
Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction


 ,
,
Abstract
:1. Introduction
2. Results and Discussion
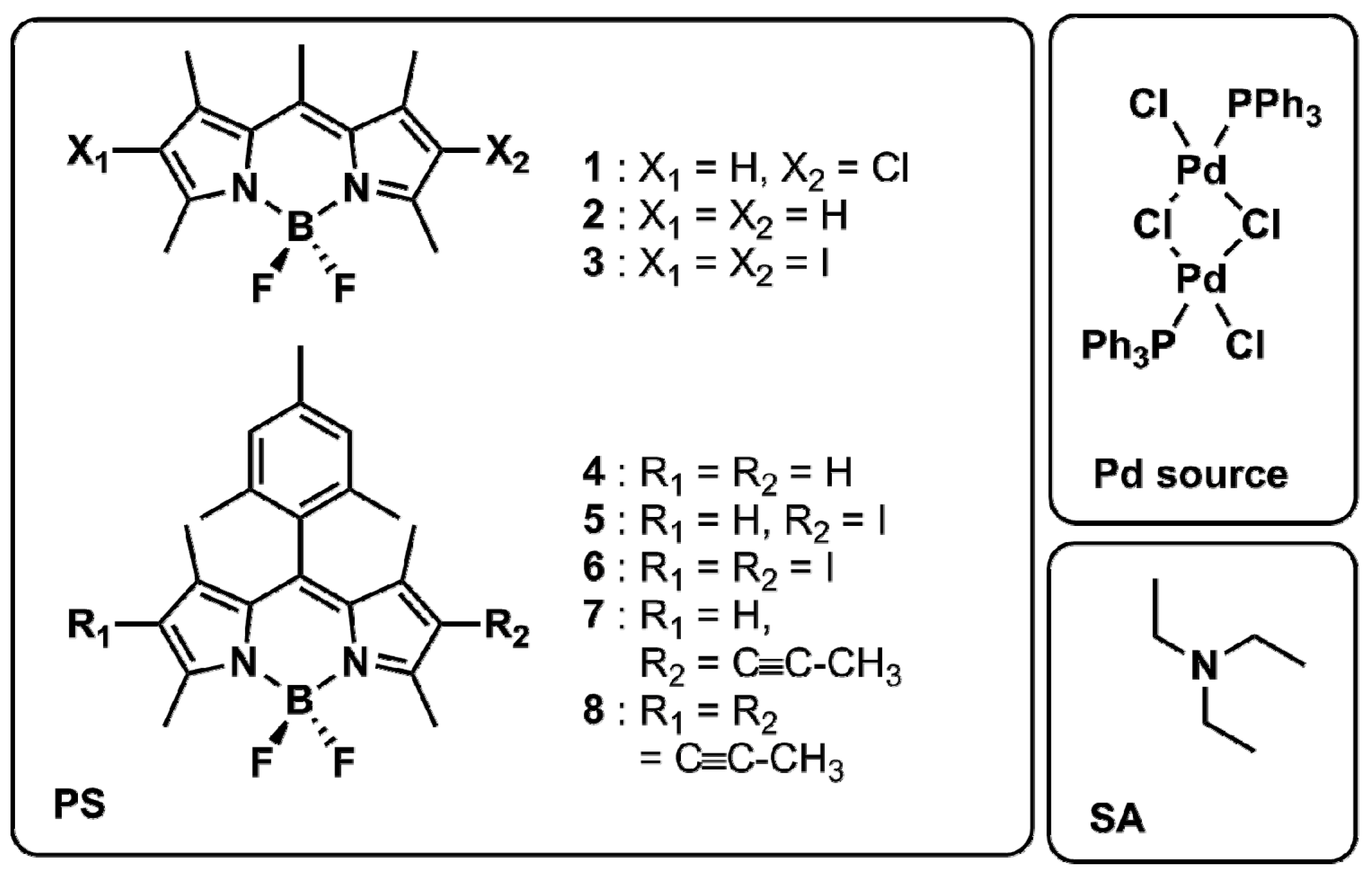
2.1. Photophysical Characterisation of the BODIPY Dyes
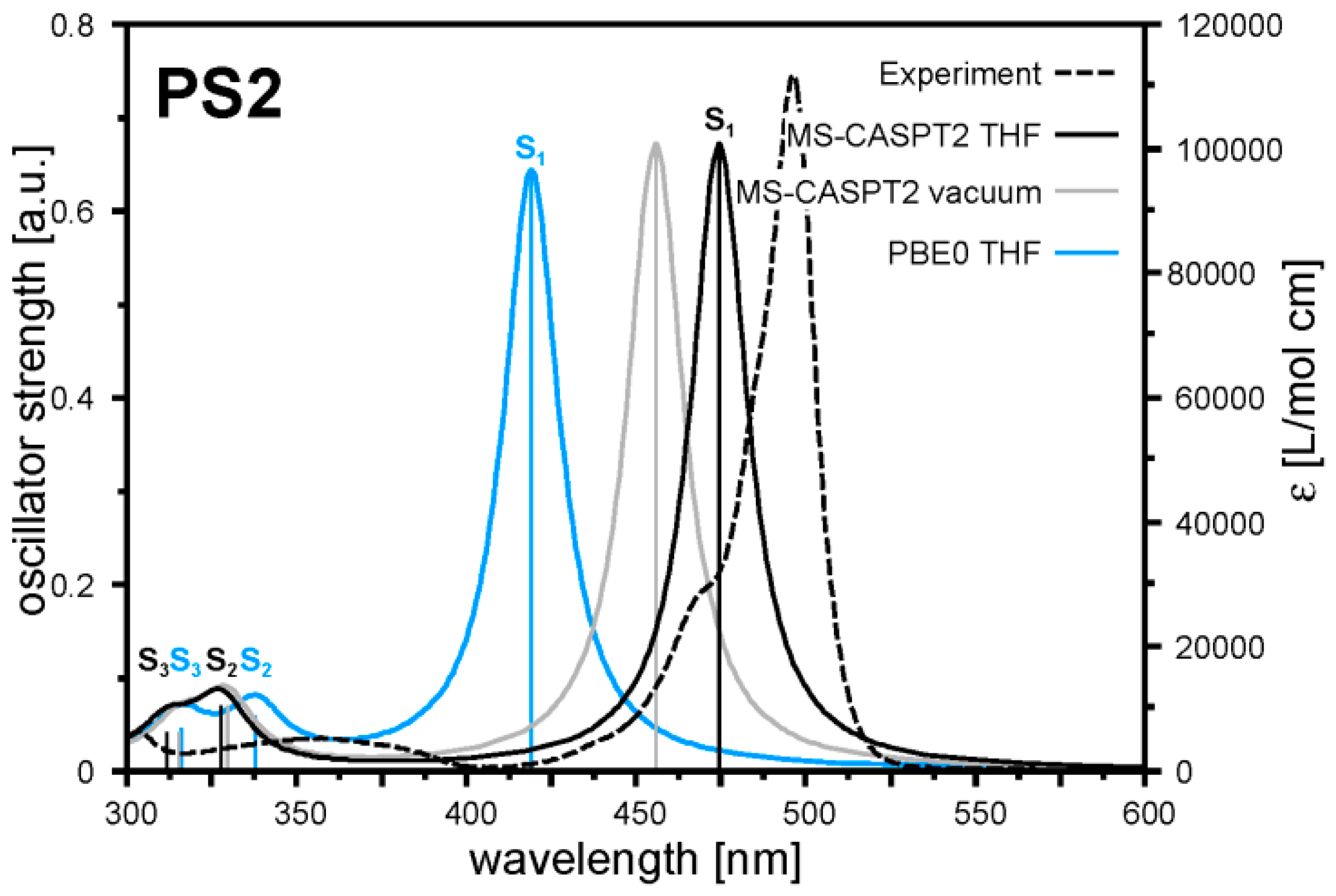
2.2. Quantum Chemical Calculations
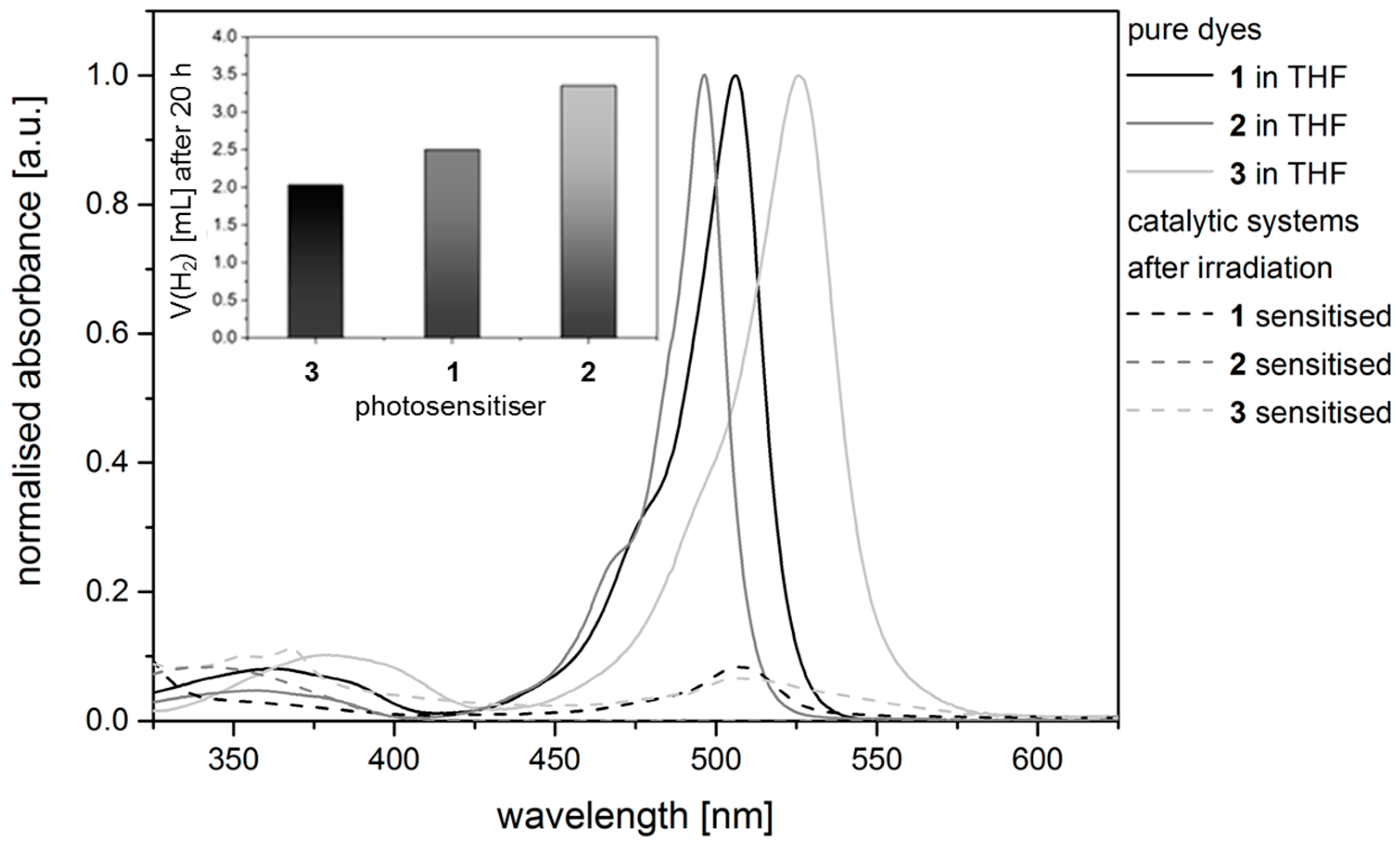
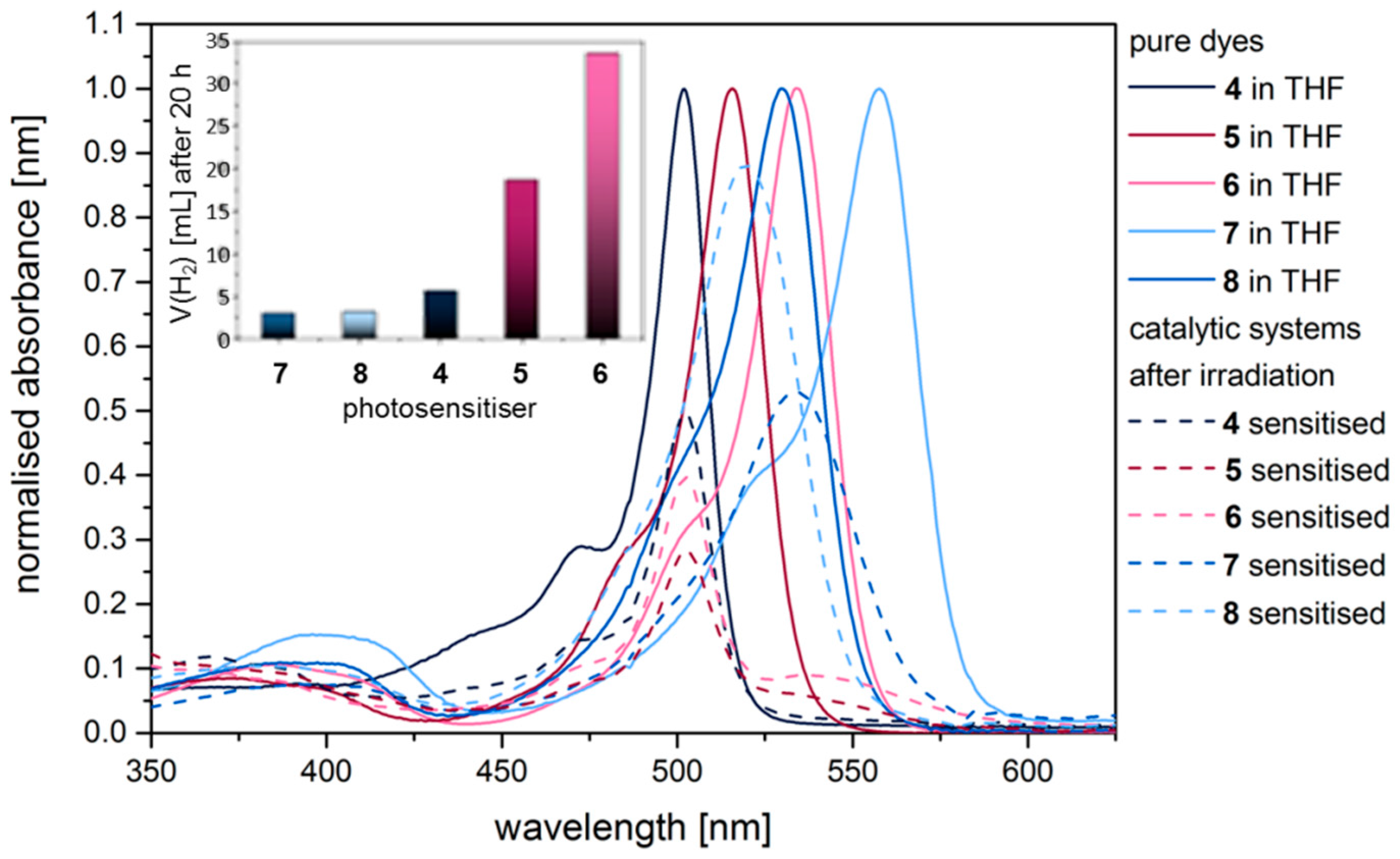
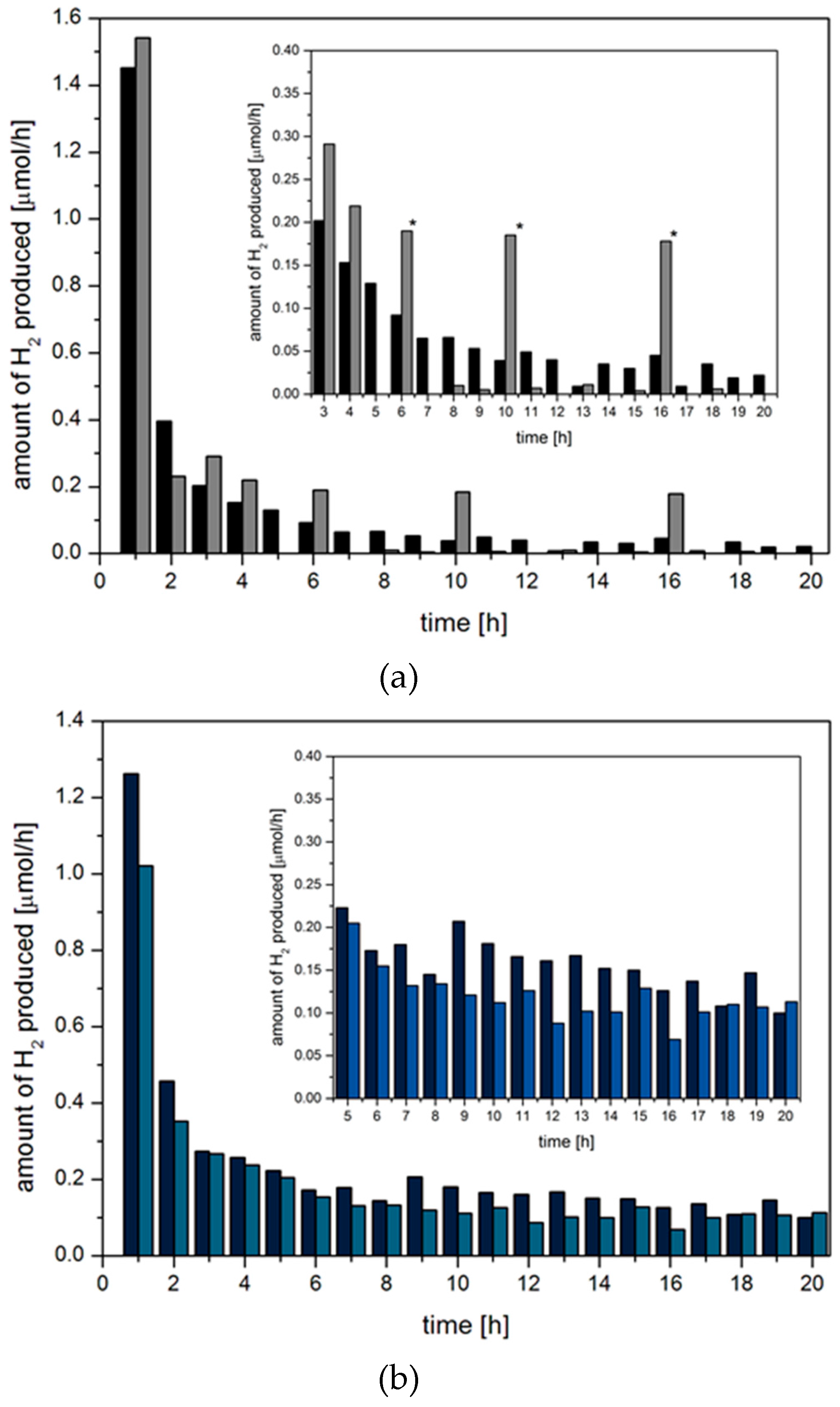
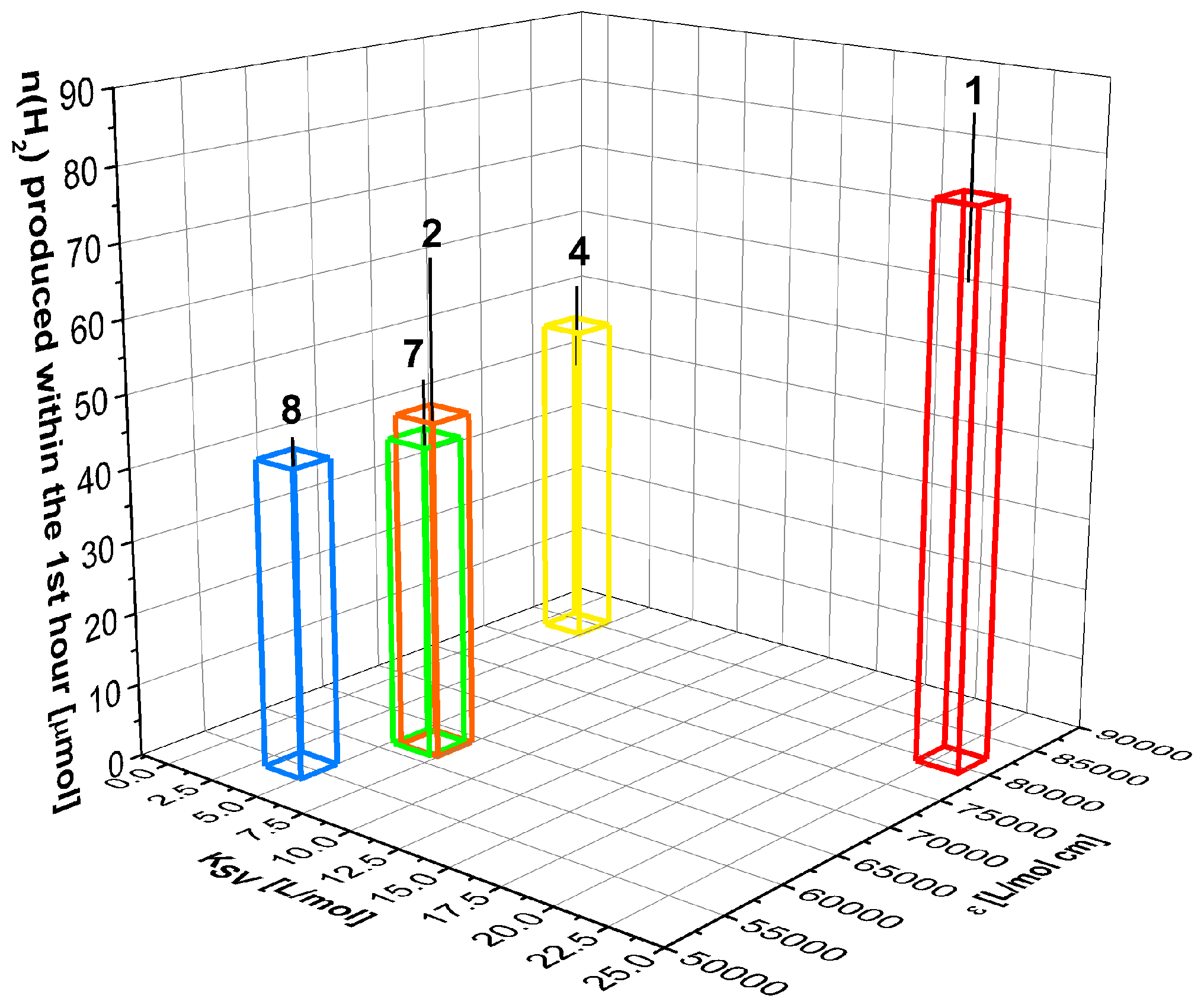
2.3. Hydrogen Evolution Experiments on BODIPY Sensitised Multicomponent Catalyst Systems
- (a)
- The Pd nanoparticle precursor [PdCl2(PPh3)]2 serves as a highly active water reduction catalyst at the onset of irradiation, but degrades during the first hour to the less active Pd nanoparticles which were confirmed via transmission electron microscopy after reaction (Scheme 1a).
- (b)
- [PdCl2(PPh3)]2 is an inactive precursor and degrades to highly-active Pd nanoparticles within the first seconds of irradiation. During the first hour of irradiation performance of these nanoparticles slightly decreases due to aggregation (Ostwald ripening) or blocking of active centres by a component present in solution (Scheme 1b).
2.4. Correlation of Photophysical Properties and Performance of BODIPY Dyes
3. Materials and Methods
3.1. General
3.2. Hydrogen Evolution Experiments
3.3. Stationary Optical Spectroscopy
3.4. Time-Dependent Emission Spectroscopy
3.5. ns Time-Resolved Transient Absorption Spectroscopy
3.6. fs Time-Resolved Transient Absorption Spectroscopy
3.7. Quantum Chemical Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Esswein, A.J.; Nocera, D.G. Hydrogen Production by Molecular Photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Eckenhoff, W.T.; Eisenberg, R. Molecular systems for light driven hydrogen production. Dalton Trans. 2012, 41, 13004–13021. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Hong, D.; Yamada, Y. Bioinspired Photocatalytic Water Reduction and Oxidation with Earth-Abundant Metal Catalysts. J. Phys. Chem. Lett. 2013, 4, 3458–3467. [Google Scholar] [CrossRef]
- Lakadamyali, F.; Kato, M.; Muresan, N.M.; Reisner, E. Selective Reduction of Aqueous Protons to Hydrogen with a Synthetic Cobaloxime Catalyst in the Presence of Atmospheric Oxygen. Angew. Chem. Int. Ed. 2012, 51, 9381–9384. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Mersch, D.; Reisner, E. Photocatalytic Hydrogen Evolution with a Hydrogenase in a Mediator-Free System under High Levels of Oxygen. Angew. Chem. Int. Ed. 2013, 52, 12313–12316. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Yang, X.; Li, J.; Zhang, F.; Sun, L. Co-sensitization of Organic Dyes for Efficient Dye-Sensitized Solar Cells. ChemSusChem 2013, 6, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, M.; Li, C.; Li, X.; Dong, J.; Sun, L. Photochemical H2 production with noble-metal-free molecular devices comprising a porphyrin photosensitizer and a cobaloxime catalyst. Chem. Commun. 2010, 46, 8806–8808. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; McNamara, W.R.; Eum, M.-S.; Holland, P.L.; Eisenberg, R. A Nickel Thiolate Catalyst for the Long-Lived Photocatalytic Production of Hydrogen in a Noble-Metal-Free System. Angew. Chem. Int. Ed. 2012, 51, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Lazarides, T.; McCormick, T.M.; Du, P.; Luo, G.; Lindley, B.; Eisenberg, R. Making Hydrogen from Water Using a Homogeneous System Without Noble Metals. J. Am. Chem. Soc. 2009, 131, 9192–9194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hong, J.; Zheng, J.; Huang, Z.; Zhou, J.; Xu, R. Nickel–Thiolate Complex Catalyst Assembled in One Step in Water for Solar H2 Production. J. Am. Chem. Soc. 2011, 133, 20680–20683. [Google Scholar] [CrossRef] [PubMed]
- Hartley, C.L.; DiRisio, R.J.; Screen, M.E.; Mayer, K.J.; McNamara, W.R. Iron Polypyridyl Complexes for Photocatalytic Hydrogen Generation. Inorg. Chem. 2016, 55, 8865–8870. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, R.P.; McCormick, T.M.; Lazarides, T.; Wilson, K.C.; Eisenberg, R.; McCamant, D.W. Intersystem Crossing in Halogenated BODIPY Chromophores Used for Solar Hydrogen Production. J. Phys. Chem. Lett. 2011, 2, 223–227. [Google Scholar] [CrossRef]
- Bartelmess, J.; Francis, A.J.; El Roz, K.A.; Castellano, F.N.; Weare, W.W.; Sommer, R.D. Light-Driven Hydrogen Evolution by BODIPY-Sensitized Cobaloxime Catalysts. Inorg. Chem. 2014, 53, 4527–4534. [Google Scholar] [CrossRef] [PubMed]
- Manton, J.C.; Long, C.; Vos, J.G.; Pryce, M.T. A photo- and electrochemical investigation of BODIPY-cobaloxime complexes for hydrogen production, coupled with quantum chemical calculations. Phys. Chem. Chem. Phys. 2014, 16, 5229–5236. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Fang, K.; Wu, J.-H.; Dai, J.-C.; Zhao, Q.-H. Noble-metal-free BODIPY-cobaloxime photocatalysts for visible-light-driven hydrogen production. Phys. Chem. Chem. Phys. 2014, 16, 23884–23894. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Fang, K.; Wu, J.-H.; Mo, J. Photocatalytic water reduction from a noble-metal-free molecular dyad based on a thienyl-expanded BODIPY photosensitizer. Chem. Commun. 2015, 51, 12361–12364. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.-G.; Lu, H.; Zhang, X.-L.; Dai, J.-C.; Wu, J.-H.; Wu, J.-J. The relationship between the boron dipyrromethene (BODIPY) structure and the effectiveness of homogeneous and heterogeneous solar hydrogen-generating systems as well as DSSCs. Phys. Chem. Chem. Phys. 2015, 17, 9716–9729. [Google Scholar] [CrossRef] [PubMed]
- Lazarides, T.; McCormick, T.M.; Wilson, K.C.; Lee, S.; McCamant, D.W.; Eisenberg, R. Sensitizing the Sensitizer: The Synthesis and Photophysical Study of BODIPY–Pt(II)(diimine)(dithiolate) Conjugates. J. Am. Chem. Soc. 2011, 133, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Sabatini, R.P.; Fu, W.-F.; Eum, M.-S.; Brennessel, W.W.; Wang, L.; McCamant, D.W.; Eisenberg, R. Light-driven generation of hydrogen: New chromophore dyads for increased activity based on BODIPY dye and Pt(diimine)(dithiolate) complexes. Proc. Natl. Acad. Sci. USA 2015, 112, E3987–E3996. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, R.P.; Lindley, B.; McCormick, T.M.; Lazarides, T.; Brennessel, W.W.; McCamant, D.W.; Eisenberg, R. Efficient Bimolecular Mechanism of Photochemical Hydrogen Production Using Halogenated Boron-Dipyrromethene (BODIPY) Dyes and a Bis(dimethylglyoxime) Cobalt(III) Complex. J. Phys. Chem. B 2016, 120, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Dura, L.; Ahrens, J.; Pohl, M.-M.; Höfler, S.; Bröring, M.; Beweries, T. Design of BODIPY Dyes as Photosensitisers in Multicomponent Catalyst Systems for Light-Driven Hydrogen Production. Chem. Eur. J. 2015, 21, 13549–13552. [Google Scholar] [CrossRef] [PubMed]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O. Ab Initio Methods in Quantum Chemistry II; Wiley-VCH: Chichester, UK, 1987. [Google Scholar]
- Finley, J.; Malmqvist, P.-Å.; Roos, B.O.; Serrano-Andrés, L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. [Google Scholar] [CrossRef]
- Lincoln, R.; Greene, L.E.; Krumova, K.; Ding, Z.; Cosa, G. Electronic Excited State Redox Properties for BODIPY Dyes Predicted from Hammett Constants: Estimating the Driving Force of Photoinduced Electron Transfer. J. Phys. Chem. A 2014, 118, 10622–10630. [Google Scholar] [CrossRef] [PubMed]
- Bonardi, L.; Ulrich, G.; Ziessel, R. Tailoring the Properties of Boron–Dipyrromethene Dyes with Acetylenic Functions at the 2,6,8 and 4-B Substitution Positions. Org. Lett. 2008, 10, 2183–2186. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Guo, H.; Wu, W.; Ji, S.; Zhao, J. Organic Triplet Sensitizer Library Derived from a Single Chromophore (BODIPY) with Long-Lived Triplet Excited State for Triplet-Triplet Annihilation Based Upconversion. J. Org. Chem. 2011, 76, 7056–7064. [Google Scholar] [CrossRef] [PubMed]
- Kölmel, D.K.; Hörner, A.; Castaneda, J.A.; Ferencz, J.A.P.; Bihlmeier, A.; Nieger, M.; Bräse, S.; Padilha, L.A. Linear and Nonlinear Optical Spectroscopy of Fluoroalkylated BODIPY Dyes. J. Phys. Chem. C 2016, 120, 4538–4545. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent bodipy dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. 2008, 120, 1184–1201. [Google Scholar] [CrossRef] [PubMed]
- Chibani, S.; Le Guennic, B.; Charaf-Eddin, A.; Laurent, A.D.; Jacquemin, D. Revisiting the optical signatures of BODIPY with ab initio tools. Chem. Sci. 2013, 4, 1950–1963. [Google Scholar] [CrossRef]
- Chibani, S.; Laurent, A.D.; Le Guennic, B.; Jacquemin, D. Improving the Accuracy of Excited-State Simulations of BODIPY and Aza-BODIPY Dyes with a Joint SOS-CIS(D) and TD-DFT Approach. J. Chem. Theory Comput. 2014, 10, 4574–4582. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. PhysChemChemPhys 2014, 16, 14334–14356. [Google Scholar]
- Momeni, M.R.; Brown, A. Why Do TD-DFT Excitation Energies of BODIPY/Aza-BODIPY Families Largely Deviate from Experiment? Answers from Electron Correlated and Multireference Methods. J. Chem. Theory Comput. 2015, 11, 2619–2632. [Google Scholar] [CrossRef] [PubMed]
- Erten-Ela, S.; Yilmaz, M.D.; Icli, B.; Dede, Y.; Icli, S.; Akkaya, E.U. A Panchromatic Boradiazaindacene (BODIPY) Sensitizer for Dye-Sensitized Solar Cells. Org. Lett. 2008, 10, 3299–3302. [Google Scholar]
- Kolemen, S.; Cakmak, Y.; Erten-Ela, S.; Altay, Y.; Brendel, J.; Thelakkat, M.; Akkaya, E.U. Solid-State Dye-Sensitized Solar Cells Using Red and Near-IR Absorbing BODIPY Sensitizers. Org. Lett. 2010, 12, 3812–3815. [Google Scholar] [CrossRef] [PubMed]
- Noskowska, M.; Sliwinska, E.; Duczmal, W. Simple fast preparation of neutral palladium(II) complexes with SnCl−3 and Cl− ligands. Trans. Met. Chem. 2003, 28, 756–759. [Google Scholar] [CrossRef]
- Duran-Sampedro, G.; Agarrabeitia, A.R.; Garcia-Moreno, I.; Costela, A.; Bañuelos, J.; Arbeloa, T.; Arbeloa, I.L.; Chiara, J.L.; Ortiz, M.J. Chlorinated BODIPYs: Surprisingly Efficient and Highly Photostable Laser Dyes. Eur. J. Org. Chem. 2012, 6335–6350. [Google Scholar] [CrossRef]
- Shah, M.; Thangaraj, K.; Soong, M.-L.; Wolford, L.T.; Boyer, J.H.; Politzer, I.R.; Pavlopoulos, T.G. Pyrromethene–BF2 complexes as laser dyes:1. Heteroatom Chem. 1990, 1, 389–399. [Google Scholar] [CrossRef]
- Nepomnyashchii, A.B.; Bröring, M.; Ahrens, J.; Bard, A.J. Synthesis, Photophysical, Electrochemical, and Electrogenerated Chemiluminescence Studies. Multiple Sequential Electron Transfers in BODIPY Monomers, Dimers, Trimers, and Polymer. J. Am. Chem. Soc. 2011, 133, 8633–8645. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, J.; Haberlag, B.; Scheja, A.; Tamm, M.; Bröring, M. Conjugated BODIPY DYEmers by Metathesis Reactions. Chem. Eur. J. 2014, 20, 2901–2912. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Jiang, F.-L.; Fortin, D.; Harvey, P.D.; Liu, Y. A reaction-based chromogenic and fluorescent chemodosimeter for fluoride anions. Chem. Comm. 2011, 47, 5503–5505. [Google Scholar] [CrossRef] [PubMed]
- Beweries, T.; Thomas, J.; Klahn, M.; Heller, D.; Schulz, A.; Rosenthal, U. Catalytic and Kinetic Studies of the Dehydrogenation of Dimethylamine Borane with an iPr Substituted Titanocene Catalyst. ChemCatChem 2011, 3, 1865–1868. [Google Scholar] [CrossRef]
- Barthelmes, K.; Kübel, J.; Winter, A.; Wächtler, M.; Friebe, C.; Dietzek, B.; Schubert, U.S. New Ruthenium Bis(terpyridine) Methanofullerene and Pyrrolidinofullerene Complexes: Synthesis and Electrochemical and Photophysical Properties. Inorg. Chem. 2015, 54, 3159–3171. [Google Scholar] [CrossRef] [PubMed]
- Siebert, R.; Akimov, D.; Schmitt, M.; Winter, A.; Schubert, U.S.; Dietzek, B.; Popp, J. Spectroscopic Investigation of the Ultrafast Photoinduced Dynamics in π-Conjugated Terpyridines. ChemPhysChem 2009, 10, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Errata: Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mennucci, B.; Cappelli, C.; Guido, C.A.; Cammi, R.; Tomasi, J. Structures and Properties of Electronically Excited Chromophores in Solution from the Polarizable Continuum Model Coupled to the Time-Dependent Density Functional Theory. J. Phys. Chem. A 2009, 113, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fdez. Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Malmqvist, P.-Å.; Pedersen, T.B.; Ghosh, A.; Roos, B.O. Cholesky Decomposition-Based Multiconfiguration Second-Order Perturbation Theory (CD-CASPT2): Application to the Spin-State Energetics of CoIII(diiminato)(NPh). J. Chem. Theory Comput. 2008, 4, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Malmqvist, P.-Å.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Igel-Mann, G.; Stoll, H.; Preuss, H. Pseudopotentials for main group elements (IIIa through VIIa). Mol. Phys. 1988, 65, 1321–1328. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
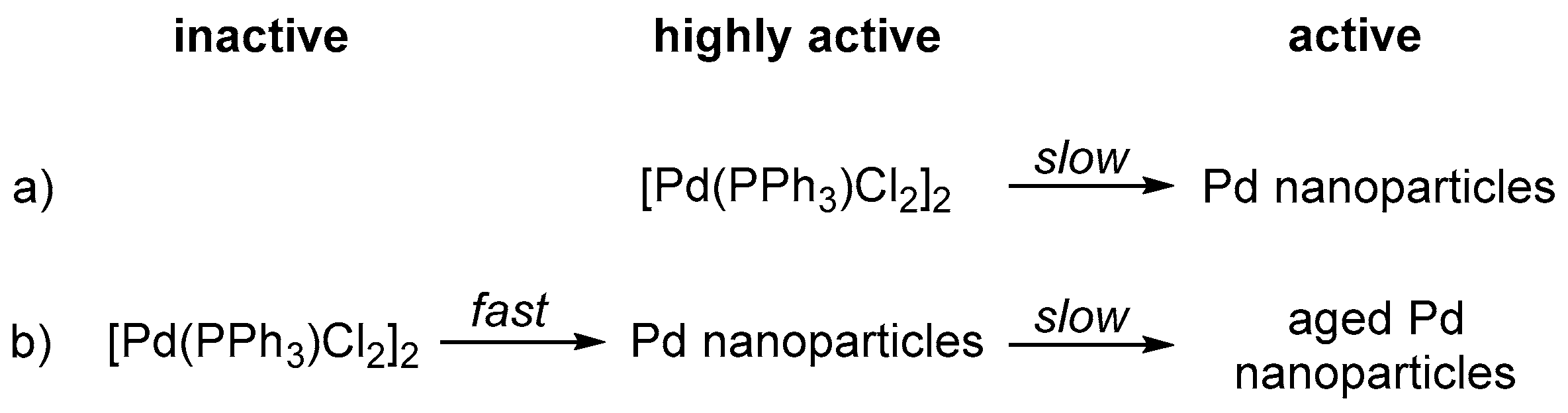{kind=link}
| PS | λabs a (nm) | E b (L/mol·cm) | λem c (nm) | Φem d (%) | KSV (L/mol) | kq (109 L/mol∙s) |
|---|---|---|---|---|---|---|
| 1 | 506 | 80,600 | 537 | 94 | 23.0 ± 0.9 | 3.8 e |
| 2 | 496 | 62,700 | 526 | 97 | 6.46 ± 0.1 | 1.2 e |
| 3 | 526 | 77,000 | 559 | 7 | 1.43 ± 0.1 | >1.4 f |
| 4 | 502 | 84,400 | 523 | 91 | 14.2 ± 0.4 | 3.1 ± 0.4 |
| 5 | 516 | 85,500 | 543 | 3 | 1.10 ± 0.1 | 3.4 ± 0.7 |
| 6 | 534 | 89,700 | 565 | 1 | 1.05 ± 0.03 | 5.5 ± 0.7 |
| 7 | 530 | 62,500 | 555 | 70 | 5.04 ± 0.2 | 1.0 ± 0.2 |
| 8 | 549 | 55,000 | 579 | 85 | 2.93 ± 0.3 | 0.6 ± 0.1 |
| PS | τem a (ns) | KSV(τ) (L/mol) | τex1 b (μs) | τex2 b (μs) | kq (109 L/mol·s) |
|---|---|---|---|---|---|
| 4 | 4.62 | 11.26 ± 0.28 | - d | - d | 2.4 ± 0.3 |
| 5 | 0.32 | 1.12 ± 0.14 | 28 | 120 | 3.5 ± 0.9 |
| 6 | 0.19 | -/- c | 37 | 150 | -/- c |
| 7 | 5.02 | 5.11 ± 0.16 | - d | - d | 1.0 ± 0.1 |
| 8 | 4.79 | -/- c | - d | - d | -/- c |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dura, L.; Wächtler, M.; Kupfer, S.; Kübel, J.; Ahrens, J.; Höfler, S.; Bröring, M.; Dietzek, B.; Beweries, T. Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction. Inorganics 2017, 5, 21. https://doi.org/10.3390/inorganics5020021
Dura L, Wächtler M, Kupfer S, Kübel J, Ahrens J, Höfler S, Bröring M, Dietzek B, Beweries T. Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction. Inorganics. 2017; 5(2):21. https://doi.org/10.3390/inorganics5020021
Chicago/Turabian StyleDura, Laura, Maria Wächtler, Stephan Kupfer, Joachim Kübel, Johannes Ahrens, Sebastian Höfler, Martin Bröring, Benjamin Dietzek, and Torsten Beweries. 2017. "Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction" Inorganics 5, no. 2: 21. https://doi.org/10.3390/inorganics5020021
APA StyleDura, L., Wächtler, M., Kupfer, S., Kübel, J., Ahrens, J., Höfler, S., Bröring, M., Dietzek, B., & Beweries, T. (2017). Photophysics of BODIPY Dyes as Readily-Designable Photosensitisers in Light-Driven Proton Reduction. Inorganics, 5(2), 21. https://doi.org/10.3390/inorganics5020021