999, 625, 1748–1751. [Google Scholar] [CrossRef]
- Holink, E.; Stewart, J.C.; Wei, P.; Stephan, D.W. Ti and Zr bidentate bis-phosphinimide complexes. Dalton Trans. 2003, 3, 3968–3974. [Google Scholar] [CrossRef]
- Holthausen, M.H.; Mallov, I.; Stephan, D.W. Phosphinimine-substituted boranes and borenium ions. Dalton Trans. 2014, 43, 15201–15211. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.; Chivers, T.; Parvez, M.; Boeré, R.T. Stable Cubic Phosphorus-Containing Radicals. Angew. Chem. Int. Ed. 2004, 43, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
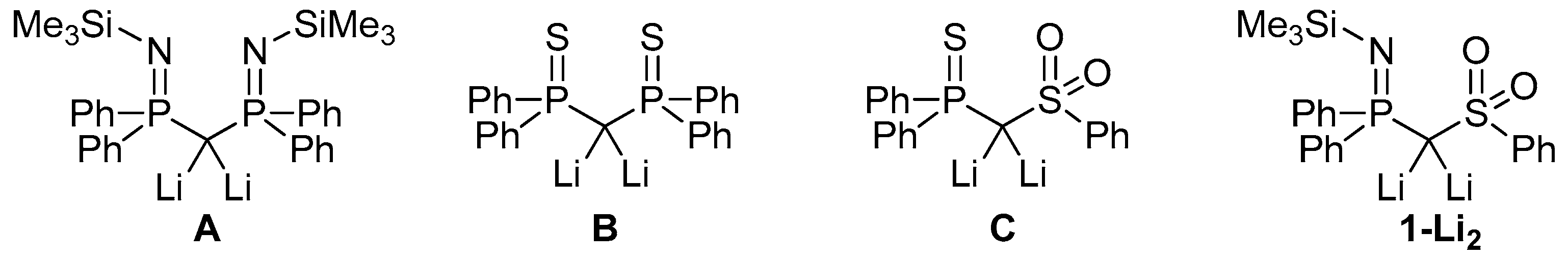{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | 1 | 1-K | 1-Li2 | C |
|---|---|---|---|---|
| C–S (Å) | 1.7807(16) | 1.638(3) | 1.608(3) | 1.613(3) |
| C–P (Å) | 1.8441(15) | 1.727(3) | 1.714(3) | 1.710(3) |
| P–N (Å) | 1.5229(13) | 1.566(2) | 1.586(3) | - |
| S–O (Å) | 1.4427(11) | 1.453(2) | 1.494(2) | 1.501(2) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feichtner, K.-S.; Gessner, V.H. Synthesis and Characterization of a Sulfonyl- and Iminophosphoryl-Functionalized Methanide and Methandiide. Inorganics 2016, 4, 40. https://doi.org/10.3390/inorganics4040040
Feichtner K-S, Gessner VH. Synthesis and Characterization of a Sulfonyl- and Iminophosphoryl-Functionalized Methanide and Methandiide. Inorganics. 2016; 4(4):40. https://doi.org/10.3390/inorganics4040040
Chicago/Turabian StyleFeichtner, Kai-Stephan, and Viktoria H. Gessner. 2016. "Synthesis and Characterization of a Sulfonyl- and Iminophosphoryl-Functionalized Methanide and Methandiide" Inorganics 4, no. 4: 40. https://doi.org/10.3390/inorganics4040040
APA StyleFeichtner, K.-S., & Gessner, V. H. (2016). Synthesis and Characterization of a Sulfonyl- and Iminophosphoryl-Functionalized Methanide and Methandiide. Inorganics, 4(4), 40. https://doi.org/10.3390/inorganics4040040