
Hetero- and Homoleptic Magnesium Triazenides


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Syntheses and Spectroscopic Characterization
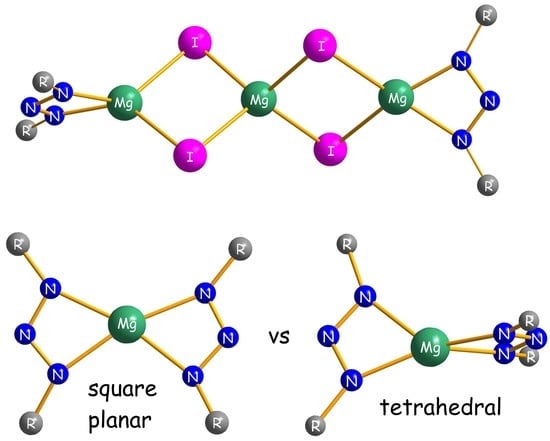
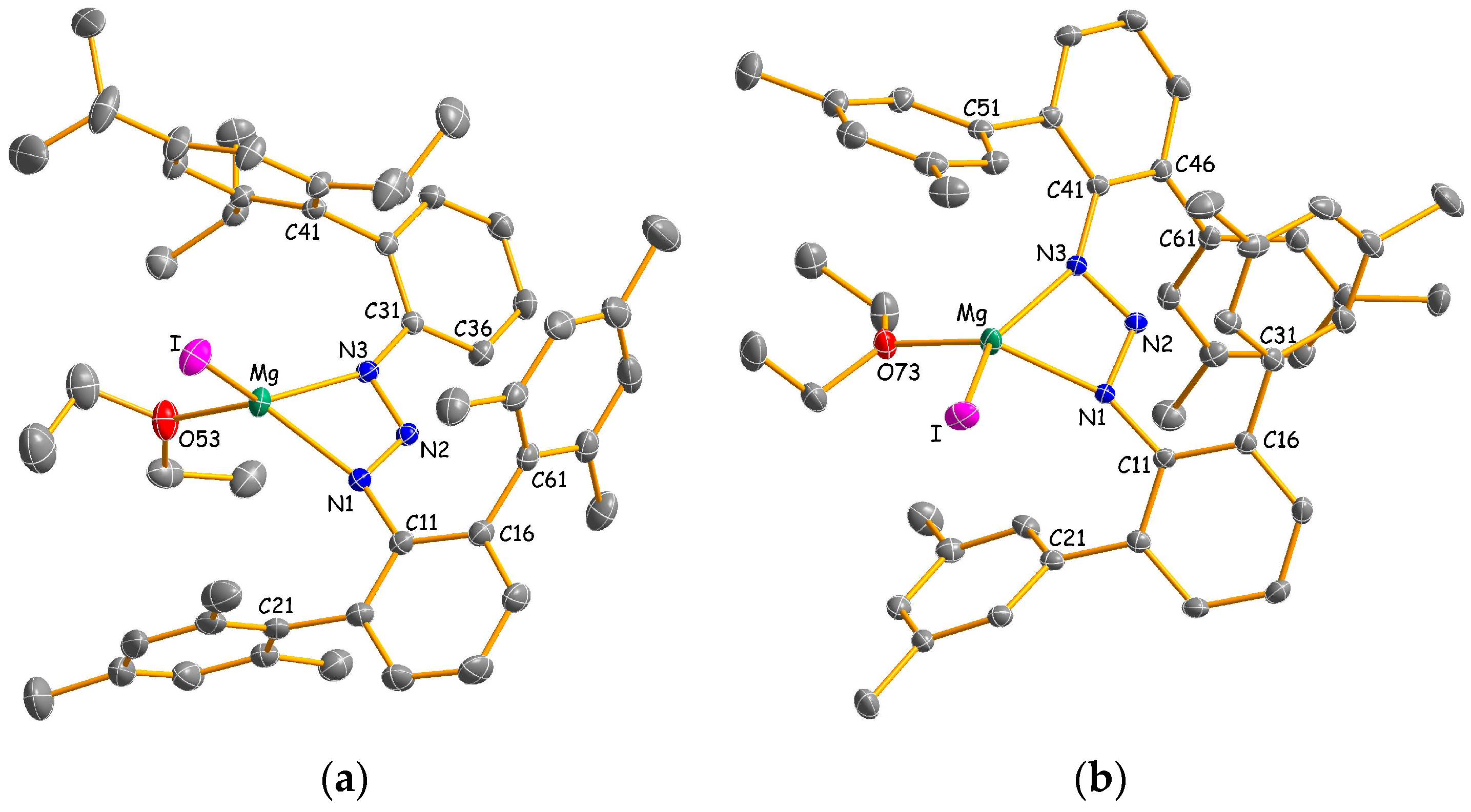
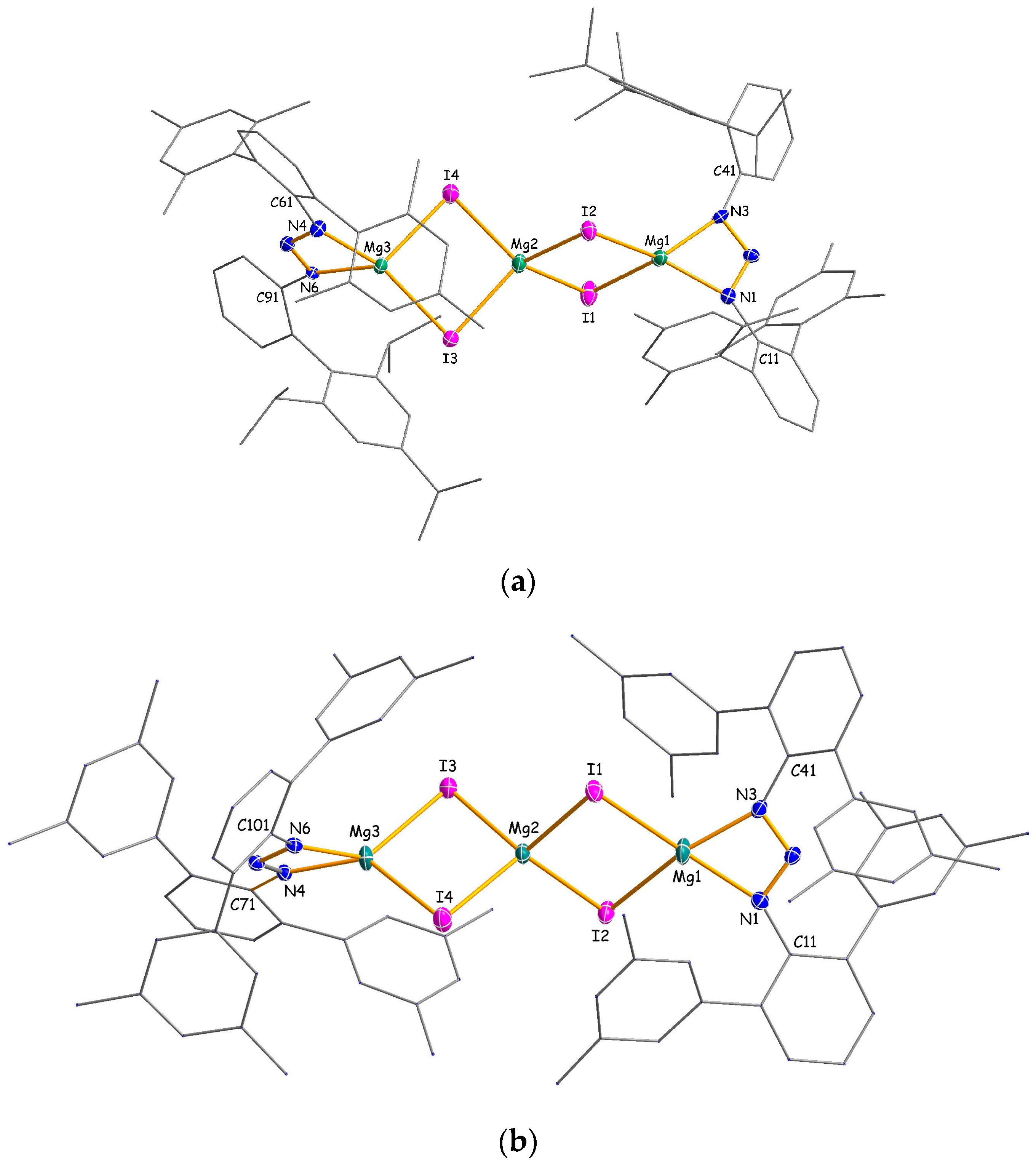
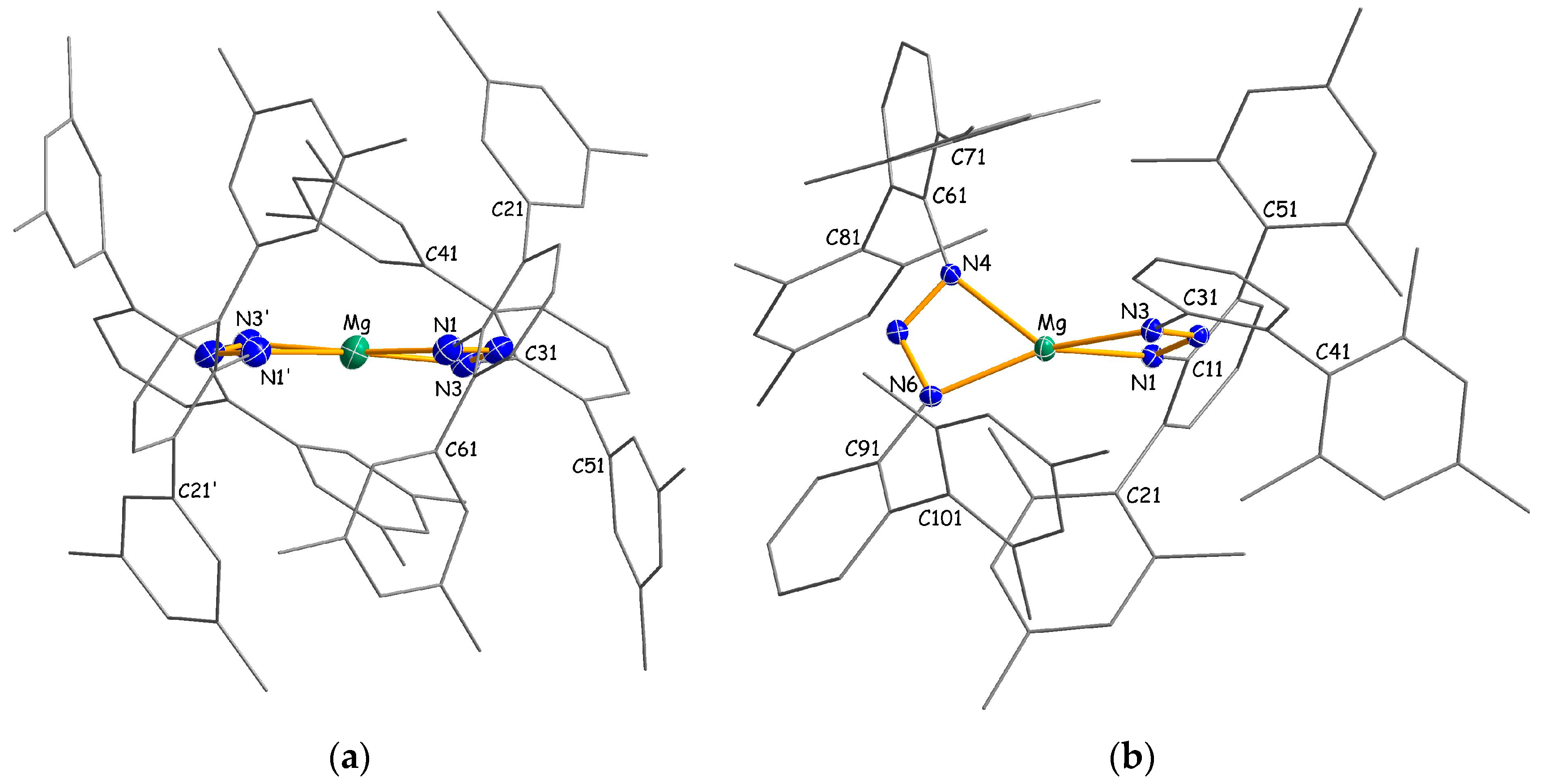
2.2. Structural Studies
3. Materials and Methods
3.1. General Procedures
3.2. Syntheses
3.2.1. Experimental Procedure for [Mg{N3(Dmp)Tph}I(OEt2)] (2a)
3.2.2. Experimental Procedure for [Mg{N3(Me4Ter)2}I(OEt2)] (2b)
3.2.3. Experimental Procedure for [Mg3{N3(Dmp)Tph}2I4] (3a)
3.2.4. Experimental Procedure for [Mg3{N3(Me4Ter)2}2I4] (3b)
3.2.5. Experimental Procedure for [Mg{N3(Me4Ter)2}2] (4b)
3.2.6. Experimental Procedure for [Mg{N3(Dmp)Mph}2] (4c)
3.3. X-Ray Crystallography
3.4. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gibson, V.C.; Spitzmesser, S.K. Advances in Non-Metallocene Olefin Polymerization Catalysis. Chem. Rev. 2003, 103, 283–315. [Google Scholar] [CrossRef] [PubMed]
- Bourget-Merle, L.; Lappert, M.F.; Severn, J.R. The Chemistry of β-Diketiminatometal Complexes. Chem. Rev. 2002, 102, 3031–3065. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.P. Application of neutral amidines and guanidines in coordination chemistry. Dalton Trans. 2006, 37, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, K.; van Koten, G. Sulfurdiimine, Triazenido, Azabutadiene and Triatomic Hetero Anion Ligands. In Comprehensive Coordination Chemistry, 1st ed.; Wilkinson, G., Gillard, R.D., McCleverty, J., Eds.; Pergamon Press: Oxford, UK, 1987; Volume 2, pp. 195–206. [Google Scholar]
- Hauber, S.-O.; Lissner, F.; Deacon, G.B.; Niemeyer, M. Stabilization of Aryl-Calcium, -Strontium, and -Barium Compounds by Designed Steric and π-Bonding Encapsulation. Angew. Chem. Int. Ed. 2005, 44, 5871–5875. [Google Scholar] [CrossRef] [PubMed]
- Hauber, S.-O.; Niemeyer, M. Stabilization of Unsolvated Europium and Ytterbium Pentafluorophenyls by π-Bonding Encapsulation through a Sterically Crowded Triazenido Ligand. Inorg. Chem. 2005, 44, 8644–8646. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Niemeyer, M. Inverse Aggregation Behavior of Alkali-Metal Triazenides. Inorg. Chem. 2006, 45, 6126–6128. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Hauber, S.-O.; Vinduš, D.; Niemeyer, M. Isostructural Potassium and Thallium Salts of Sterically Crowded Triazenes: A Structural and Computational Study. Inorg. Chem. 2008, 47, 4401–4412. [Google Scholar] [CrossRef] [PubMed]
- Balireddi, S.; Niemeyer, M. A sterically crowded triazene: 1,3-Bis(3,5,3′′,5′′-tetramethyl-1,1′:3′;1′′-ter-phenyl-2′-yl)triazene. Acta Crystallogr. 2007, E63, o3525. [Google Scholar] [CrossRef]
- Lee, H.S.; Niemeyer, M. Homoleptic Heavy Alkaline Earth and Europium Triazenides. Inorg. Chem. 2010, 49, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Hauber, S.-O.; Seo, J.W.; Niemeyer, M. Halogenomercury Salts of Sterically Crowded Triazenides—Convenient Starting Materials for Redox-Transmetallation Reactions. Z. Anorg. Allg. Chem. 2010, 636, 750–757. [Google Scholar] [CrossRef]
- Lee, H.S.; Niemeyer, M. Sterically crowded triazenides as novel ancillary ligands in copper chemistry. Inorg. Chim. Acta 2011, 374, 163–170. [Google Scholar] [CrossRef]
- Hinz, A.; Schulz, A.; Villinger, A.; Wolter, J.-M. Cyclo-Pnicta-triazanes: Biradicaloids or Zwitterions? J. Am. Chem. Soc. 2015, 137, 3975–3980. [Google Scholar] [CrossRef] [PubMed]
- Westhusin, S.; Gantzel, P.; Walsh, P.J. Synthesis and Crystal Structures of Magnesium and Calcium Triazenide Complexes. Inorg. Chem. 1998, 37, 5956–5959. [Google Scholar] [CrossRef]
- Kalden, D.; Krieck, S.; Görls, H.; Westerhausen, M. 1,3-Bis(2,4,6-trimethylphenyl)triazenides of potassium, magnesium, calcium, and strontium. Dalton Trans. 2015, 44, 8089–8099. [Google Scholar] [CrossRef] [PubMed]
- Nimitsiriwat, N.; Gibson, V.C.; Marshall, E.L.; Takolpuckdee, P.; Tomov, A.K.; White, A.J.P.; Williams, D.J.; Elsegood, M.R.J.; Dale, S.H. Mono-versus Bis-chelate Formation in Triazenide and Amidinate Complexes of Magnesium and Zinc. Inorg. Chem. 2007, 46, 9988–9997. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bonyhady, S.J.; Jones, C.; Nembenna, S.; Stasch, A.; Edwards, A.J.; McIntyre, G.J. β-Diketiminate-Stabilized Magnesium(I) Dimers and Magnesium(II) Hydride Complexes: Synthesis, Characterization, Adduct Formation, and Reactivity Studies. Chem. Eur. J. 2010, 16, 938–955. [Google Scholar] [CrossRef] [PubMed]
- Stasch, A. Synthesis of a Dimeric Magnesium(I) Compound by an MgI/MgII Redox Reaction. Angew. Chem. Int. Ed. 2014, 53, 10200–10203. [Google Scholar] [CrossRef] [PubMed]
- Moxey, G.J.; Blake, A.J.; Lewis, W.; Kays, D.L. Alkaline Earth Complexes of a Sterically Demanding Guanidinate Ligand. Eur. J. Inorg. Chem. 2015, 2015, 5892–5902. [Google Scholar] [CrossRef]
- MacNeil, C.S.; Johnson, K.R.D.; Hayes, P.G.; Boeré, R.T. Crystal structure of a dimeric β-diketiminate magnesium complex. Acta Crystallogr. 2016, E72, 1754–1756. [Google Scholar] [CrossRef] [PubMed]
- Boutland, A.J.; Dange, D.; Stasch, A.; Maron, L.; Jones, C. Two-Coordinate Magnesium(I) Dimers Stabilized by Super Bulky Amido Ligands. Angew. Chem. Int. Ed. 2016, 55, 9239–9243. [Google Scholar] [CrossRef] [PubMed]
- Brogan, M.A.; Blake, A.J.; Wilson, C.; Gregory, D.H. Magnesium diiodide, MgI2. Acta Crystallogr. 2003, C59, i136–i138. [Google Scholar] [CrossRef]
- Akishin, P.A.; Spiridonov, V.P. Electron-diffraction investigation of magnesium iodide molecular structure. Zhurnal Fizicheskoi Khimii 1958, 32, 1682–1683. [Google Scholar]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 9, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Boeré, R.T.; Cole, M.L.; Junk, P.C. The syntheses and structures of some main group complexes of the sterically hindered N,N′-bis(2,6-diisopropylphenyl)-4-toluamidinate ligand. New J. Chem. 2005, 29, 128–134. [Google Scholar] [CrossRef]
- Moxey, G.J.; Ortu, F.; Goldney Sidley, L.; Strandberg, H.N.; Blake, A.J.; Lewis, W.; Kays, D.L. Synthesis and characterisation of magnesium complexes containing sterically demanding N,N′-bis(aryl)amidinate ligands. Dalton Trans. 2014, 43, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.; El-Kaderi, H.M.; Heeg, M.J.; Winter, C.H. Synthesis, structure, and properties of magnesium complexes containing cyclopentadienyl and amidinate ligand sets. J. Organomet. Chem. 2003, 682, 224–232. [Google Scholar] [CrossRef]
- Sadique, A.R.; Heeg, M.J.; Winter, C.H. Monomeric and Dimeric Amidinate Complexes of Magnesium. Inorg. Chem. 2001, 40, 6349–6355. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.A.R.; Arnold, J. Synthesis and characterization of a series of sterically-hindered amidines and their lithium and magnesium complexes. J. Chem. Soc. Dalton Trans. 2002, 2890–2899. [Google Scholar] [CrossRef]
- Sedai, B.; Heeg, M.J.; Winter, C.H. Magnesium complexes containing β-ketiminate and β-diketiminate ligands with dimethylamino substituents on the ligand core nitrogen atoms. J. Organomet. Chem. 2008, 693, 3495–3503. [Google Scholar] [CrossRef]
- El-Kaderi, H.M.; Xia, A.; Heeg, M.J.; Winter, C.H. Factors that Influence π-versus η2-Coordination of β-Diketiminato Ligands in Magnesium Complexes. Organometallics 2004, 23, 3488–3495. [Google Scholar] [CrossRef]
- Caro, C.F.; Hitchcock, P.B.; Lappert, M.F. Monomeric magnesium 1-azaallyl and β-diketiminato complexes derived from the bis(trimethylsilyl)methyl ligand: The X-ray structure of the four-coordinate planar magnesium complex [Mg{N(R)C(But)C(H)R}2] and of [Mg({N(R)C(Ph)}2CH)2]. Chem. Commun. 1999, 1433–1434. [Google Scholar] [CrossRef]
- Byrn, M.P.; Curtis, C.J.; Goldberg, I.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Strouse, C.E. Porphyrin sponges: Structural systematics of the host lattice. J. Am. Chem. Soc. 1991, 113, 6549–6557. [Google Scholar] [CrossRef]
- Byrn, M.P.; Curtis, C.J.; Hsiou, Y.; Khan, S.I.; Sawin, P.A.; Tendick, S.K.; Terzis, A.; Strouse, C.E. Porphyrin sponges: Conservative of host structure in over 200 porphyrin-based lattice clathrates. J. Am. Chem. Soc. 1993, 115, 9480–9497. [Google Scholar] [CrossRef]
- Mizuguchi, J. Crystal Structure of Magnesiumphthalocyanine and Its Polarized Reflection Spectra. J. Phys. Chem. A 2001, 105, 1121–1124. [Google Scholar] [CrossRef]
- Janczak, J.; Kubiak, R. X-ray single crystal investigations of magnesium phthalocyanine. The 4+1 coordination of the Mg ion and its consequence. Polyhedron 2001, 20, 2901–2909. [Google Scholar] [CrossRef]
- Chandra, T.; Kraft, B.J.; Huffman, J.C.; Zaleski, J.M. Synthesis and Structural Characterization of Porphyrinic Enediynes: Geometric and Electronic Effects on Thermal and Photochemical Reactivity. Inorg. Chem. 2003, 42, 5158–5172. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Djukic, J.-P. Cation-Cation “Attraction”: When London Dispersion Attraction Wins over Coulomb Repulsion. Inorg. Chem. 2011, 50, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Schreiner, P.R. Steric Crowding Can Stabilize a Labile Molecule: Solving the Hexaphenylethane Riddle. Angew. Chem. Int. Ed. 2011, 50, 12639–12642. [Google Scholar] [CrossRef] [PubMed]
- Harder, S.; Naglav, D.; Schwerdtfeger, P.; Nowik, I.; Herber, R.H. Metal Atom Dynamics in Superbulky Metallocenes: A Comparison of (CpBIG)2Sn and (CpBIG)2Eu. Inorg. Chem. 2014, 53, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Franta, U. Synthese, Strukturchemie und Untersuchungen von Aluminium, Ytterbium und Lithiumkomplexen mit Sterisch Anspruchsvollen Triazenido-Liganden; Staatsexamensarbeit; Johannes Gutenberg-Universität: Mainz, Germany, 2011. [Google Scholar]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, A46, 194–201. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON-98; Utrecht University: Utrecht, The Netherlands, 1998. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound 1 | γ (°) | τ4 | Ref. |
|---|---|---|---|
| Triazenides | |||
| [Mg{N3(Me4Ter)2}2] 4b | 9.6 | 0.20 | |
| [Mg{N3((Dmp)Mph)2}2] 4c | 83.6 | 0.51 | |
| Amidinates | |||
| [Mg{DipN{C(pTol)}NDip}2] | 13.3 | 0.10 | [26] |
| [Mg{DipN{C(Me)}NDip}2] 2 | 54.1/54.9 | 0.40/0.41 | [16] |
| [Mg{DipN{C(cHex)}NDip}2] | 61.3 | 0.45 | [27] |
| [Mg{DipN{C(3,5-Me2C6H3)}NDip}2] | 76.4 | 0.56 | [27] |
| [Mg{MesN{C(tBu)}NMes}2] | 80.3 | 0.57 | [28] |
| [Mg{tBuN{C(Ph)}NtBu}2] | 89.5 | 0.58 | [29] |
| [Mg{iPrN{C(Dmp)}NiPr}2] | 88.1 | 0.60 | [30] |
| Guanidinates | |||
| [Mg{MesN{C(NcHex)}NMes}2] | 8.6 | 0.06 | [20] |
| β-Diketiminates | |||
| [Mg(HC{C(Me)N(NiPr2)}2)2] | 89.5 | 0.83 | [31] |
| [Mg(HC{C(Me)N(iPr)}2)2] | 88.9 | 0.88 | [32] |
| [Mg(HC{C(Me)N(tBu)}2)2] | 88.4 | 0.92 | [32] |
| [Mg(HC{C(Ph)N(SiMe3)}2)2] | 89.0 | 0.92 | [33] |
| Compound 1 | Cn | Mg–N | N–Mg–N | Mg–N–C | Ref. |
|---|---|---|---|---|---|
| [Mg{N3(Dmp)Tph}I(OEt2)] 2a | 4 | 2.102 | 61.5 | 151.4 | |
| [Mg{N3(Me4Ter)2}I(OEt2)] 2b | 4 | 2.098 | 61.0 | 147.3 | |
| [Mg3I4{N3(Dmp)Tph)}2] 3a | 4 | 2.074 | 62.4 | 152.0 | |
| [Mg3I4{N3(Me4Ter)2}2] 3b | 4 | 2.070 | 61.8 | 148.9 | |
| [Mg{N3(Me4Ter)2}2] 4b | 4 | 2.128 | 61.0 | 151.3 | |
| [Mg{N3(Dmp)Mph)}2] 4c | 4 | 2.086 | 61.6 | 145.5 | |
| [Mg{N3(Dmp)Tph}I(thf)] | 4 | 2.093 | 61.9 | 147.0 | [11] |
| [Mg{N3(Dip)2}2(OEt2)] | 5 | 2.137 | 60.2 | 150.6 | [16] |
| [Mg(nBu){N3(Mes)2}(tmeda)] | 5 | 2.202 | 58.2 | 150.3 | [15] |
| [Mg{N3(pTol)2}2(thf)2] | 6 | 2.183 | 58.8 | 149.6 | [14] |
| [Mg{N3(Mes)2}2(thf)2] | 6 | 2.181 | 59.1 | 150.2 | [15] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinduš, D.; Niemeyer, M. Hetero- and Homoleptic Magnesium Triazenides. Inorganics 2017, 5, 33. https://doi.org/10.3390/inorganics5020033
Vinduš D, Niemeyer M. Hetero- and Homoleptic Magnesium Triazenides. Inorganics. 2017; 5(2):33. https://doi.org/10.3390/inorganics5020033
Chicago/Turabian StyleVinduš, Denis, and Mark Niemeyer. 2017. "Hetero- and Homoleptic Magnesium Triazenides" Inorganics 5, no. 2: 33. https://doi.org/10.3390/inorganics5020033
APA StyleVinduš, D., & Niemeyer, M. (2017). Hetero- and Homoleptic Magnesium Triazenides. Inorganics, 5(2), 33. https://doi.org/10.3390/inorganics5020033