
Backbone-Substituted β-Ketoimines and Ketoiminate Clusters: Transoid Li2O2 Squares and D2-Symmetric Li4O4 Cubanes. Synthesis, Crystallography and DFT Calculations



Abstract
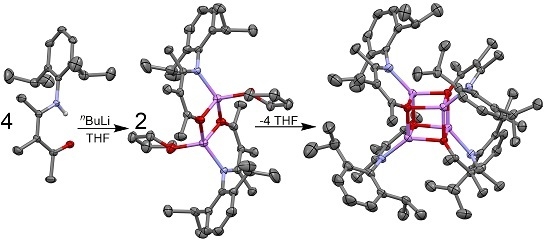
:
1. Introduction
2. Results and Discussion
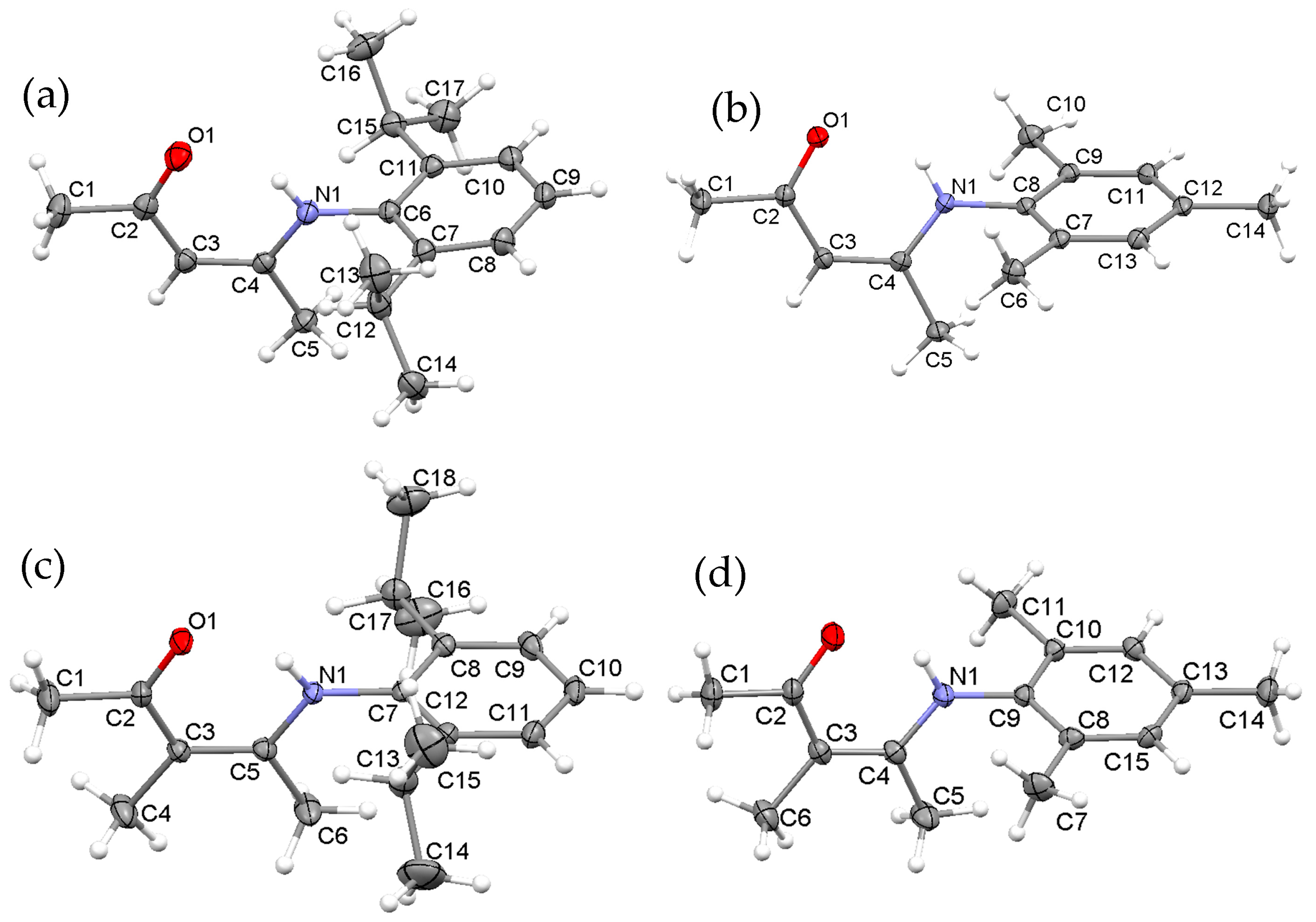
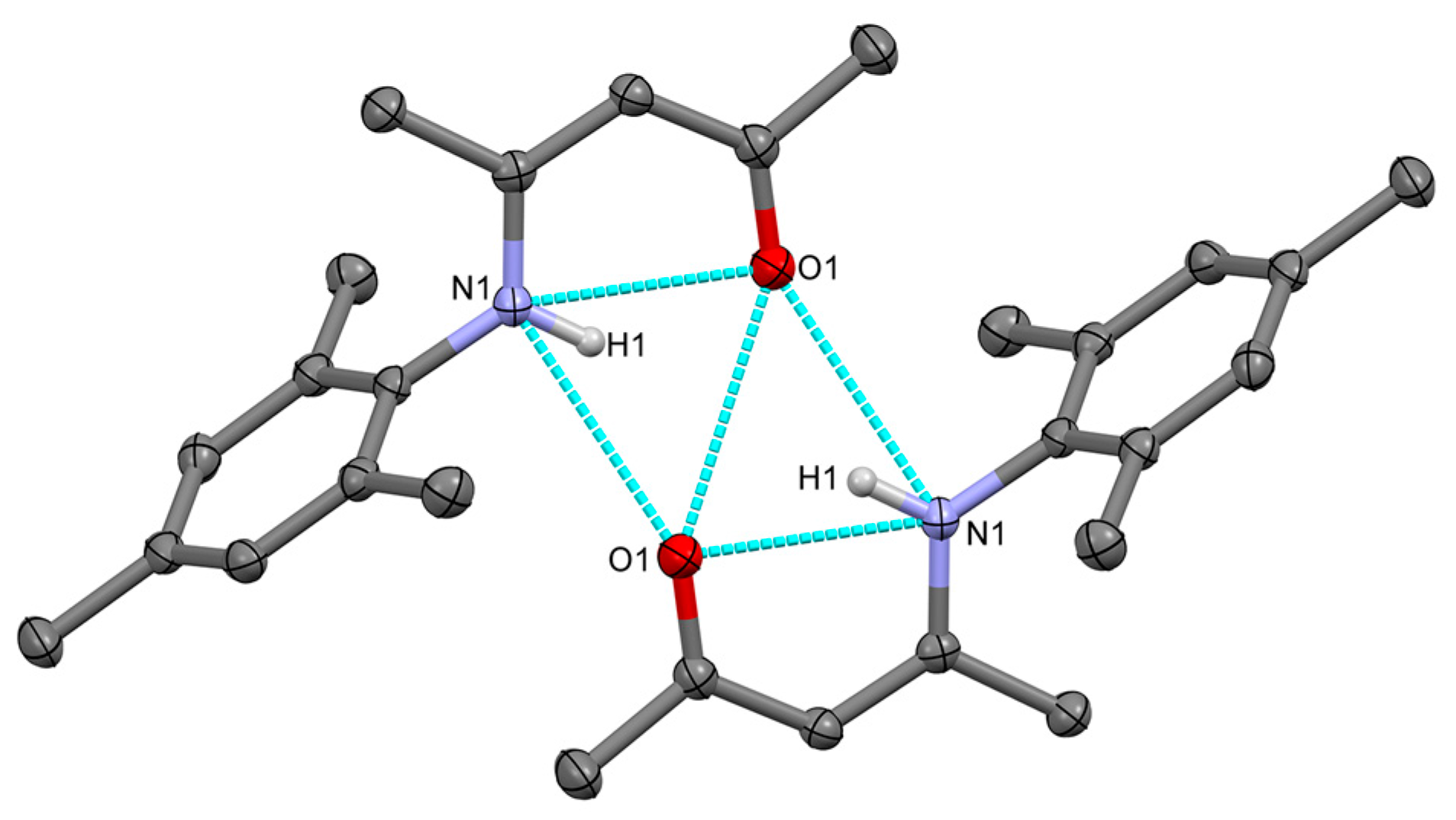
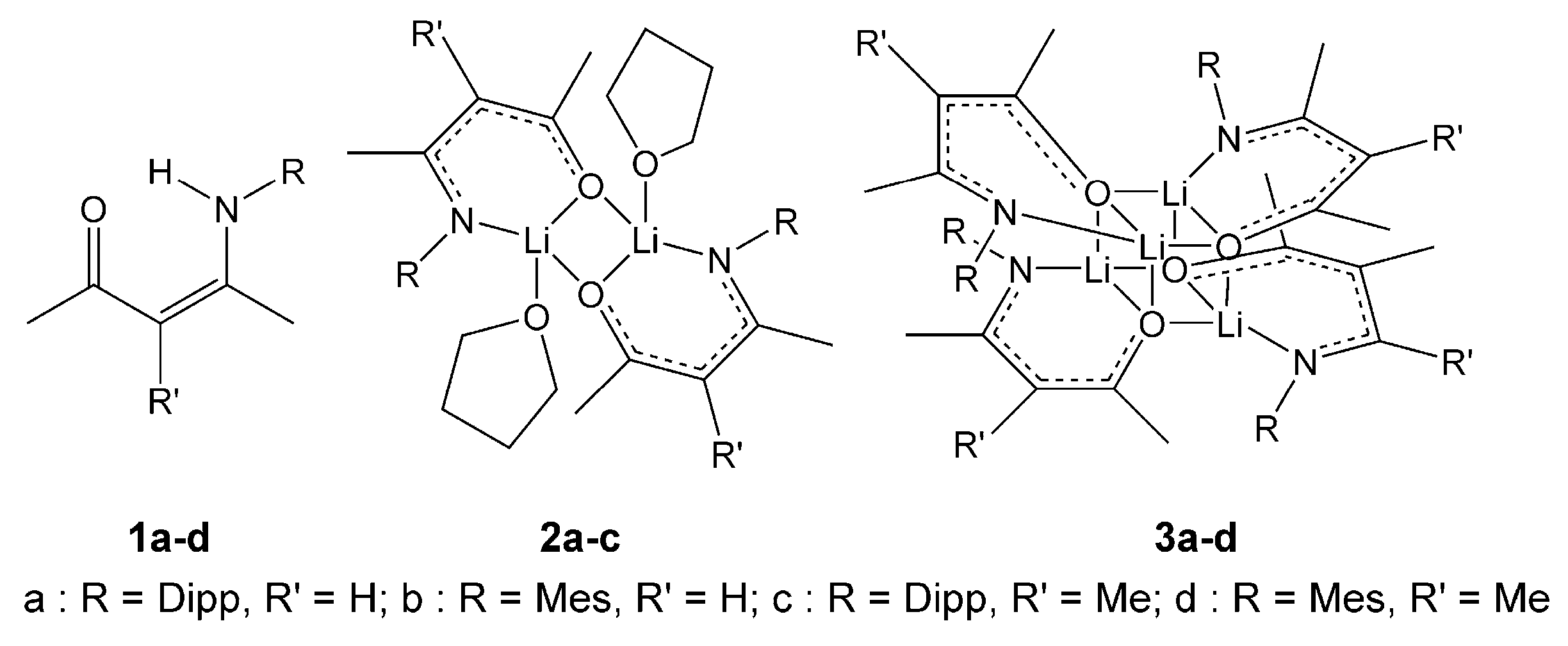
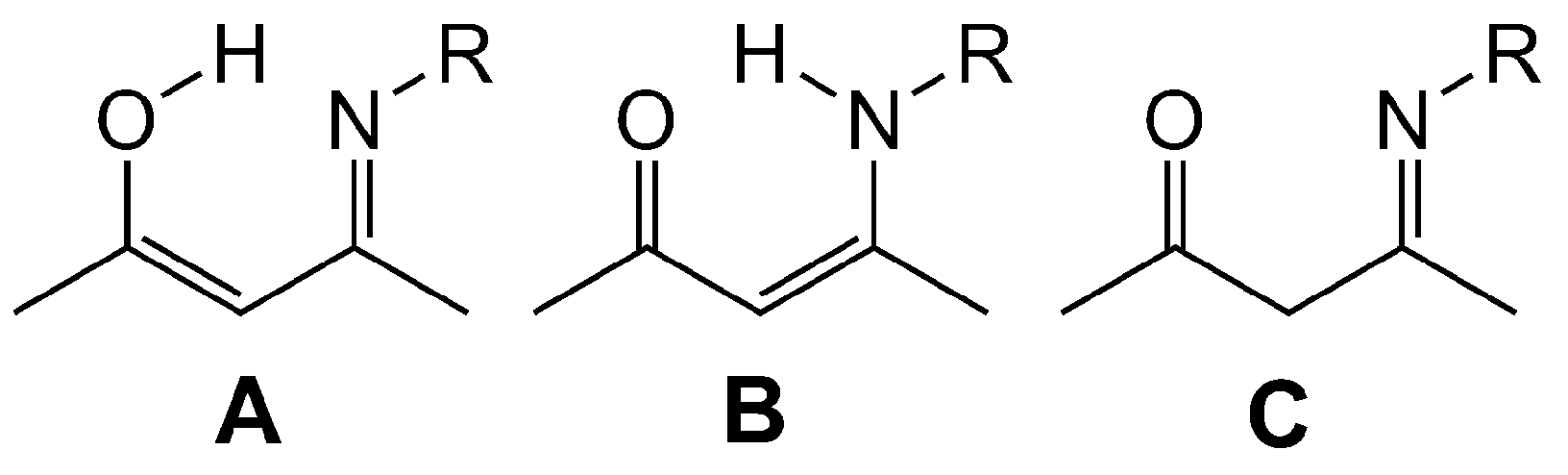
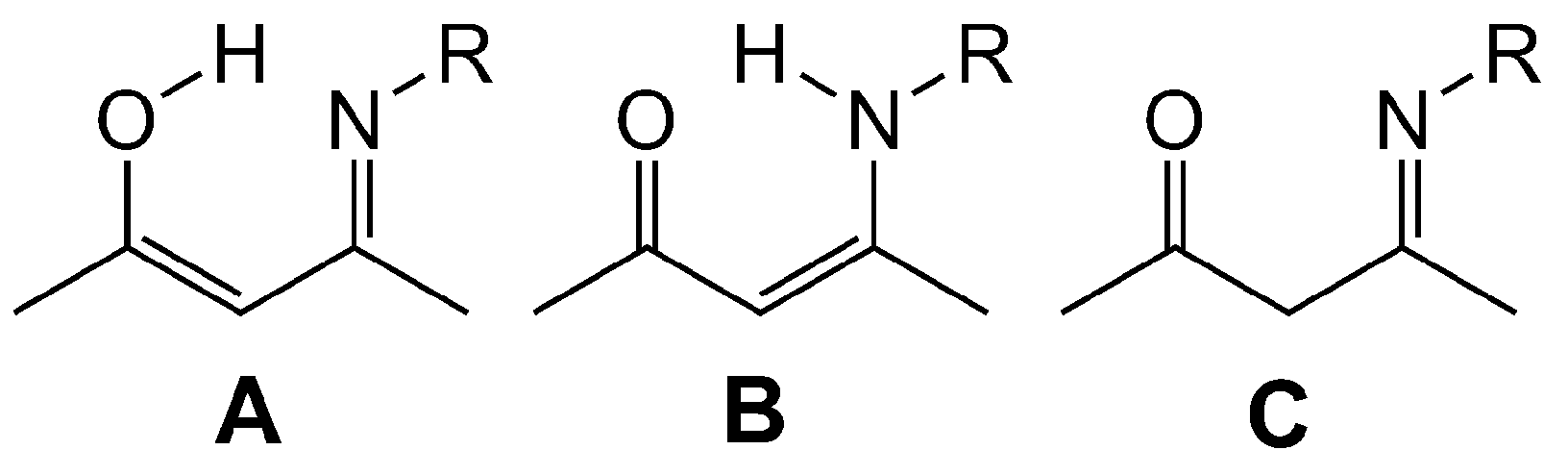
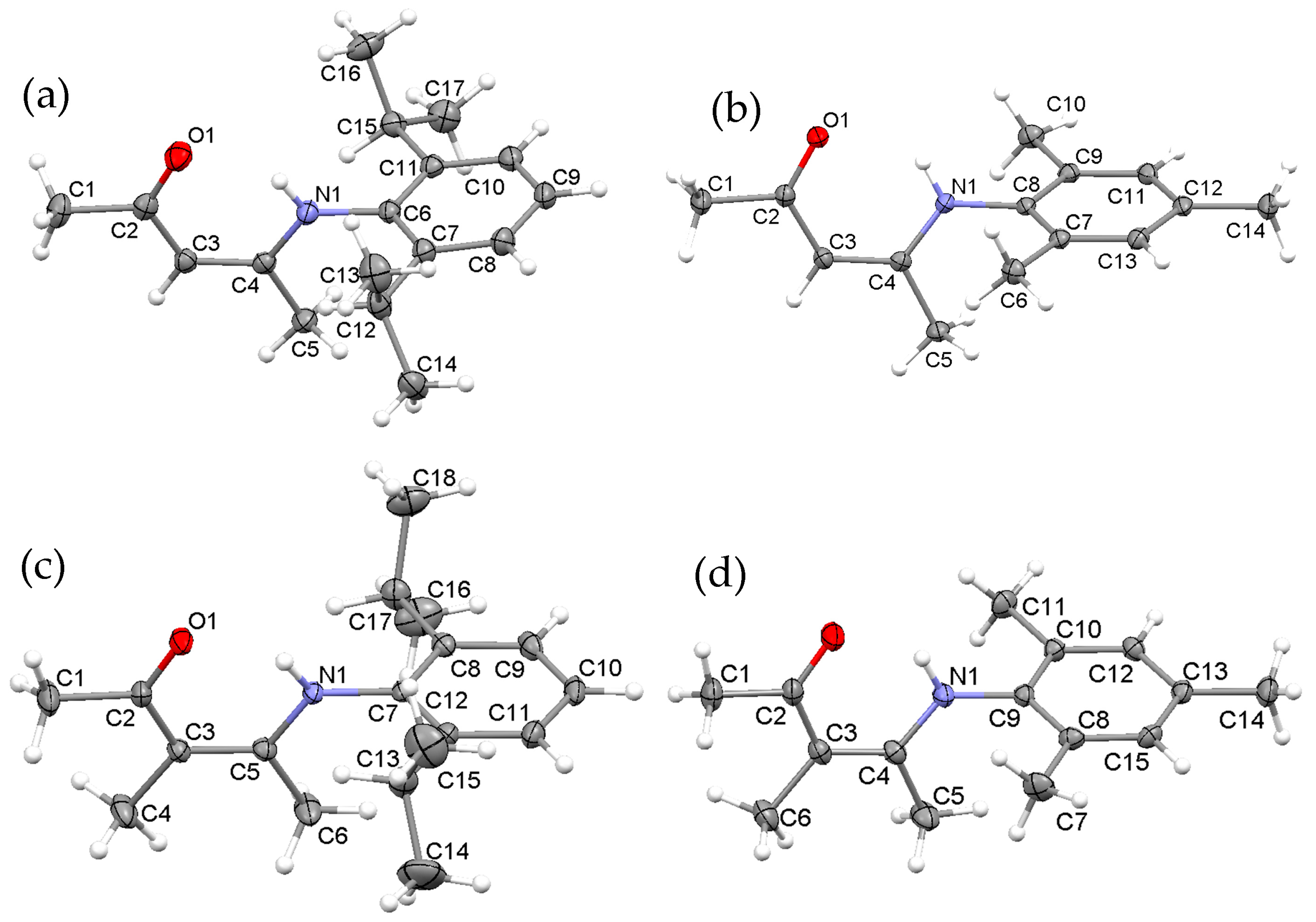
2.1. Synthesis, Structures and Tautomers of β-Ketoimines
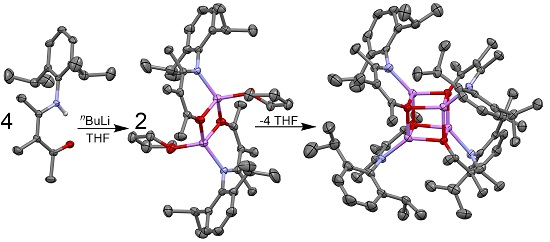
2.2. Synthesis and Structures of Ketoiminate Lithium Complexes
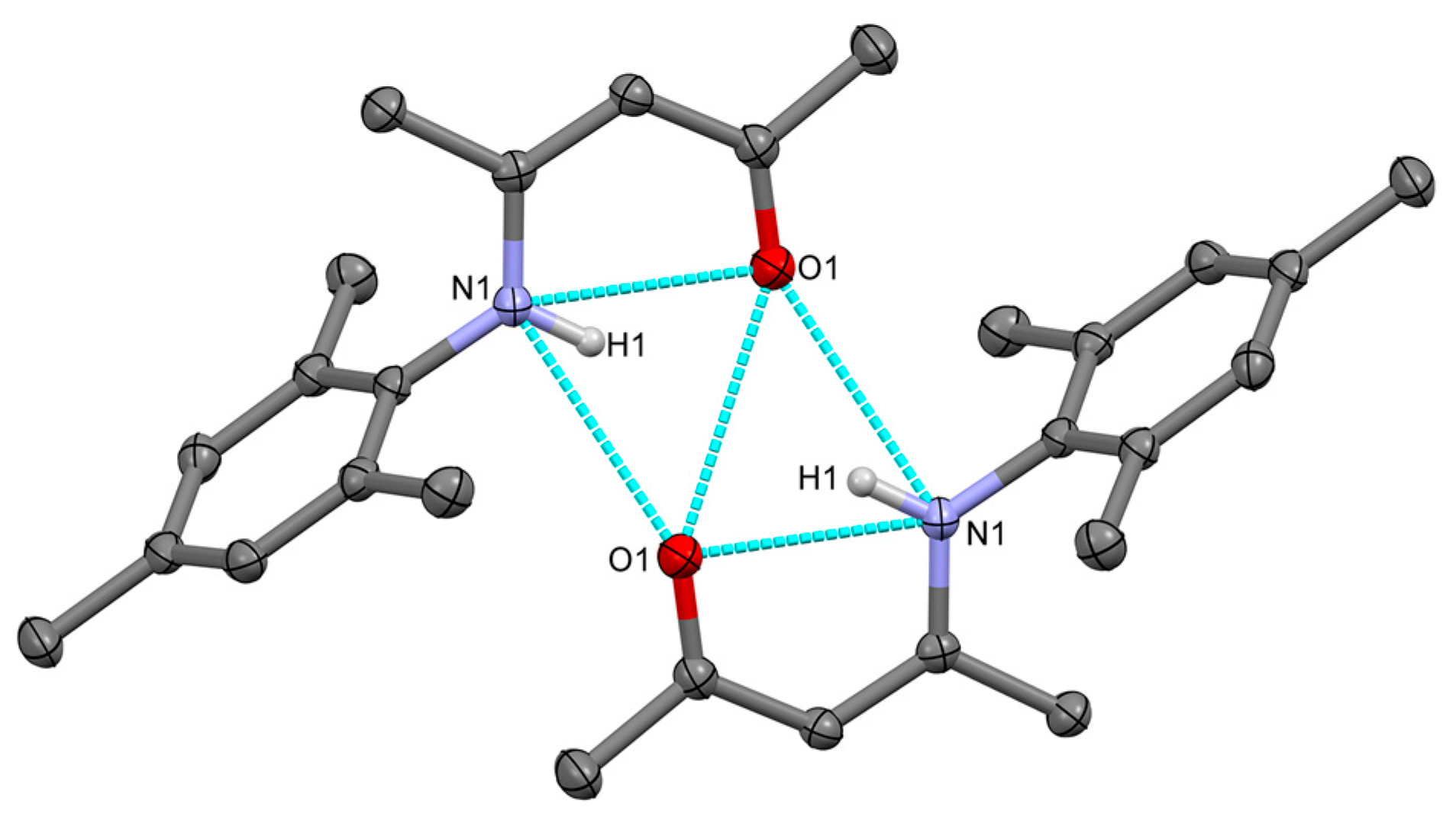
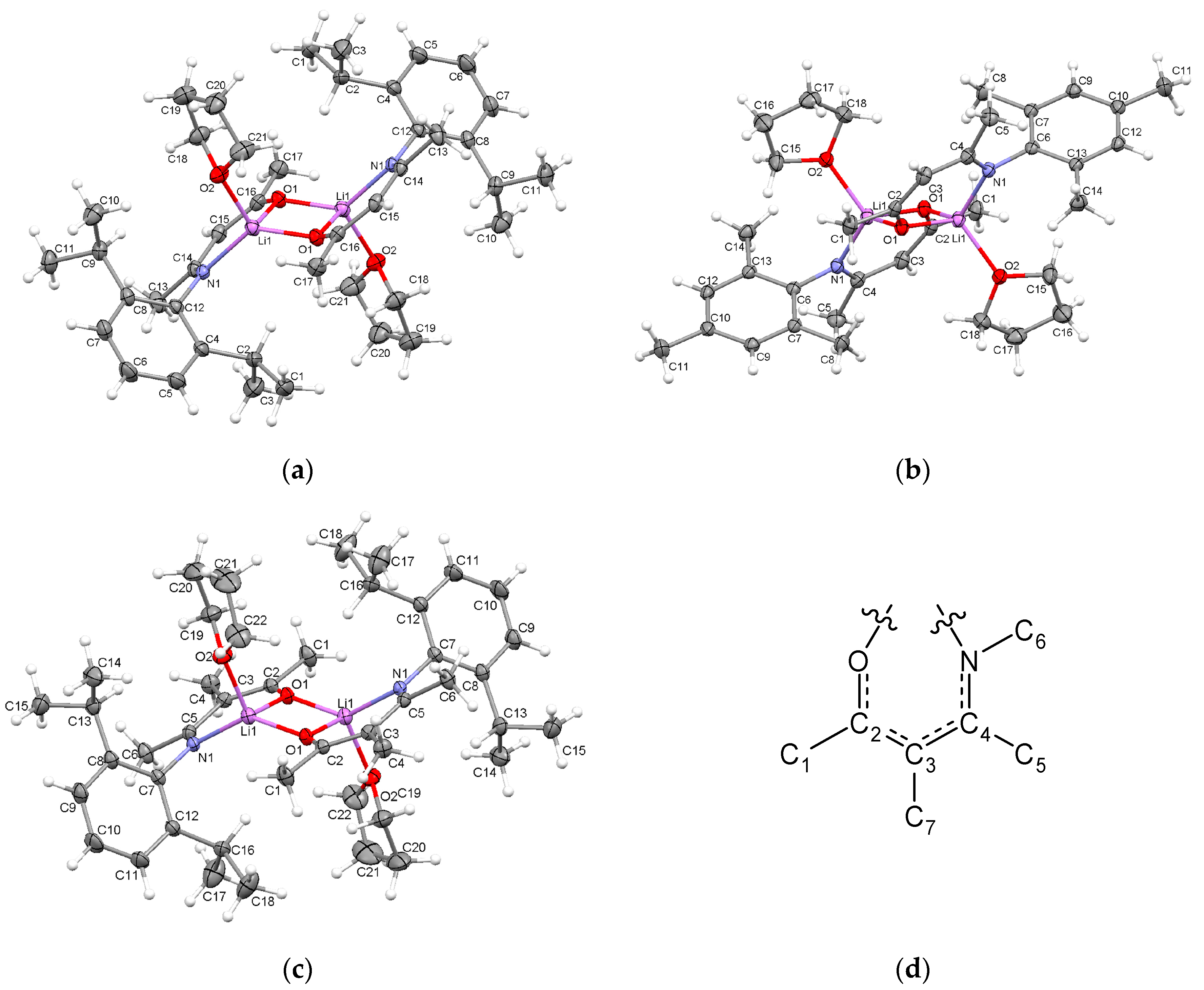
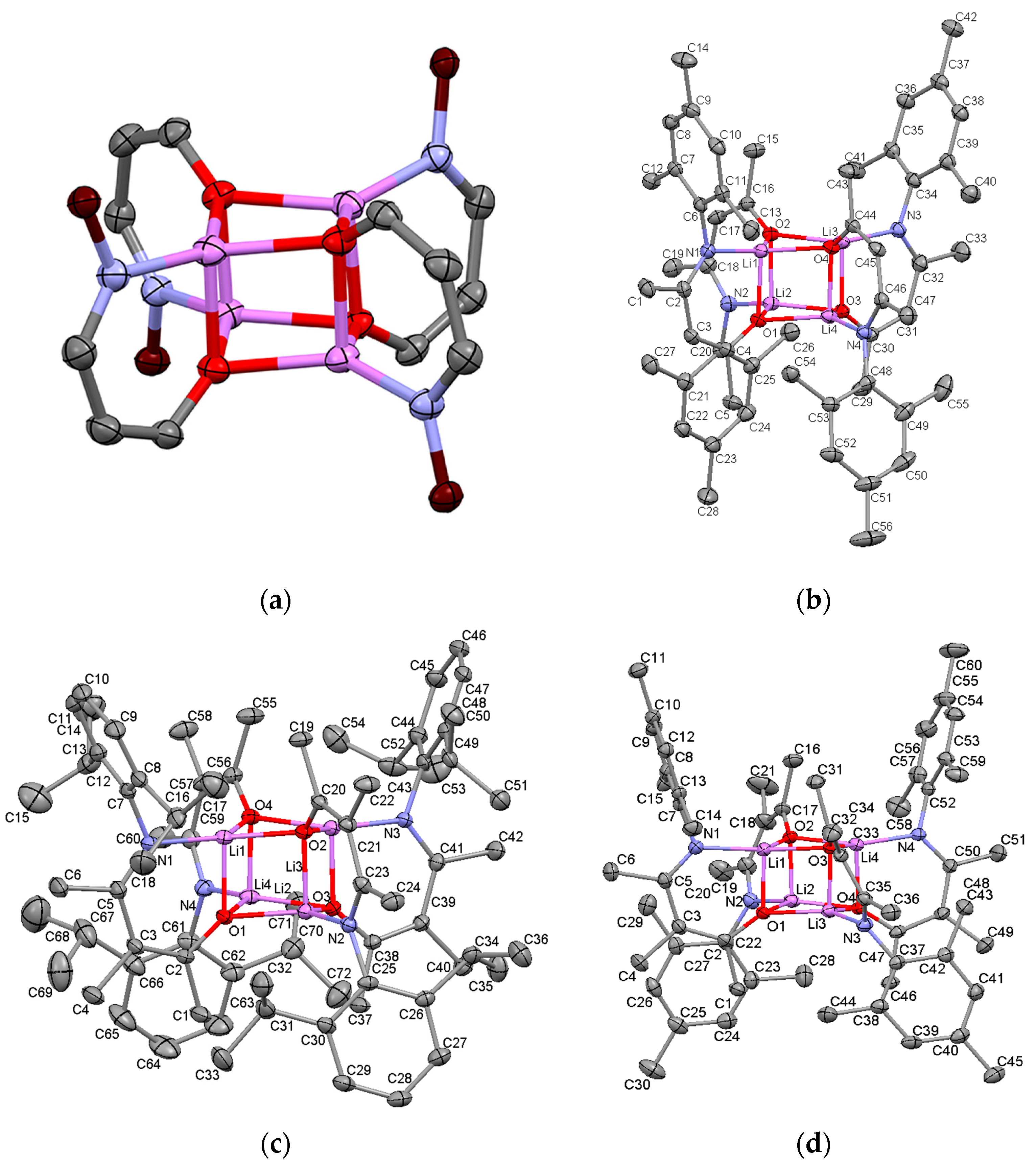
2.2.1. Transoid Li2O2 Clusters
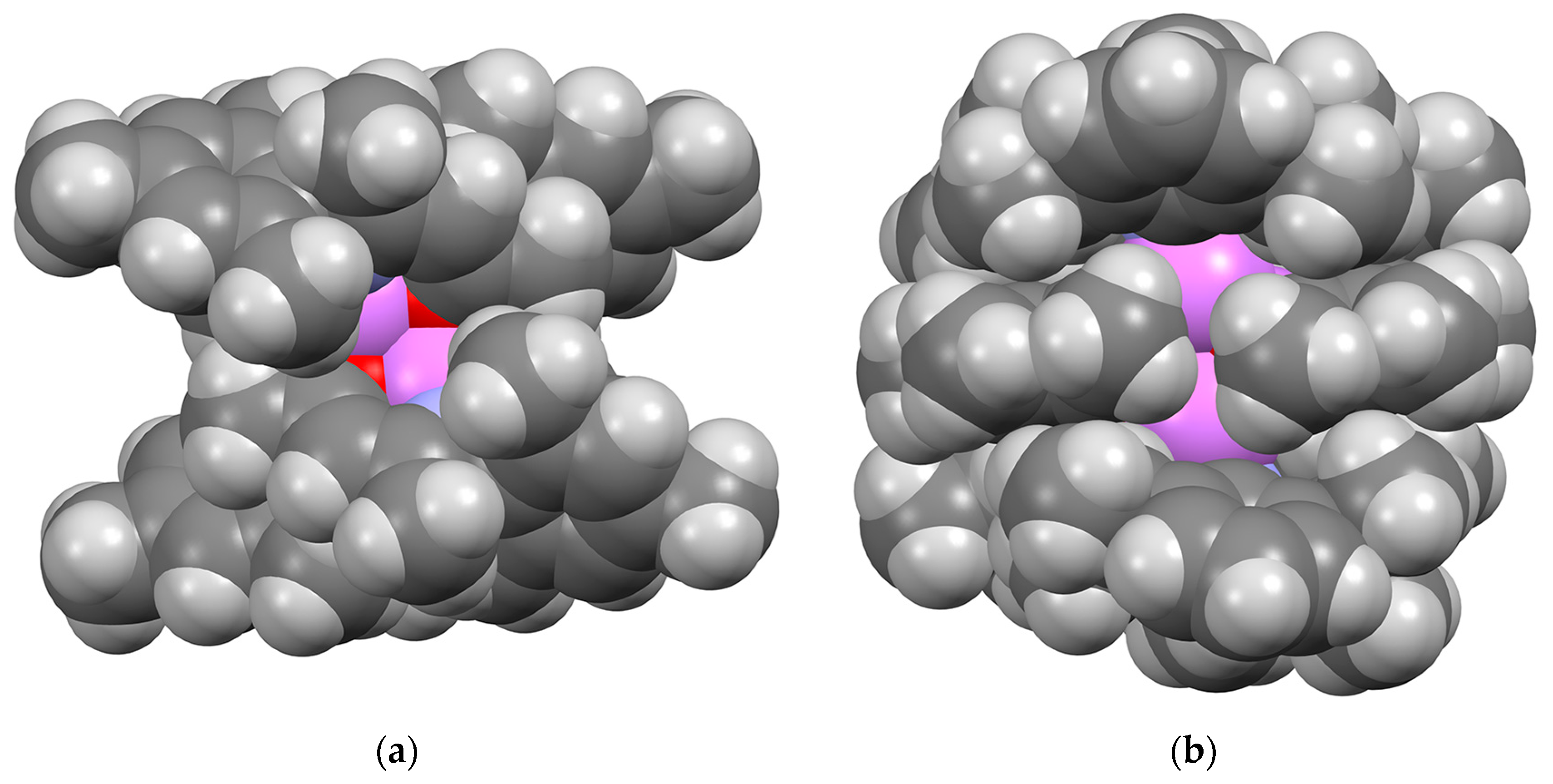
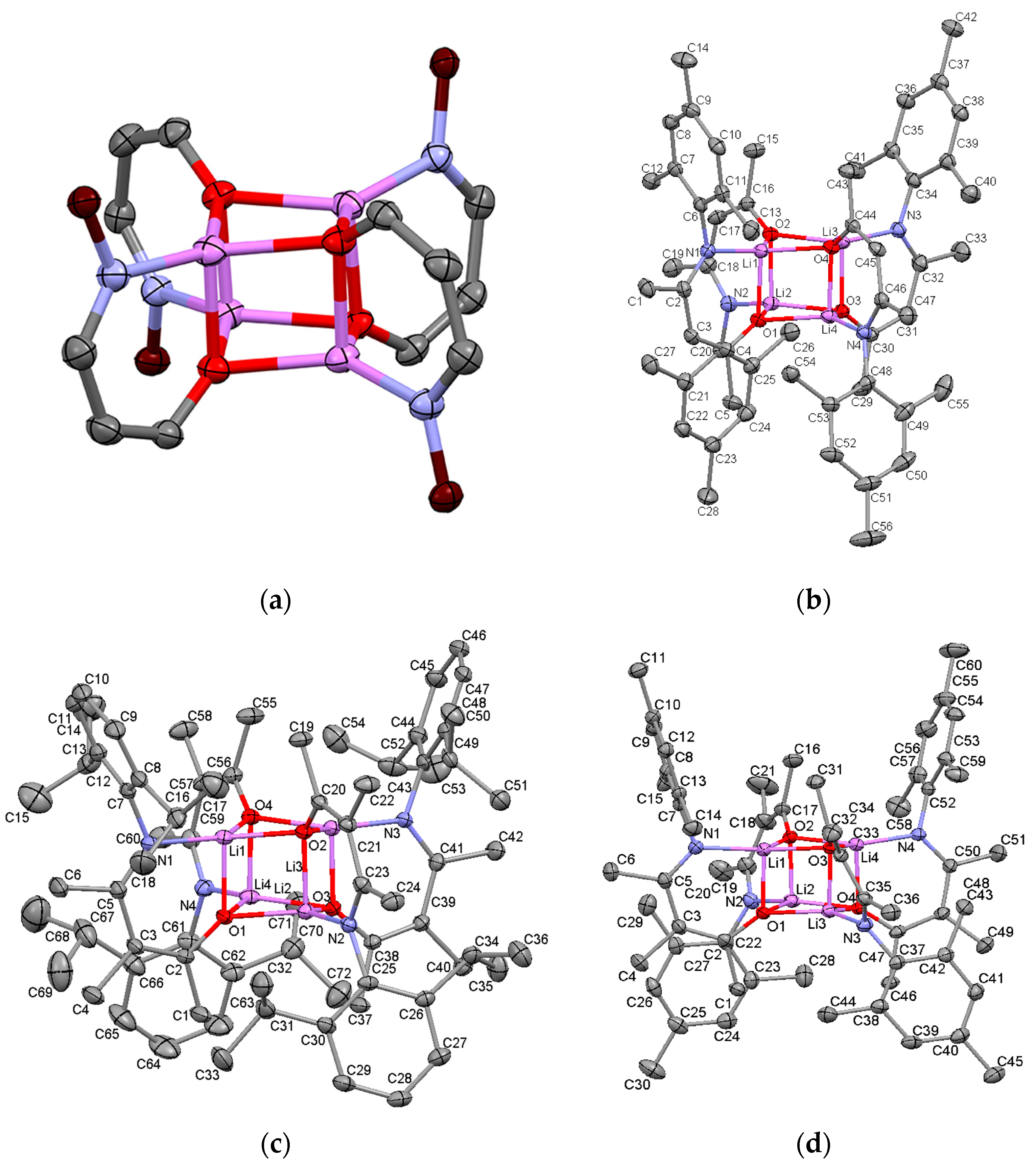
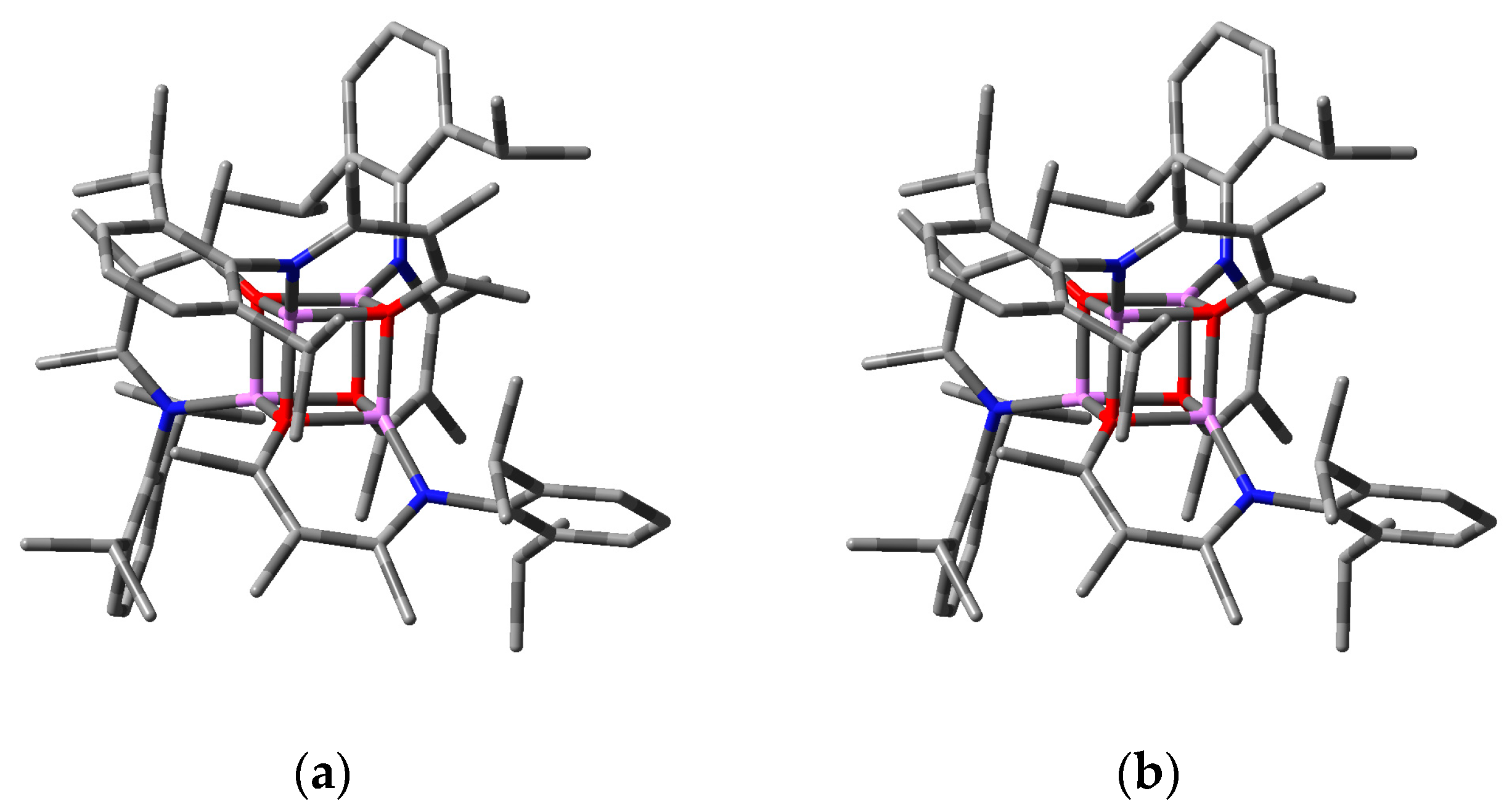
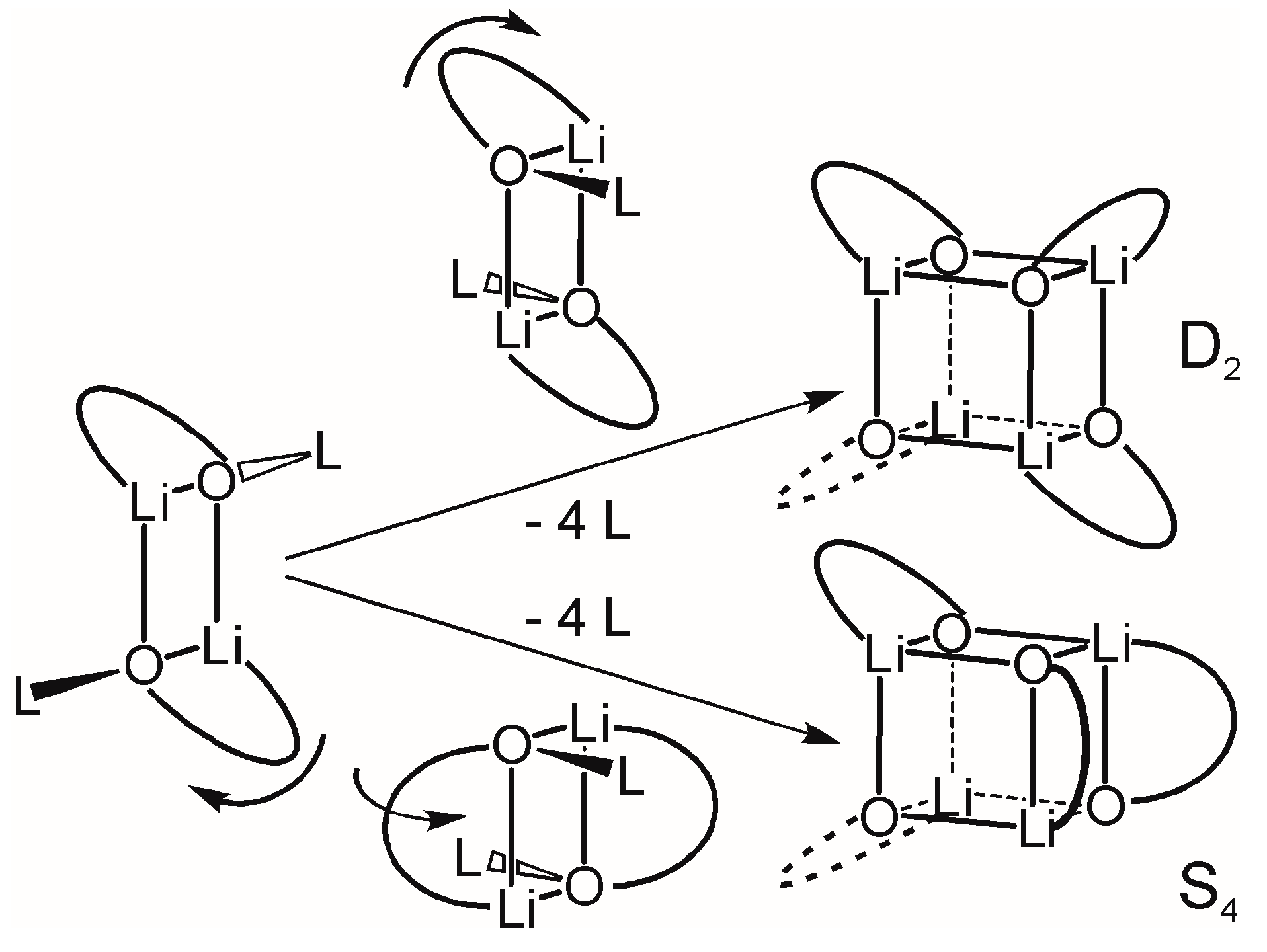
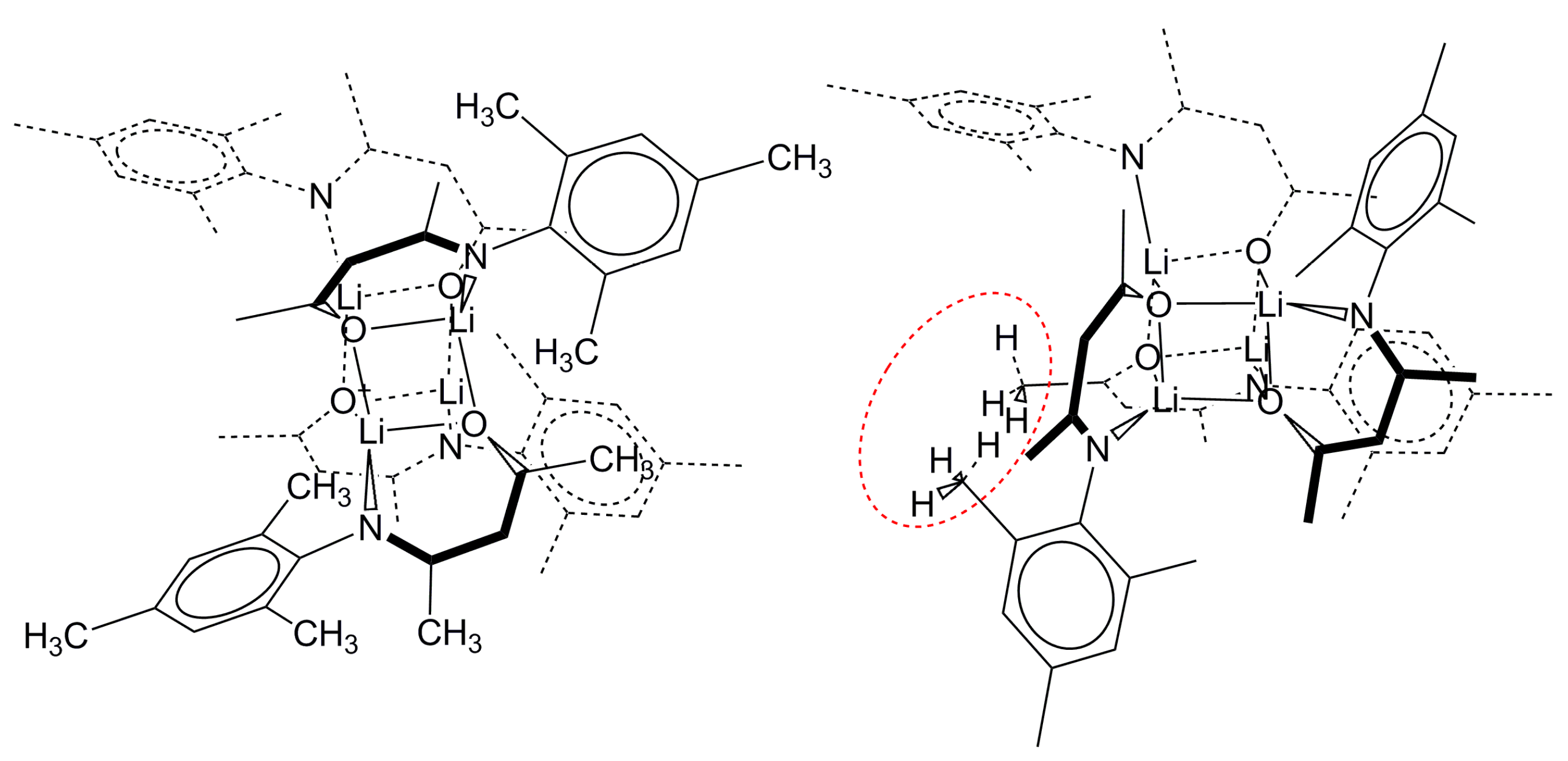
2.2.2. “Tetrameric” Cuboidal Clusters
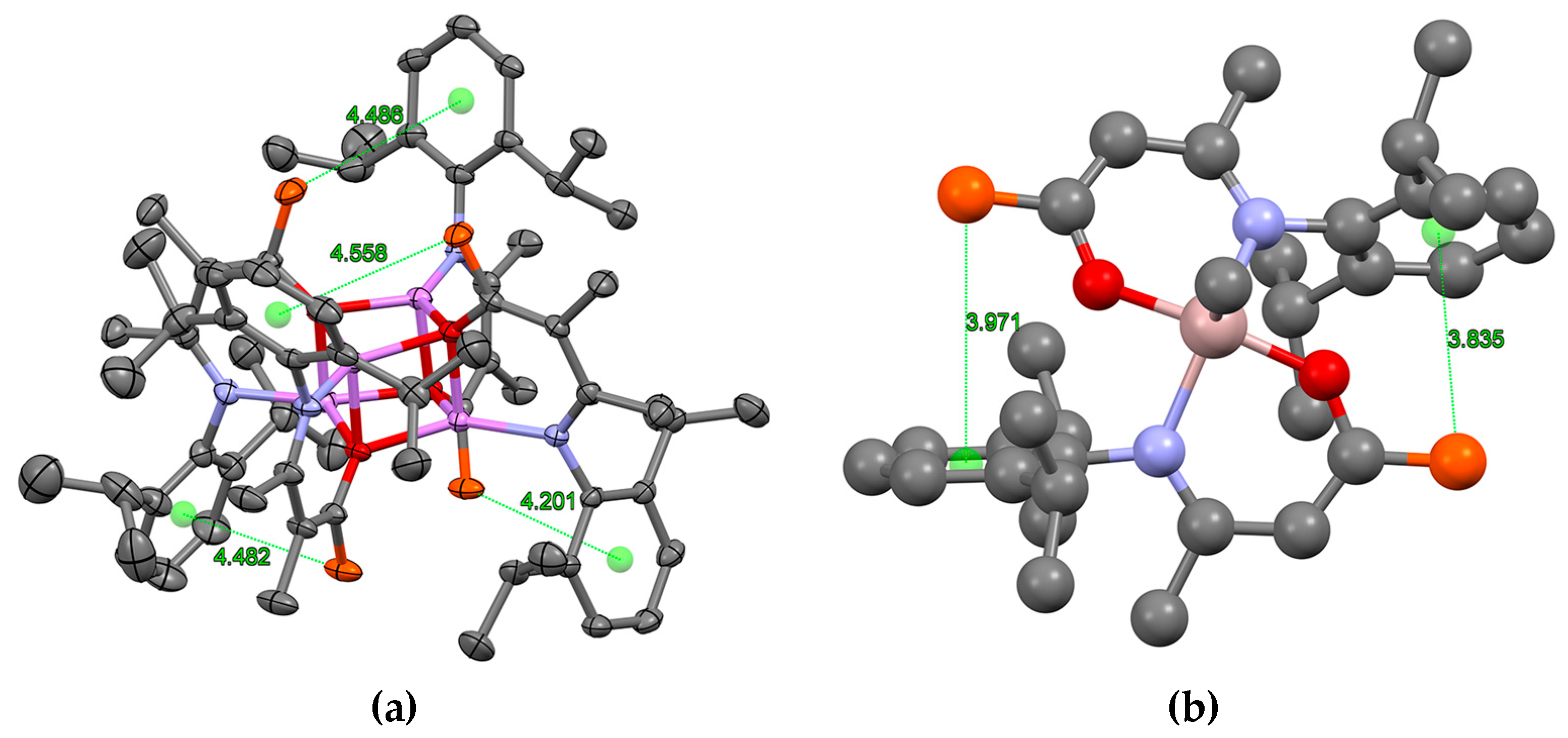
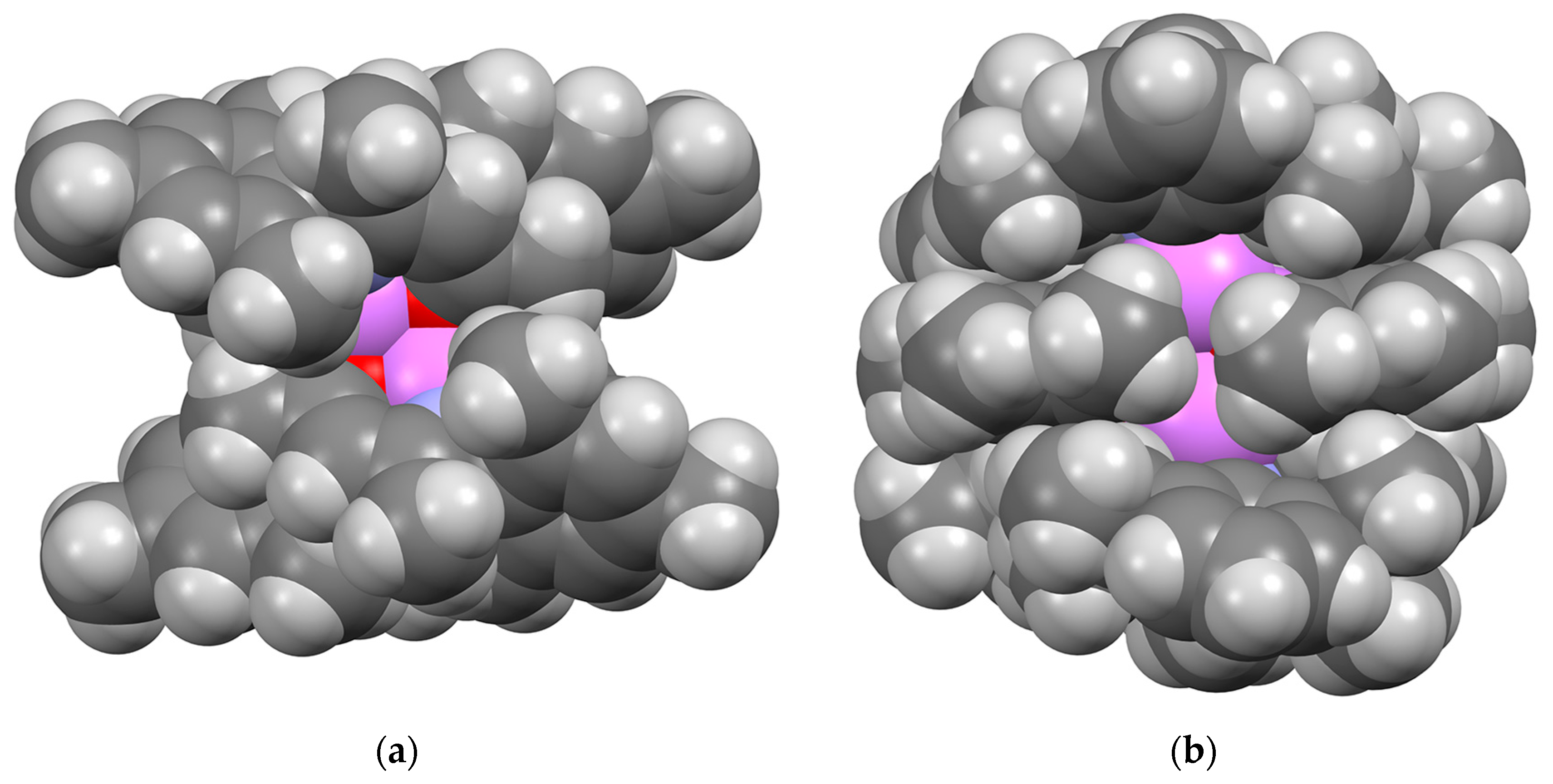
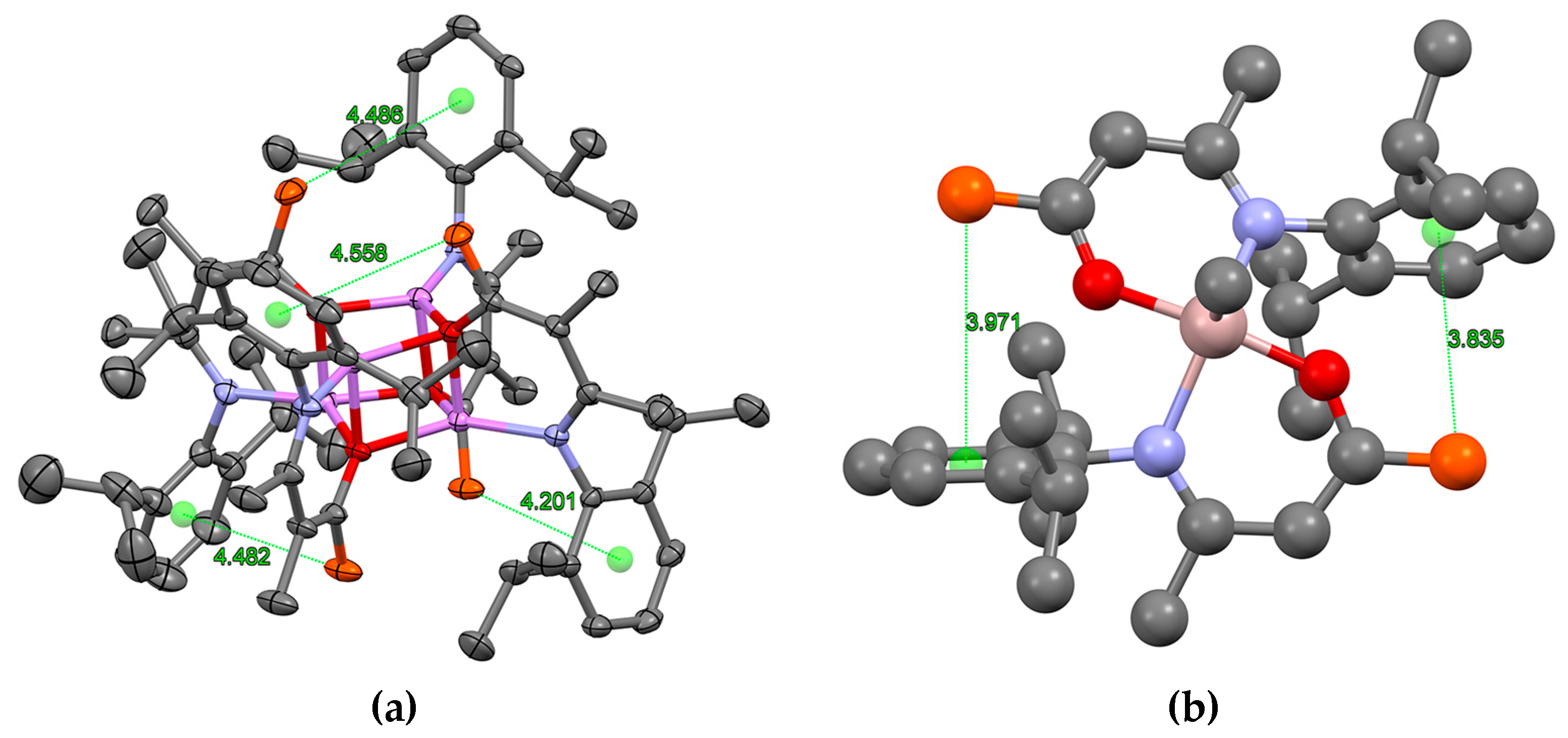
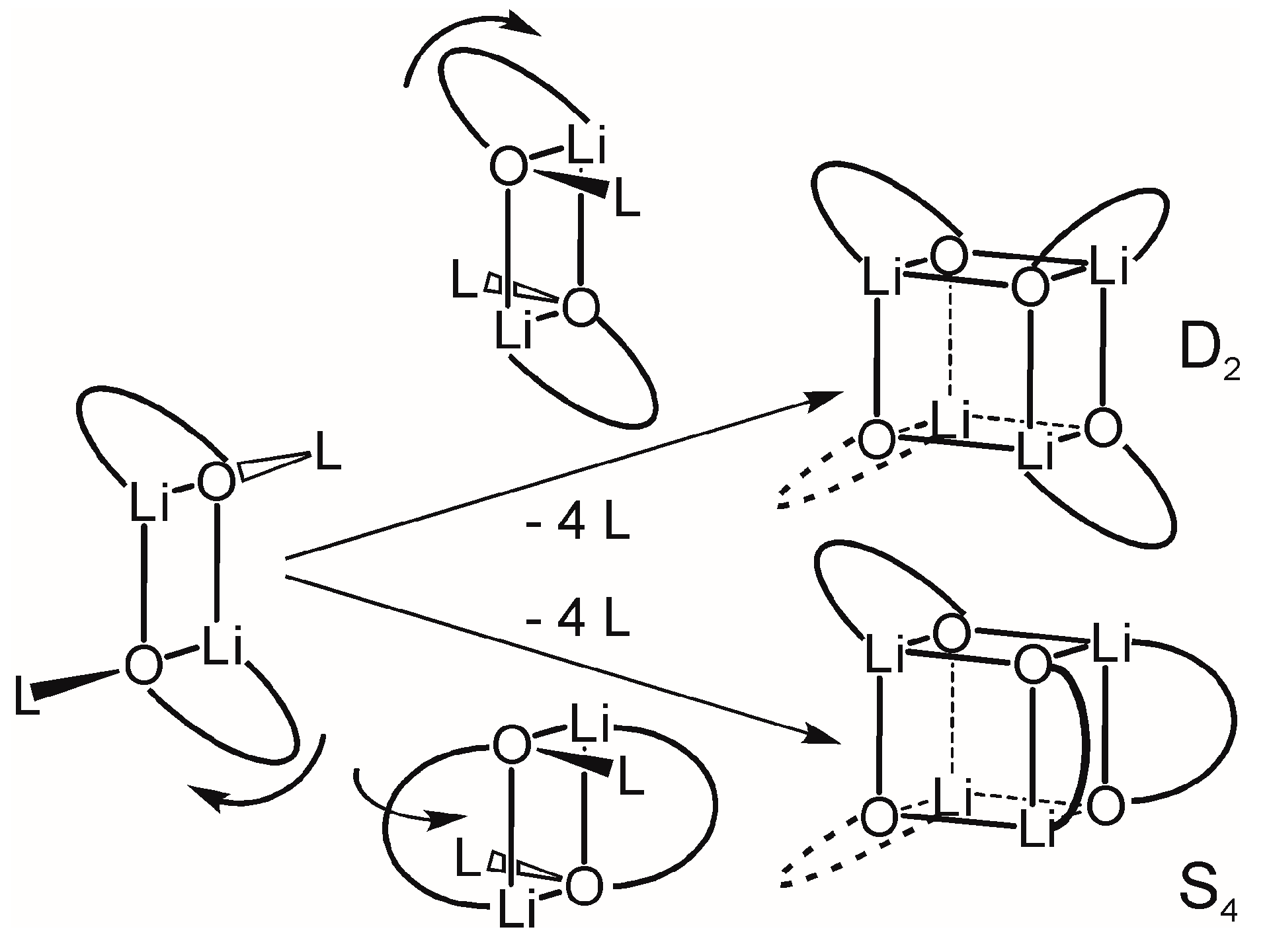
2.3. Formation and Isomer Selection of Li4O4 Clusters
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of Ketoimines
3.2.1. 4-((2,6-Diisopropylphenyl)amino)-3-methyl-3-methyl-pent-3-en-2-one 1c
3.2.2. 4-((2,4,6-Trimethylphenyl)amino)-3-methyl-pent-3-en-2-one 1d
3.3. Synthesis of the Ketoimide Lithium Complexes
3.3.1. Lithium Ketoiminate Complex 2a
3.3.2. Lithium Ketoiminate Complex 2b
3.3.3. Lithium Ketoiminate Complex 2c
3.3.4. Lithium Ketoiminate Complex 3b Toluene Solvate
3.3.5. Lithium Ketoiminate Complex 3c Solvate
3.3.6. Lithium Ketoiminate Complex 3d
3.4. X-ray Crystallography
3.5. Computation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1a | 1b | 1c | 1d |
|---|---|---|---|---|
| Formula | C17H25NO | C14H19NO | C18H27NO | C15H21NO |
| FW, amu | 259.38 | 217.30 | 273.41 | 231.33 |
| T, K | 173(2) | 173(2) | 173(2) | 173(2) |
| λ, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | orthorhombic | monoclinic | orthorhombic | monoclinic |
| Space group | Pccn | P21/n | P212121 | C2/c |
| Cell: a, Å | 12.3641(12) | 10.0068(6) | 6.7423(4) | 18.8762(17) |
| b, Å | 16.3858(16) | 9.8961(6) | 13.9706(8) | 8.0097(7) |
| c, Å | 15.4945(15) | 12.7933(8) | 18.0631(10) | 19.0037(17) |
| α, ° | 90 | 90 | 90 | 90 |
| β, ° | 90 | 99.1380(10) | 90 | 107.5430(10) |
| γ, ° | 90 | 90 | 90 | 90 |
| V, Å3 | 3139.1(5) | 1250.82(13) | 1701.44(17) | 2739.6(4) |
| Z | 8 | 4 | 4 | 8 |
| Dcalc, g/cm3 | 1.098 | 1.154 | 1.067 | 1.112 |
| μ, mm−1 | 0.067 | 0.072 | 0.065 | 0.069 |
| F(000) | 1136 | 472 | 600 | 1008 |
| Cryst. size, mm3 | 0.54 × 0.41 × 0.20 | 0.27 × 0.25 × 0.14 | 0.42 × 0.20 × 0.14 | 0.43 × 0.34 × 0.16 |
| θmin, max, ° | 2.06, 26.73 | 2.41, 27.10 | 1.84, 27.40 | 2.25, 27.48 |
| h min, max | −15, 15 | −12, 12 | −8, 8 | −24, 24 |
| k min, max | −20, 20 | −12, 12 | −18, 18 | −10, 10 |
| l min, max | −19, 19 | −16, 16 | −23, 23 | −24, 24 |
| Rflall | 40165 | 17205 | 24505 | 19053 |
| Rflindep, Rint | 3338, 0.0561 | 2762, 0.0254 | 2224, 0.0285 | 3153, 0.0240 |
| Compl., θ, ° | 100, 26.73 | 99.9, 27.10 | 100, 25.25 | 99.9, 27.48 |
| Abs corr. | semi-empirical from equivalents | |||
| Max/min trans. | 0.9868, 0.9646 | 0.9914, 0.9378 | 0.9916, 0.9279 | 0.9918, 0.9169 |
| Data | 3338 | 2762 | 2224 | 3153 |
| Restraints | 0 | 0 | 0 | 0 |
| Parameters | 182 | 153 | 191 | 163 |
| GOF | 1.032 | 1.047 | 1.031 | 1.053 |
| R1 (I > 2σI) | 0.0428 | 0.0462 | 0.0407 | 0.0465 |
| wR2 (all data) | 0.1201 | 0.1349 | 0.1172 | 0.1402 |
| Max, min, e∙Å−3 | 0.20, −0.21 | 0.26, −0.31 | 0.20, −0.18 | 0.26, −0.22 |
| Parameter | 2a | 2b | 2c |
|---|---|---|---|
| Formula | C42H64Li2N2O4 | C36H52Li2N2O4 | C44H68Li2N2O4 |
| FW, amu | 674.83 | 590.68 | 702.88 |
| T, K | 173(2) | 173(2) | 173(2) |
| λ, Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | triclinic | monoclinic | monoclinic |
| Space group | P | P21/c | P21/n |
| Cell: a, Å | 9.4060(8) | 8.0148(5) | 20.843(5) |
| b, Å | 10.7931(9)) | 15.0628(9) | 10.495(2) |
| c, Å | 11.9959(10) | 14.5918(9) | 21.365(5) |
| α, ° | 69.0400(10 | 90 | 90 |
| β, ° | 73.6160(10) | 103.8550(10) | 112.886(2) |
| γ, ° | 67.0160(10) | 90 | 90 |
| V, Å3 | 1032.50(15) | 1710.35(18) | 4305.7(17) |
| Z | 1 | 2 | 4 |
| Dcalc, g/cm3 | 1.085 | 1.147 | 1.084 |
| μ, mm−1 | 0.067 | 0.073 | 0.067 |
| F(000) | 368 | 640 | 1536 |
| Cryst. size, mm3 | 0.36 × 0.20 × 0.12 | 0.39 × 0.18 × 0.11 | 0.46 × 0.19 × 0.17 |
| θmin, max, ° | 1.84, 26.03 | 1.97, 27.48 | 1.75, 27.48 |
| h min, max | −11, 11 | −10, 10 | −27, 27 |
| k min, max | −13, 13 | −19, 19 | −13, 13 |
| l min, max | −14, 14 | −18, 18 | −27, 27 |
| Rflall | 10854 | 19537 | 60577 |
| Rflindep, Rint | 4040, 0.0245 | 3919, 0.0373 | 9865, 0.0250 |
| Compl.,%; θ, ° | 99.6, 25.25 | 100.0, 25.25 | 99.9, 27.48 |
| Abs corr. | semi-empirical from equivalents | ||
| Max/min trans. | 0.9918, 0.9136 | 0.9916, 0.8956 | 0.9918, 0.9266 |
| Data | 4040 | 3919 | 9865 |
| Restraints | 0 | 0 | 0 |
| Parameters | 232 | 204 | 483 |
| GOF | 1.035 | 1.025 | 1.030 |
| R1 (I > 2σI) | 0.0464 | 0.0495 | 0.0453 |
| wR2 (all data) | 0.1274 | 0.1409 | 0.1285 |
| Max, min, e∙Å−3 | 0.23, −0.19 | 0.24, −0.25 | 0.32, −0.21 |
| Parameter | 3b | 3c | 3d |
|---|---|---|---|
| Formula | C56H72Li4N4O4∙C7H8 | C72H104Li4N4O4 | C60H80Li4N4O4 |
| FW, amu | 985.07 | 1117.35 | 949.04 |
| T, K | 173(2) | 173(2) | 173(2) |
| λ, Å | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | orthorhombic | monoclinic | monoclinic |
| Space group | P212121 | P21/n | P21/n |
| Cell: a, Å | 13.7530(9) | 14.3398(14) | 13.0543(11) |
| b, Å | 18.3062(13) | 27.504(3) | 26.647(2) |
| c, Å | 24.0904(17) | 19.8735(19) | 16.4965(14) |
| α, ° | 90 | 90 | 90 |
| β, ° | 90 | 107.4870(10) | 91.2180(10) |
| γ, ° | 90 | 90 | 90 |
| V, Å3 | 6065.1(7) | 7475.9(13) | 5737.1(8) |
| Z | 4 | 4 | 4 |
| Dcalc, g/cm3 | 1.079 | 0.993 | 1.099 |
| μ, mm−1 | 0.065 | 0.059 | 0.067 |
| F(000) | 2120 | 2432 | 2048 |
| Cryst. size, mm3 | 0.48 × 0.40 × 0.24 | 0.69 × 0.27 × 0.13 | 0.40 × 0.22 × 0.22 |
| θmin, max, ° | 1.69, 27.48 | 1.66, 25.03 | 1.75, 27.48 |
| h min, max | −17, 17 | −17, 17 | −16, 16 |
| k min, max | −23, 23 | −32, 32 | −34, 34 |
| l min, max | −31, 31 | −23, 23 | −21, 21 |
| Measured rfl | 70155 | 88683 | 82309 |
| Indep. rfl, Rint | 7604 0.0372 | 13200, 0.0921 | 13161, 0.0572 |
| Compl.,%; θ, ° | 100.0, 25.25 | 100.0, 25.03 | 100.0, 27.48 |
| Abs corr. | semi-empirical from equivalents | ||
| Max/min trans. | 0.9842, 0.9692 | 0.9918, 0.8738 | 0.9843, 0.9016 |
| Data | 7604 | 13200 | 13161 |
| Restraints | 84 | 0 | 0 |
| Parameters | 762 | 785 | 673 |
| GOF | 1.039 | 0.985 | 1.011 |
| R1 (I > 2σI) | 0.0386 | 0.0654 | 0.0532 |
| wR2 (all data) | 0.1077 | 0.1786 | 0.1478 |
| Max, min, e∙Å−3 | 0.21, −0.16 | 0.25, −0.19 | 0.26, −0.19 |
References
- Cho, M.H.; Yoon, J.S.; Lee, I.M. Efficient bimodal ring-opening polymerization of ε-caprolactone catalyzed by titanium complexes with N-alkoxy-β-ketoiminate ligands. Bull. Korean Chem. Soc. 2007, 28, 2471–2476. [Google Scholar]
- Wang, L.Y.; Wu, Q. Ethylene polymerization catalyzed by Pd-based β-ketoiminato complexes/MAO systems. Eur. Polym. J. 2007, 43, 3970–3975. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, Z.; Qi, R.; Yao, Y.; Zhang, Y.; Shen, Q.; Cheng, Y. Synthesis, reactivity, and characterization of sodium and rare-earth metal complexes bearing a dianionic N-aryloxo-functionalized β-ketoiminate ligand. Inorg. Chem. 2008, 47, 9828–9835. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Cho, M.H.; Kwon, I.H.; Jun, I.C.; Lee, I.M.; Jin, M.J. Palladium phosphanyl-β-ketoiminate complexes as highly active catalysts for Heck coupling reaction. Catal. Commun. 2008, 9, 1517–1520. [Google Scholar] [CrossRef]
- Lim, S.W.; Lee, J.C.; Shon, D.S.; Lee, W.I.; Lee, I.M. A study on the development of chemical vapor deposition precursors. 4. Syntheses and characterization of new N-alkoxo-β-ketoiminate complexes of niobium and tantalum. Chem. Mater. 2002, 14, 1548–1554. [Google Scholar] [CrossRef]
- Lim, S.W.; Choi, B.; Min, Y.S.; Lee, S.S. A study on the development of CVD precursors V—Syntheses and characterization of new N-alkoxy-β-ketoiminate complexes of titanium. J. Organomet. Chem. 2004, 689, 224–237. [Google Scholar] [CrossRef]
- Yu, R.C.; Hung, C.H.; Huang, J.H.; Lee, H.Y.; Chen, J.T. Four- and five-coordinate aluminum ketiminate complexes: Synthesis, characterization, and ring-opening polymerization. Inorg. Chem. 2002, 41, 6450–6455. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.C.; Chen, I.C.; Chang, J.C.; Lee, M.T.; Hu, C.H.; Hung, C.H.; Lee, H.M.; Huang, J.H. Hydroalumination of carbon dioxide, carbon disulfide, and phenyl isocyanate with an aluminum ketiminate compound. Eur. J. Inorg. Chem. 2004, 24, 4898–4906. [Google Scholar] [CrossRef]
- Ouattara, T.S.; Butcher, R.J.; Matthews, J.S. Synthesis and characterization of bis[4-N-(cyclohexylimino)-2-pentanonato]magnesium(II). J. Coord. Chem. 2005, 58, 461–465. [Google Scholar] [CrossRef]
- Jones, D.; Roberts, A.; Cavell, K.; Keim, W.; Englert, U.; Skelton, B.; White, A. Synthesis of new adducts and co-ordination complexes of zirconium and titanium containing β-aminoketone ligands. Crystal structures of isostructural adducts MCl4∙2PriHNCMe=CHCMe=O (M = Ti or Zr) and the complex [Zr(PhNCMe =CHCMe=O)2Cl2]. J. Chem. Soc. Dalton Trans. 1998, 2, 255–262. [Google Scholar] [CrossRef]
- Hsu, S.; Chang, J.; Lai, C.; Hu, C.; Lee, H.M.; Lee, G.; Peng, S.; Huang, J. Terminal titanium-ligand multiple bonds. Cleavages of CdO and CdS double bonds with Ti imido complexes. Inorg. Chem. 2004, 43, 6786–6792. [Google Scholar] [CrossRef] [PubMed]
- Brehon, M.; Cope, E.K.; Mair, F.S.; Nolan, P.; O’Brien, J.E.; Pritchard, R.G.; Wilcock, D.J. Structural studies of lithiated enaminones: the 1-oxa-5-azapentadienyllithium fluxional heterocubane [(PriNCMeCHCMeOLi)4] and its dimeric hexamethylphosphoric triamide complex [{PriNCMeCHCMeOLi∙OP(NMe2)3}2]. J. Chem. Soc. Dalton Trans. 1997, 19, 3421–3425. [Google Scholar] [CrossRef]
- Olejník, R.; Padělková, Z.; Horáček, M.; Růžiča, A. Structure of β-diketiminates and β-aminoketones made from anisidines or chloroanilines: tin and lithium complexes. Main Group Met. Chem. 2012, 35, 13–27. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, H.-X.; Huang, D.; Zhang, Y.; Yao, Y.-M. A facile route to lithium complexes supported by β-ketoiminate ligands and their reactivity. J. Organomet. Chem. 2014, 749, 7–12. [Google Scholar] [CrossRef]
- Clegg, W.; Dale, S.H.; Graham, D.V.; Harrington, R.W.; Hevia, E.; Hogg, L.M.; Kennedy, A.R.; Mulvey, R.E. Structural variations in bimetallic sodium–magnesium and sodium–zinc ketimides, and a sodium–zinc alkide–alkoxide–amide: Connections to ring-stacking, ring-laddering, and inverse crown concepts. Chem. Commun. 2007, 16, 1641–1643. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.; Schleyer, P.v.R.; Snaith, R. Structures of organonitrogen–lithium compounds: Recent patterns and perspectives in organolithium chemistry. Adv. Inorg. Chem. 1991, 37, 47–142. [Google Scholar]
- Mulvey, R.E. Meldola Medal Lecture. Ring-stacking and ring-laddering in organonitrogenlithium compounds: The development of concepts with wide applicability throughout lithium structural chemistry. Chem. Soc. Rev. 1991, 20, 167–209. [Google Scholar] [CrossRef]
- Downward, A.; Chivers, T. Applications of the laddering principle—A two-stage approach to describe lithium heterocarboxylates. Eur. J. Inorg. Chem. 2001, 9, 2193–2201. [Google Scholar] [CrossRef]
- Neculai, D.; Neculai, A.M.; Roesky, H.W.; Magul, J.; Bunkòczi, G. Synthesis and structure of a new fluorinated β-ketoiminato ligand and its lithium derivative. J. Fluor. Chem. 2002, 118, 131–134. [Google Scholar] [CrossRef]
- Hsu, S.-H.; Li, C.-Y.; Chiu, Y.-W.; Chiu, M.-C.; Lien, Y.-L.; Kuo, P.-C.; Lee, H.M.; Huang, J.-H.; Cheng, C.-P. Synthesis and characterization of Cu(I) and Cu(II) complexes containing ketiminate ligands. J. Organomet. Chem. 2007, 692, 5421–5428. [Google Scholar] [CrossRef]
- Lyashenko, G.; Saischek, G.; Judmaier, M.E.; Volpe, M.; Baumgartner, J.; Belaj, F.; Jancik, V.; Herbst-Irmer, R.; Mosch-Zanetti, N.C. Oxo-molybdenum and oxo-tungsten complexes of Schiff bases relevant to molybdoenzymes. Dalton Trans. 2009, 29, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Granum, D.M.; Riedel, P.J.; Crawford, J.A.; Mahle, T.K.; Wyss, C.M.; Begej, A.K.; Arulsamy, N.; Pierce, B.S.; Mehn, M.P. Synthesis and characterization of sterically encumbered β-ketoiminate complexes of iron(II) and zinc(II). Dalton Trans. 2011, 40, 5881–5890. [Google Scholar] [CrossRef] [PubMed]
- Kakaliou, L.; Scanlon, W.J., IV; Qian, B.; Baek, S.W.; Smith, M.R., III; Motry, D.H. Five- and six-coordinate group 4 compounds stabilized by β-ketiminate and diketiminate ligands: Syntheses and comparisons between solid-state and solution structures. Inorg. Chem. 1999, 38, 5964–5977. [Google Scholar] [CrossRef] [PubMed]
- Rat, C.; Comsa, C.; Silvestru, C. Dichlorido[(Z)-4-(2,6-diisopropylanilino)pent-3-en-2-one]dimethyltin(IV). Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, m130. [Google Scholar] [CrossRef] [PubMed]
- Comşa, C.; Cristea, A.; Varga, R.A.; Silvestru, C. Organotin(IV) complexes of β-ketimines. Crystal and molecular structure of OC(Me)CHC(Me)NHR-4 [R = C6H3iPr2-2′,6′; C6H4Me-4′], Bu2SnCl2(L) and [{Me2SnCl}2(L)]2 [L = OC(Me)CHC(Me)NH(C6H3iPr2-2′,6′)-4]. Rev. Roum. Chim. 2010, 55, 811–822. [Google Scholar]
- Lugo, A.F.; Richards, A.F. Ketiminate-supported LiCl cages and group 13 complexes. Eur. J. Inorg. Chem. 2010, 13, 2025–2035. [Google Scholar] [CrossRef]
- Lesikar, L.A.; Gushwa, A.F.; Richards, A.F. Synthesis, characterization, and steric hindrance comparisons of selected transition and main group metal β-ketoiminato complexes. J. Organomet. Chem. 2008, 693, 3245–3255. [Google Scholar] [CrossRef]
- Lugo, A.F.; Richards, A.F. Ketiminato and ketimine Co, Eu, Cu and Fe complexes. Inorg. Chim. Acta 2010, 363, 2104–2112. [Google Scholar] [CrossRef]
- Boeré, R.T.; Klassen, V.; Wolmershäuser, G. Synthesis of some very bulky N,N′-disubstituted amidines and initial studies of their coordination chemistry. J. Chem. Soc. Dalton Trans. 1998, 24, 2341–2345. [Google Scholar] [CrossRef]
- Boeré, R.T.; Klassen, V.; Wolmershäuser, G. Superamidines 2. Synthesis of the bulky ligand N,N′-bis-(2,6-diisopropylphenyl)-trifluoroacetamidine and its molybdenum carbonyl complex. Can. J. Chem. 2000, 78, 583–589. [Google Scholar] [CrossRef]
- Boeré, R.E.; Boeré, R.T.; Masuda, J.; Wolmershäuser, G. Preparation, X-ray structure, and dynamic solution behaviour of N,N′,N″-tris(2,6-diisopropylphenyl)-guanidine, and its reaction with molybdenum carbonyl. Can. J. Chem. 2000, 78, 1613–1619. [Google Scholar] [CrossRef]
- Boeré, R.T.; Cole, M.L.; Junk, P.C.; Masuda, J.D.; Wolmershäuser, G. An N,P-disubstituted-2-aminophosphaalkene and lithium and potassium complexes of the deprotonated “phosphaamidinate” anion. Chem. Commun. 2004, 22, 2564–2565. [Google Scholar] [CrossRef] [PubMed]
- Boeré, R.T.; Cole, M.L.; Junk, P.C. The syntheses and structures of some main group complexes of the sterically hindered N,N′-bis(2,6-diisopropylphenyl)-4-toluamidinate ligand. New J. Chem. 2005, 29, 128–134. [Google Scholar] [CrossRef]
- Boeré, R.T.; Roemmele, T.L.; Suduweli Kondage, S.; Zhou, J.; Parvez, M. Five related N′-(2,2,2-trichloroethanimidoyl)benzene-1-carboximidamides. Acta Cryst. 2011, C67, o273–o277. [Google Scholar]
- Roemmele, T.L.; Boeré, R.T. 4-Methyl-N′-(2,2,2-trichloroethanimidoyl)benzene-1-carboximidamide. Acta Cryst. 2011, E67, o3137. [Google Scholar] [CrossRef] [PubMed]
- Vitanova, D.V.; Hampel, F.; Hultzsch, K.C. Rare earth metal complexes based on β-diketiminato and novel linked bis(β-diketiminato) ligands: Synthesis, structural characterization and catalytic application in epoxide/CO2-copolymerization. J. Organomet. Chem. 2005, 690, 5182–5197. [Google Scholar] [CrossRef]
- Boeré, R.T.; Gietz, T. Butylbis[μ-4-(2,4,6-trimethylphenylamino)pent-3-en-2-onato][4-(2,4,6-trimethylphenylamino)pent-3-en-2-onato]dimagnesium. Acta Cryst. 2009, E65, m1137–m1138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-H.; Yin, L.; Wang, Y.-M. A general and efficient method for the preparation of β-enamino ketones and esters catalyzed by indium tribromide. Adv. Synth. Catal. 2006, 348, 184–190. [Google Scholar] [CrossRef]
- Dudek, G.O.; Holm, R.H. Nuclear magnetic resonance studies of keto-enol equilibria. III. α,β-unsaturated-β-ketoamines. J. Am. Chem. Soc. 1962, 84, 2691–2696. [Google Scholar] [CrossRef]
- Benito-Garagorri, D.; Kirchner, K.; Mereiter, K. CCDC 274968: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2006. [Google Scholar] [CrossRef]
- Wright, J.A. CCDC 661234: Experimental Crystal Structure Determination; Cambridge Crystallographic Data Centre: Cambridge, UK, 2008. [Google Scholar] [CrossRef]
- Lin, T.-H.; Das, K.; Datta, A.; Leu, W.-J.; Hsiao, H.-C.; Lin, C.-H.; Guh, J.-H.; Huang, J.-H. Synthesis and characterization of ruthenium compounds incorporating keto-amine ligands. The applications of catalytic transfer hydrogenation and cancer cell inhibition. J. Organomet. Chem. 2016, 807, 22–28. [Google Scholar] [CrossRef]
- Boeré, R.T.; Zhang, Y. Extremely bulky triarylphosphines incorporating 2,6-diisopropylphenyl substituents; consideration of steric shielding and steric pressure. J. Organomet. Chem. 2005, 690, 2651–2657. [Google Scholar] [CrossRef]
- Zhang, L.; Brookhart, M.; White, P.S. Synthesis, characterization, and ethylene polymerization activities of neutral nickel(II) complexes derived from anilino-substituted enone ligands bearing trifluoromethyl and trifluoroacetyl substituents. Organometallics 2006, 25, 1868–1874. [Google Scholar] [CrossRef]
- Kascheres, C.; Negri, G.; Gambardella, M.T.P.; Santos, R.H.A. X-ray crystal structure and AM1 optimized sctructure for 4-methylamino-3-diphenylacetyl-3-penten-2-one. J. Braz. Chem. Soc. 1994, 5, 31–37. [Google Scholar] [CrossRef]
- Kabak, M.; Elmali, A.; Elerman, Y. Tautomeric properties, conformations and structure of N-(2-hydroxyphenyl)-4-amino-3-penten-2-on. J. Mol. Struct. 1998, 470, 295–300. [Google Scholar] [CrossRef]
- Lide, D.R. Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Raissi, H.; Bakavol, M.; Jimenez-Fabian, I.; Tajabadi, J.; Mdoshfeghi, E.; Jalbout, A.F. Effect of substitution on the intramolecular hydrogen bonding of 4-amino-3-penten-2-one: Ab initio, AIM and NBO studies. J. Mol. Struct. THEOCHEM 2007, 847, 47–51. [Google Scholar] [CrossRef]
- APEX2, SAINT-Plus and SADABS; Bruker AXS Inc.: Madison, WI, USA.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Müller, P. (Ed.) Crystal Structure Refinement: A Crystallographer’s Guide to SHELXL; OUP: Oxford, UK, 2006. [Google Scholar]
- Spek, A.L. PLATON—A multipurpose crystallographic tool. Acta Cryst. 1990, A46, C34. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]


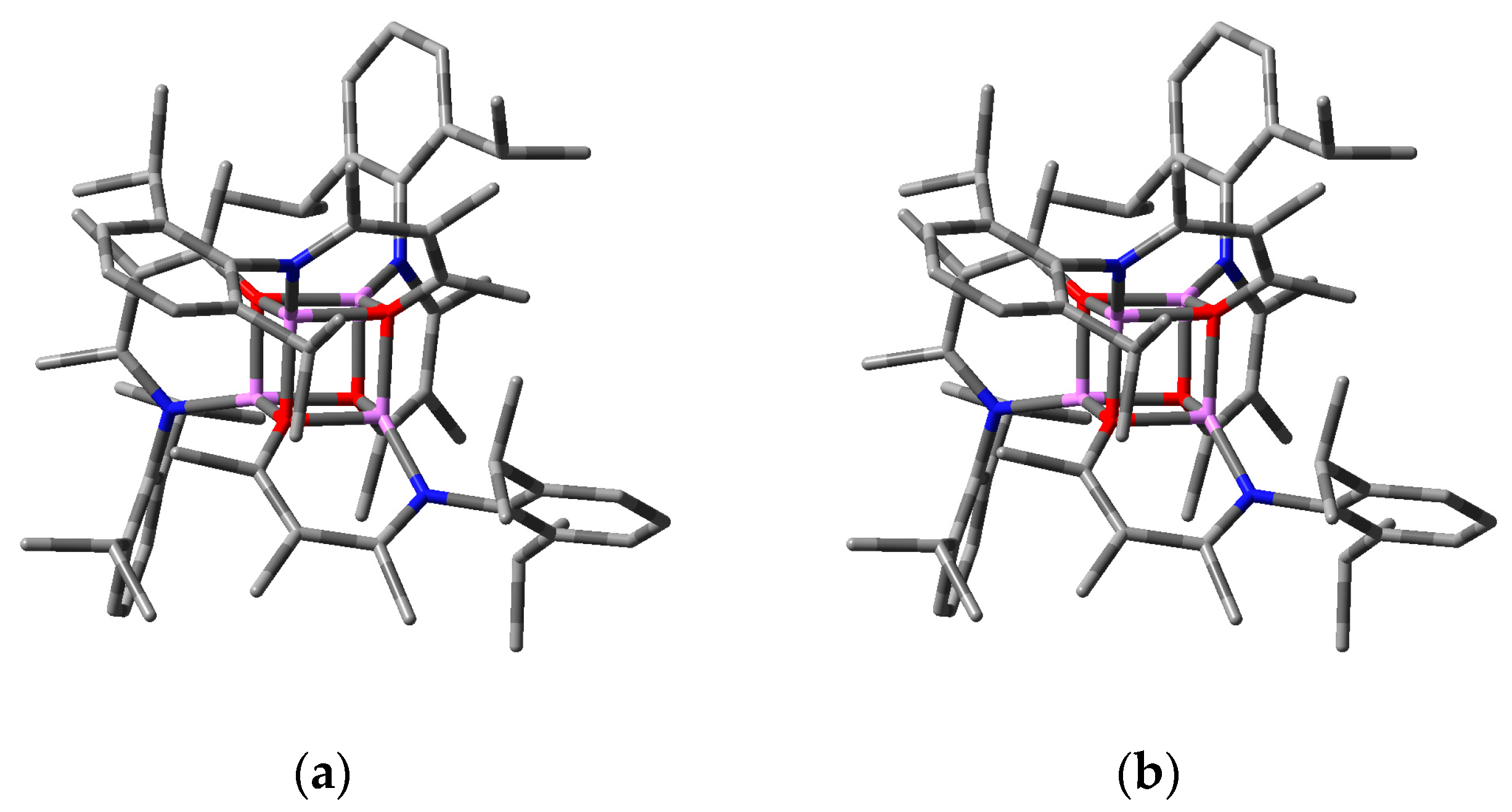
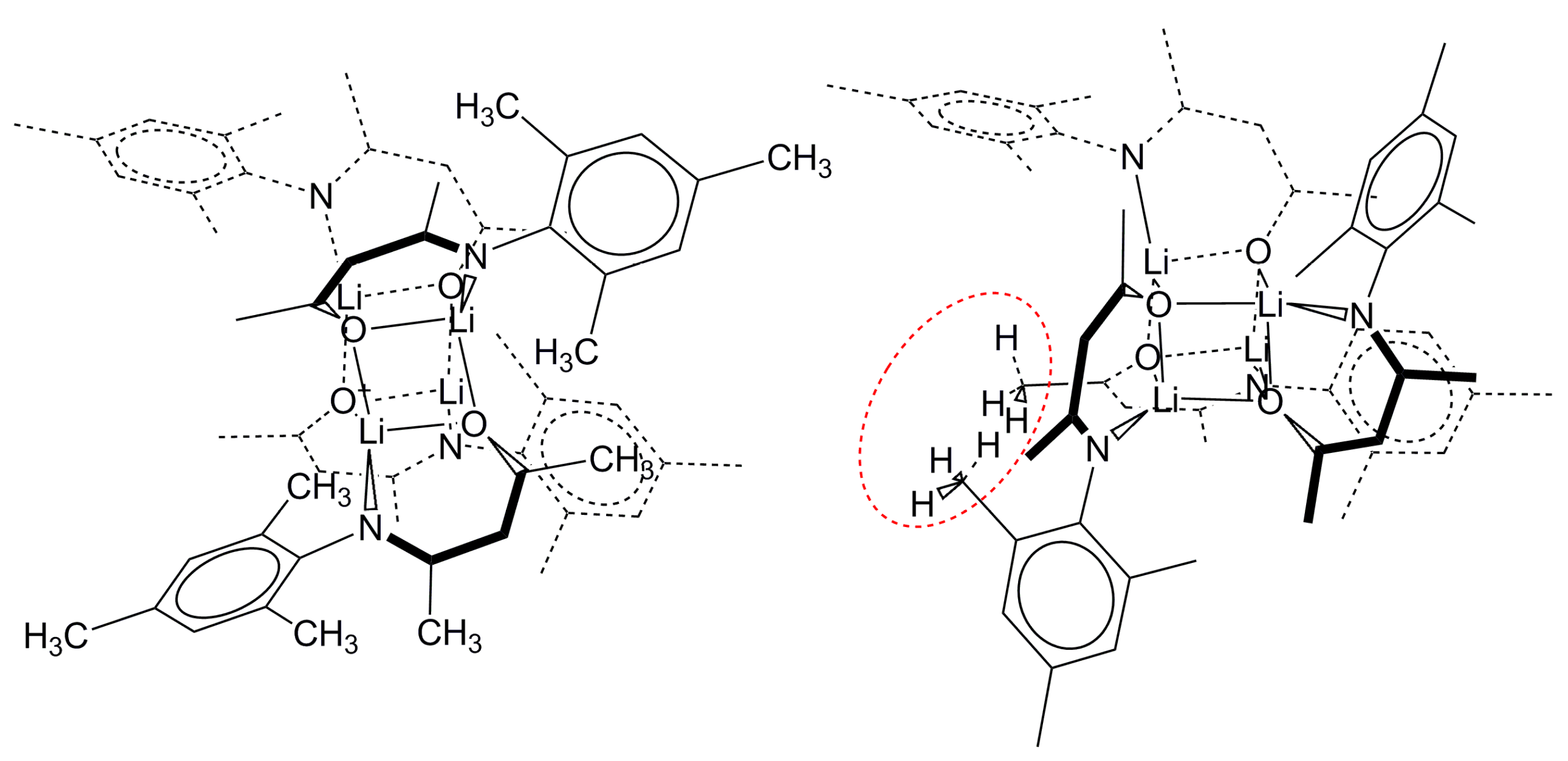
| Parameter | 1a | 1b | 1c | 1d | ||||
|---|---|---|---|---|---|---|---|---|
| X-ray | Calc. | X-ray | Calc. | X-ray | Calc. | X-ray | Calc. | |
| O–C2 | 1.2506(19) | 1.251 | 1.2408(18) | 1.250 | 1.248(2) | 1.252 | 1.2489(17) | 1.252 |
| C2–C3 | 1.421(2) | 1.438 | 1.426(2) | 1.438 | 1.426(2) | 1.450 | 1.430(2) | 1.450 |
| C3–C4 | 1.376(2) | 1.385 | 1.3827(19) | 1.385 | 1.388(2) | 1.395 | 1.3872(19) | 1.395 |
| C4–N | 1.3406(19) | 1.354 | 1.3395(18) | 1.385 | 1.349(2) | 1.358 | 1.3462(17) | 1.358 |
| C7–C3 | 1.512(2) | 1.517 | 1.512(19) | 1.517 | ||||
| O1–C2–C3 | 123.16(14) | 123.50 | 122.99(13) | 123.47 | 123.69(16) | 123.98 | 123.35(12) | 123.99 |
| C2–C3–C4 | 123.22(14) | 123.04 | 123.26(13) | 123.07 | 120.18(14) | 120.40 | 120.59(12) | 120.46 |
| C3–C4–N | 120.46(13) | 120.78 | 121.85(13) | 120.99 | 120.96(16) | 120.99 | 121.93(12) | 120.91 |
| C4–N–C6 | 127.46(13) | 126.85 | 124.92(12) | 126.46 | 120.8(13) | 128.03 | 126.29(11) | 128.37 |
| N–H | 0.885(19) | 1.030 | 0.860(19) | 1.031 | 0.92(2) | 1.032 | 0.898(17) | 1.033 |
| N∙∙∙O | 2.6139(17) | 2.647 | 2.6571(16) | 2.652 | 2.5485(18) | 2.590 | 2.5823(15) | 2.591 |
| N–H∙∙∙O | 141.9(16) | 138.8 | 133.7(16) | 138.2 | 140.9(19) | 140.1 | 140.0(15) | 140.4 |
| N∙∙∙O′ | 2.9840(16) | - | ||||||
| N–H∙∙∙O′ | 136.9(16) | - | ||||||
| Parameter | 2a | 2b | 2c | 2d | |||
|---|---|---|---|---|---|---|---|
| X-ray | Calc. | X-ray | Calc. | X-ray | Calc. | Calc. | |
| O–C2 | 1.2860(19) | 1.290 | 1.283(2) | 1.291 | 1.284(1) | 1.289 | 1.292 |
| C2–C3 | 1.372(2) | 1.390 | 1.377(2) | 1.390 | 1.385(4) | 1.403 | 1.402 |
| C3–C4 | 1.431(2) | 1.437 | 1.432(2) | 1.435 | 1.450(1) | 1.451 | 1.451 |
| C4–N | 1.301(2) | 1.312 | 1.306(2) | 1.312 | 1.307(0) | 1.317 | 1.315 |
| C7–C3 | 1.523(2) | 1.524 | 1.523 | ||||
| Li–N | 2.021(3) | 2.065 | 2.016(3) | 2.022 | 2.022(2) | 2.041 | 2.007 |
| Li–O chelate | 1.899(3) | 1.912 | 1.888(3) | 1.902 | 1.868(1) | 1.882 | 1.881 |
| Li–O bridge | 1.917(3) | 1.939 | 1.948(3) | 1.960 | 1.903(7) | 1.947 | 1.947 |
| Li–O(THF) | 1.988(3) | 2.040 | 1.991(3) | 2.016 | 1.98(2) | 2.037 | 2.026 |
| O–C2–C3 | 125.34(16) | 125.76 | 125.57(16) | 125.81 | 125.47(4) | 125.47 | 125.32 |
| C2–C3–C4 | 128.68(16) | 128.79 | 128.08(16) | 128.20 | 123.66(11) | 124.11 | 123.75 |
| C3–C4–N | 122.86(15) | 123.90 | 123.40(16) | 123.50 | 123.66(15) | 124.15 | 123.82 |
| C4–N–C6 | 120.58(14) | 121.48 | 118.78(14) | 122.10 | 120.76(9) | 121.76 | 122.27 |
| O–Li–N | 95.96(13) | 96.58 | 96.49(13) | 97.03 | 92.28(6) | 92.98 | 92.99 |
| Li–O–Li | 85.89(13) | 85.76 | 86.52(13) | 85.57 | 88.1(15) | 86.73 | 85.74 |
| O–Li–O | 94.11(13) | 94.24 | 93.48(13) | 94.43 | 92.0(15) | 93.27 | 94.26 |
| O–Li–O(THF) | 107.6(11) | 107.81 | 112(9) | 111.02 | 108(3) | 108.96 | 112.06 |
| C4–N–Li | 121.68(14) | 119.82 | 121.33(14) | 121.29 | 122.92(8) | 122.59 | 123.92 |
| C2–O–Li | 123.68(14) | 123.27 | 124.39(14) | 123.70 | 126.68(19) | 127.04 | 127.13 |
| Parameter | 3a | 3b | 3c | 3d | |||||
|---|---|---|---|---|---|---|---|---|---|
| D2 | X-ray | D2 | X-ray | D2 | S4 | X-ray | D2 | S4 | |
| O–C2 | 1.305 | 1.303(2) | 1.306 | 1.310(2) | 1.310 | 1.307 | 1.310(3) | 1.308 | 1.308 |
| C2–C3 | 1.378 | 1.366(2) | 1.382 | 1.370(3) | 1.392 | 1.395 | 1.374(1) | 1.392 | 1.394 |
| C3–C4 | 1.443 | 1.440(2) | 1.442 | 1.460(7) | 1.464 | 1.461 | 1.454(2) | 1.458 | 1.457 |
| C4–N | 1.311 | 1.302(3) | 1.308 | 1.309(2) | 1.314 | 1.318 | 1.305(4) | 1.312 | 1.314 |
| C7–C3 | 1.524(4) | 1.525 | 1.525 | 1.522(4) | 1.524 | 1.524 | |||
| Li–N | 2.078 | 1.988(4) | 2.002 | 2.012(5) | 2.056 | 2.036 | 1.968(12) | 1.983 | 2.003 |
| Li–O chelate | 1.914 | 1.924(5) | 1.931 | 1.874(3) | 1.878 | 1.881 | 1.903(15) | 1.897 | 1.887 |
| Li–O bridge | 2.026 | 1.976(10) | 1.997 | 2.01(4) | 2.046 | 2.049 | 1.98(2) | 2.014 | 2.036 |
| O–C2–C3 | 125.1 | 125.15(16) | 125.3 | 124.8(4) | 124.9 | 124.91 | 125.2(3) | 125.3 | 125.5 |
| C2–C3–C4 | 129.1 | 127.82(37) | 128.9 | 124.0(3) | 124.1 | 125.32 | 123.9(6) | 124.4 | 124.7 |
| C3–C4–N | 124.3 | 122.77(24) | 123.6 | 123.5(3) | 124.8 | 124.96 | 123.3(3) | 123.8 | 124.1 |
| C4–N–C6 | 120.7 | 120.29(98) | 121.5 | 119.1(11) | 120.8 | 120.58 | 121(1) | 122.0 | 121.4 |
| O–Li–N | 96.0 | 94.77(71) | 97.3 | 91.3(4) | 92.3 | 96.08 | 92.7(5) | 93.9 | 94.7 |
| Li–O–Li | 87.7 | 85.7(19) | 85.8 | 86(3) | 86.5 | 87.37 | 84.8(14) | 85.4 | 85.0 |
| O–Li–O | 91.9 | 94(2) | 94.0 | 94(3) | 93.2 | 92.57 | 95.0(15) | 94.4 | 94.0 |
| C4–N–Li | 120.3 | 124.1(6) | 121.7 | 125.7(4) | 123.6 | 119.91 | 123.9(9) | 124.8 | 122.7 |
| C2–O–Li | 125.1 | 125.1(8) | 123.1 | 130.2(4) | 130.0 | 126.04 | 125(3) | 127.2 | 125.9 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gietz, T.; Boeré, R.T. Backbone-Substituted β-Ketoimines and Ketoiminate Clusters: Transoid Li2O2 Squares and D2-Symmetric Li4O4 Cubanes. Synthesis, Crystallography and DFT Calculations. Inorganics 2017, 5, 30. https://doi.org/10.3390/inorganics5020030
Gietz T, Boeré RT. Backbone-Substituted β-Ketoimines and Ketoiminate Clusters: Transoid Li2O2 Squares and D2-Symmetric Li4O4 Cubanes. Synthesis, Crystallography and DFT Calculations. Inorganics. 2017; 5(2):30. https://doi.org/10.3390/inorganics5020030
Chicago/Turabian StyleGietz, Twyla, and René T. Boeré. 2017. "Backbone-Substituted β-Ketoimines and Ketoiminate Clusters: Transoid Li2O2 Squares and D2-Symmetric Li4O4 Cubanes. Synthesis, Crystallography and DFT Calculations" Inorganics 5, no. 2: 30. https://doi.org/10.3390/inorganics5020030
APA StyleGietz, T., & Boeré, R. T. (2017). Backbone-Substituted β-Ketoimines and Ketoiminate Clusters: Transoid Li2O2 Squares and D2-Symmetric Li4O4 Cubanes. Synthesis, Crystallography and DFT Calculations. Inorganics, 5(2), 30. https://doi.org/10.3390/inorganics5020030