
[Bis(Trimethylsilyl)Methyl]Lithium and -Sodium: Solubility in Alkanes and Complexes with O- and N- Donor Ligands


Abstract
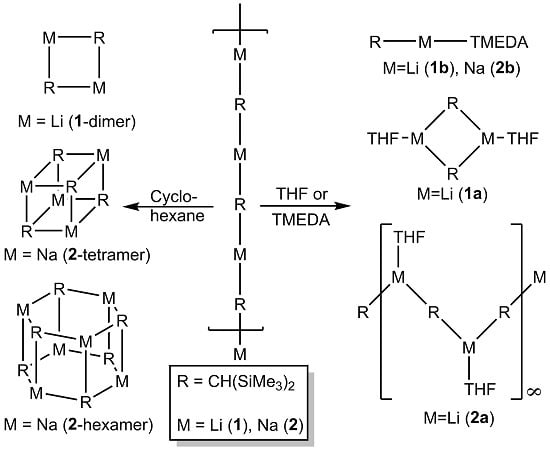
:
1. Introduction
2. Results and Discussion
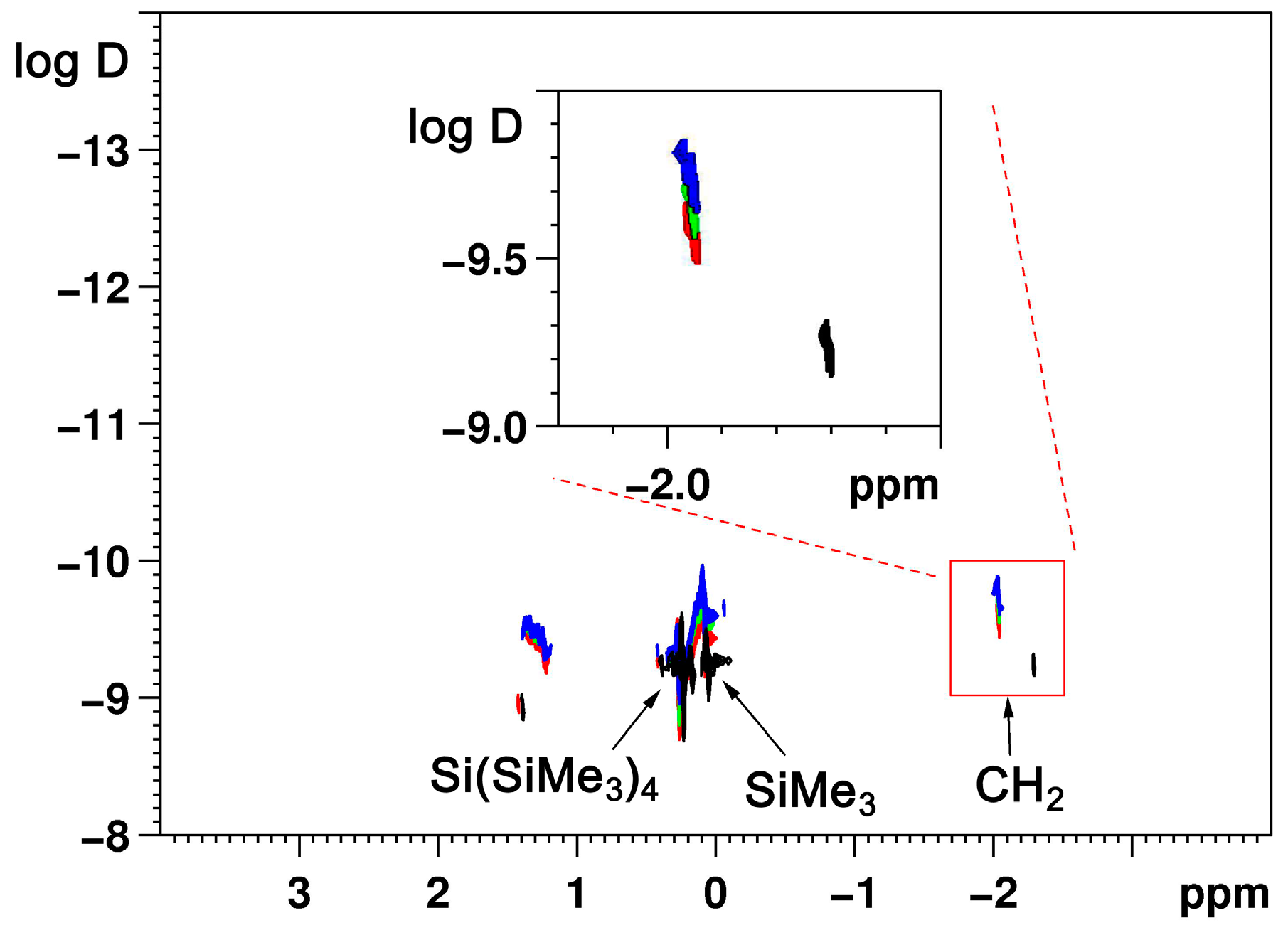
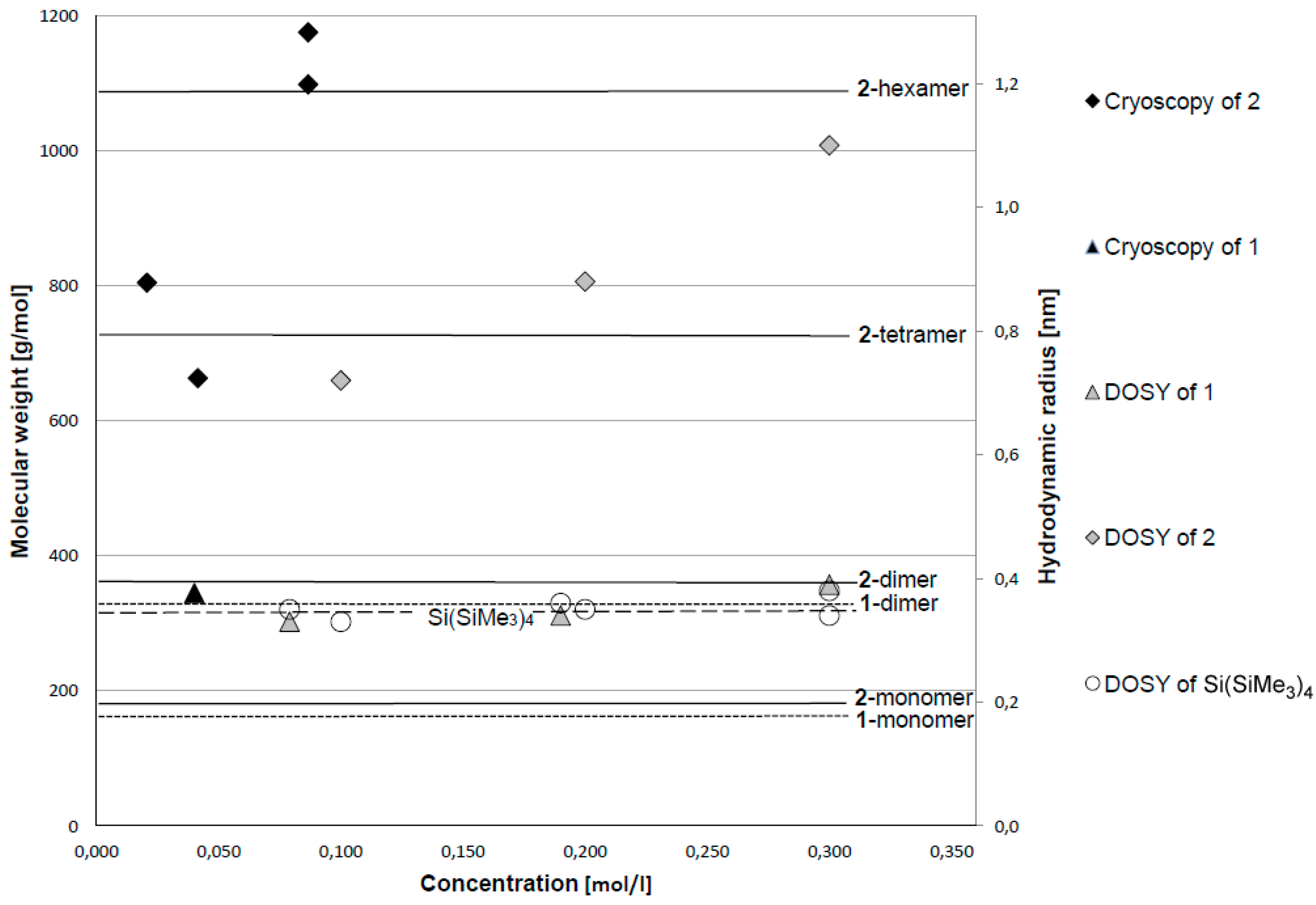
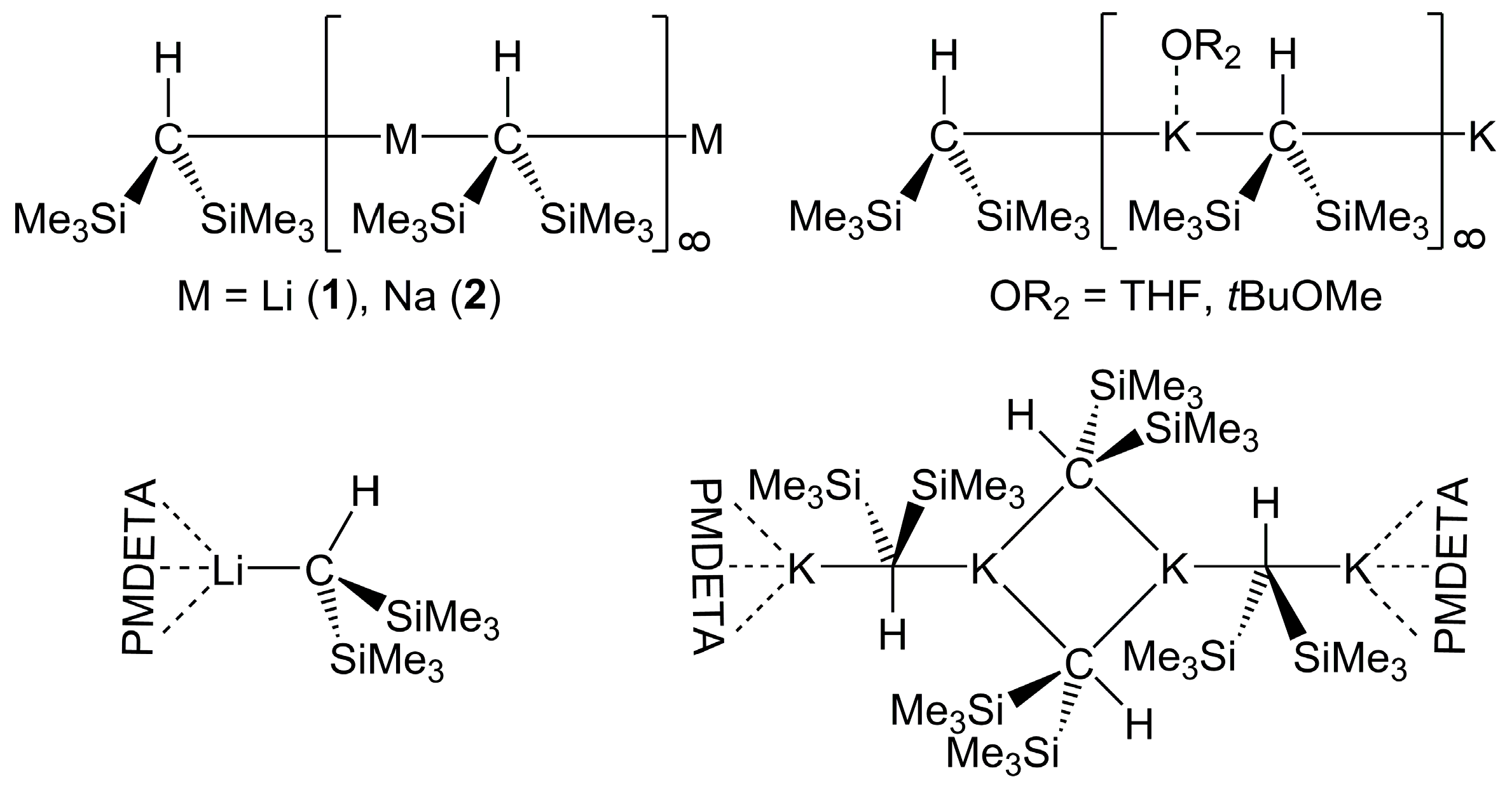
2.1. Bis(Trimethylsilyl)Methyllithium 1 and -Sodium 2 in Solution
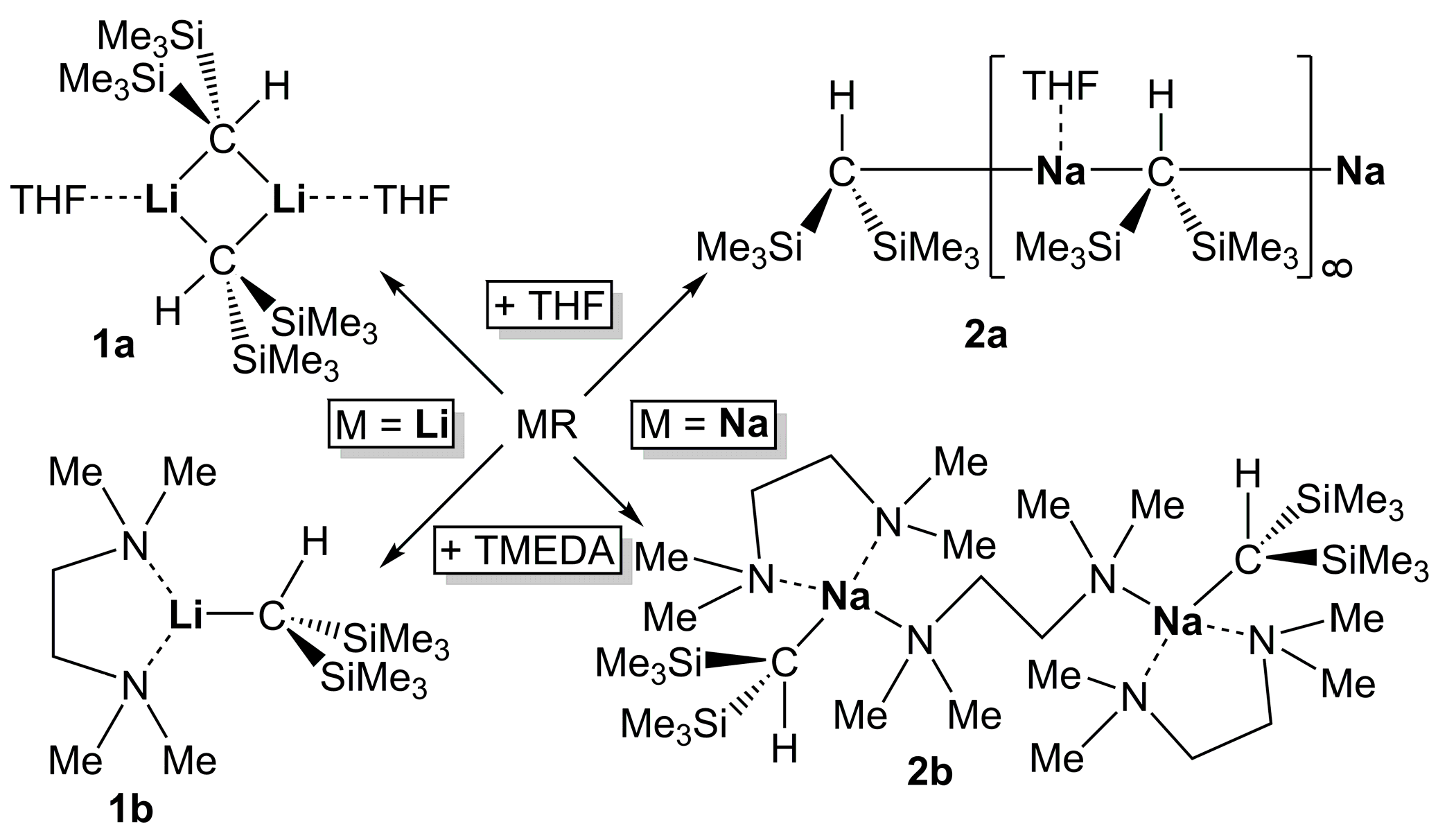
2.2. Formation of Complexes of Compounds 1 and 2 with O- and N- Donors
2.3. NMR-Spectroscopy of Complexes of Compounds 1 and 2 with O- and N- Donors
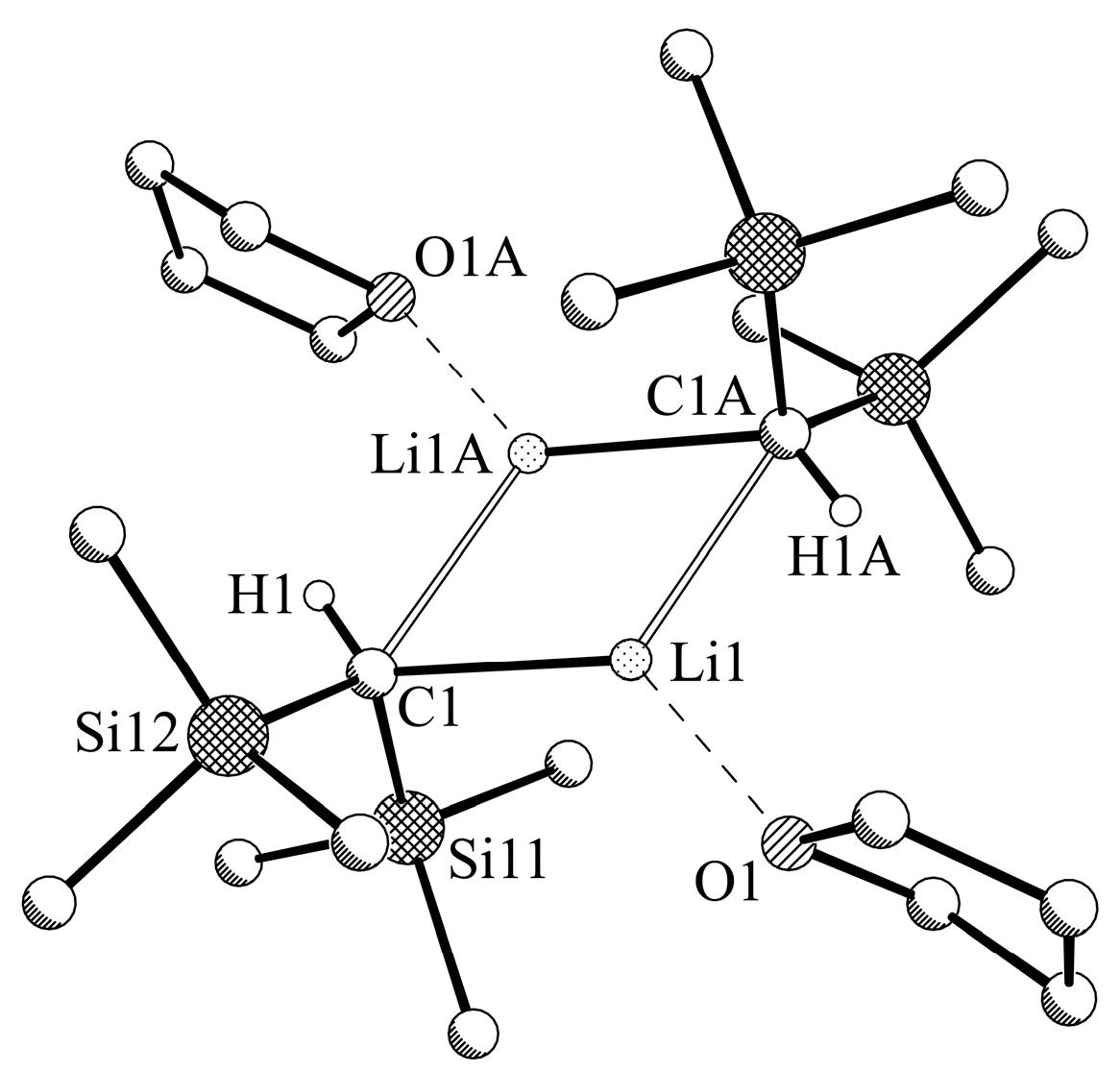
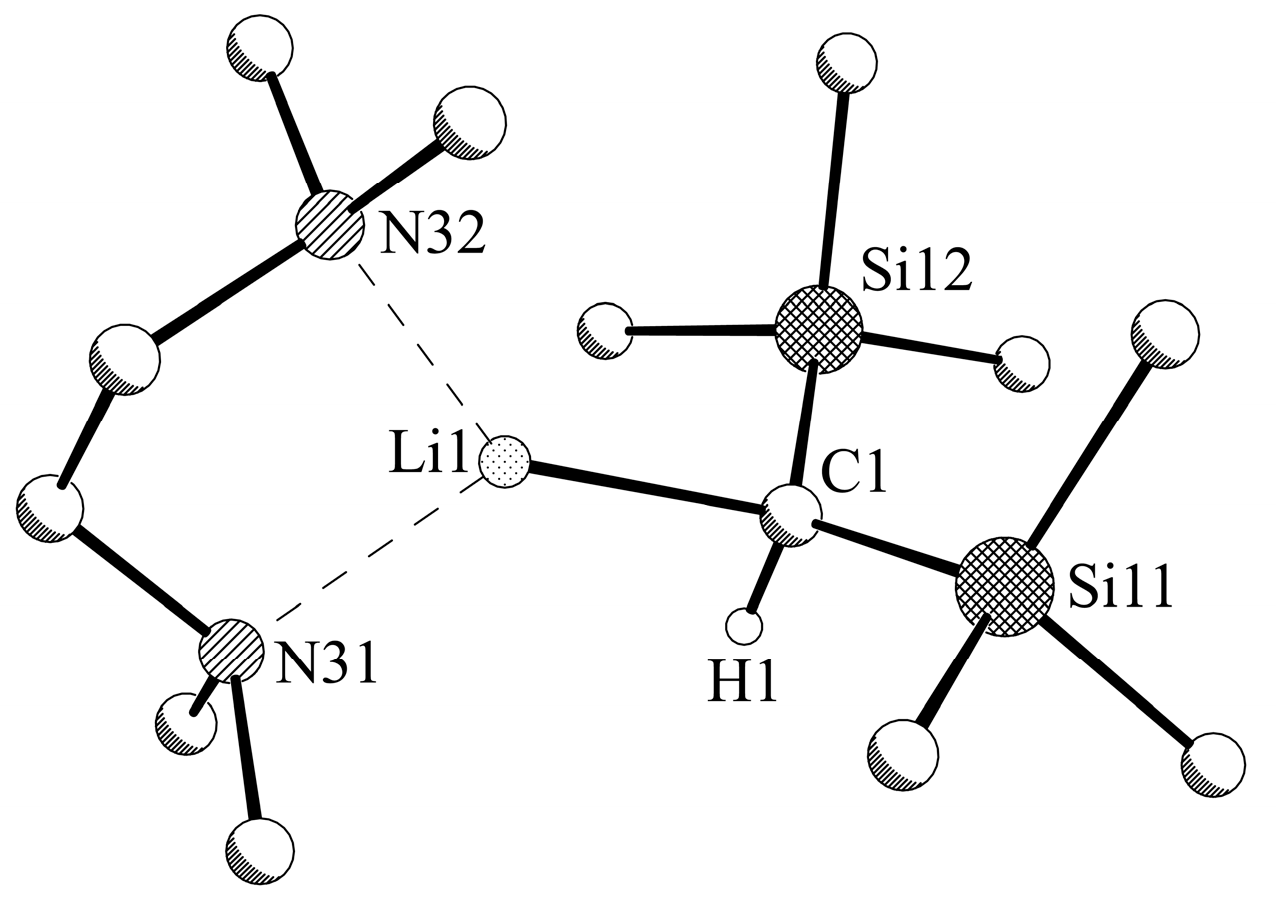
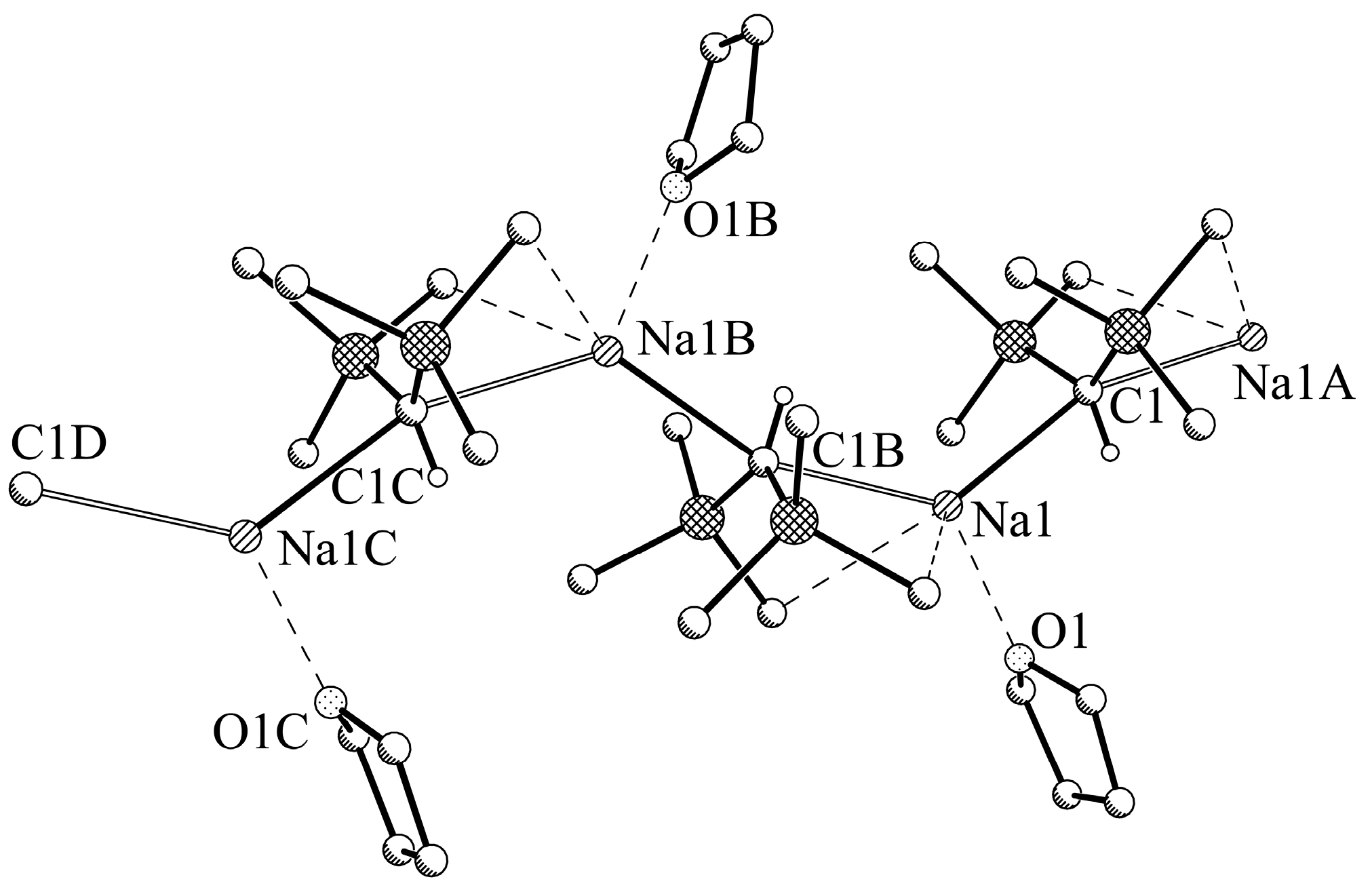
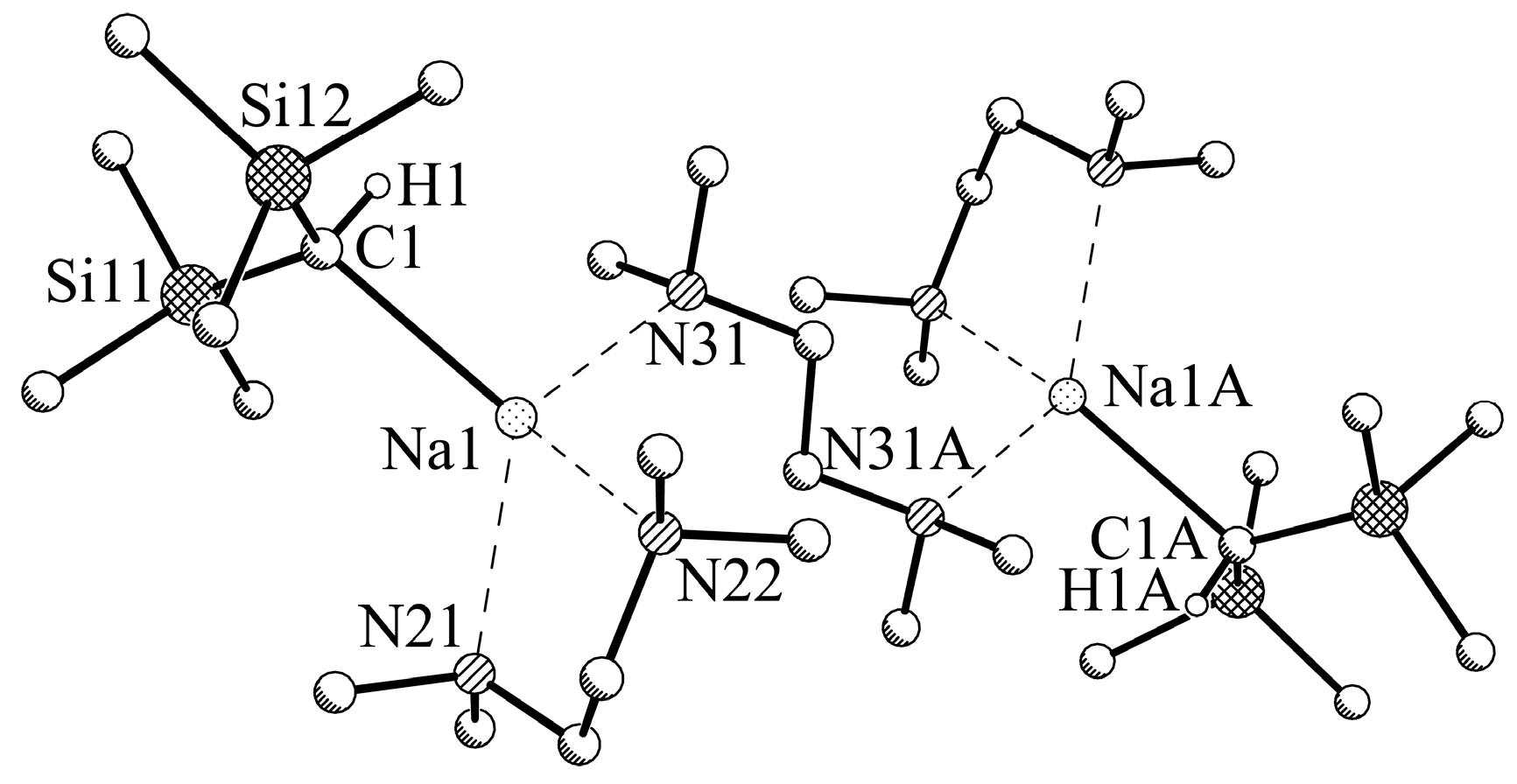
2.4. X-ray Crystallographic Measurements of Compounds 1a,b and 2a,b
3. Materials and Methods
3.1. General Procedures
3.2. Syntheses
3.2.1. Experimental Procedure for [LiCH(SiMe3)2-THF] (1a)
3.2.2. Experimental Procedure for [LiCH(SiMe3)2-TMEDA] (1b)
3.2.3. Experimental Procedure for [NaCH(SiMe3)2-THF] (2a)
3.2.4. Experimental Procedure for [NaCH(SiMe3)2-TMEDA] (2b)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Clayden, J. Organolithiums: Selectivity for Synthesis; Elsevier Oxford: Kidlington, UK, 2002. [Google Scholar]
- Schlosser, M. Organometallics in Synthesis—A Manual, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Gessner, V.H.; Däschlein, C.; Strohmann, C. Structure formation principles and reactivity of organolithium compounds. Chemistry 2009, 15, 3320–3334. [Google Scholar] [CrossRef] [PubMed]
- Seiferth, D. Alkyl and Aryl derivatives of the Alkali metals: Strong bases and reactive nucleophiles. 2. Wilhelm Schlenk’s Organoalkali-metal chemistry. The metal displacement and the transmetalation reactions. metalation of weakly acidic hydrocarbons. Superbases. Organometallics 2009, 28, 2–33. [Google Scholar] [CrossRef]
- Harrison-Marchand, A.; Mongin, F. Mixed AggregAte (MAA): A single concept for all dipolar organometallic aggregates. 1. Structural data. Chem. Rev. 2013, 113, 7470–7562. [Google Scholar] [CrossRef] [PubMed]
- Reich, H.J. Role of organolithium aggregates and mixed aggregates in organolithium mechanisms. Chem. Rev. 2013, 113, 7130–7178. [Google Scholar] [CrossRef] [PubMed]
- Collum, D.B. Is N,N,N',N'-tetramethylethylenediamine a good ligand for lithium? Acc. Chem. Res. 1992, 25, 448–454. [Google Scholar] [CrossRef]
- Clegg, W.; Conway, B.; Kennedy, A.R.; Klett, J.; Mulvey, R.E.; Russo, L. Synthesis and structures of [(Trimethylsilyl)methyl]sodium and -potassium with bi- and tridentate N-donor ligands. Eur. J. Inorg. Chem. 2011, 721–726. [Google Scholar] [CrossRef]
- Davidson, P.J.; Harris, D.H.; Lappert, M.F. Subvalent Group 4B metal alkyls and amides. Part I. The synthesis and physical properties of kinetically stable bis[bis(trimethylsilyl)methyl]-germanium(II), -tin(II), and -lead(II). J. Chem. Soc. Dalton Trans. 1976, 21, 2268–2274. [Google Scholar] [CrossRef]
- Atwood, J.L.; Fjeldberg, T.; Lappert, M.F.; Luong-Thi, N.T.; Shakir, R.; Thorne, A.J. Molecular structures of bis(trimethylsilyl)methyl-lithium[(LiR)n, R = CH(SiMe3)2] in the vapour (gas-phase electron diffraction: A monomer, n = 1) and the crystal (X-Ray: A polymer, n = ∞). J. Chem. Soc. Chem. Commun. 1984, 17, 1163–1165. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Lappert, M.F.; Leung, W.-P.; Liu, D.S.; Tian, S. Synthesis and structures of the heavier alkali metal alkyls; the X-ray structures of [Na(μ-R)]∞, and [Rb(μ-R)(pmdeta)]2[R=CH(SiMe3)2, pmdeta=(Me2NCH2CH2)2NMe]. J. Chem. Soc. Chem. Commun. 1993, 18, 1386–1387. [Google Scholar] [CrossRef]
- Pakuro, N.I.; Arest-Yakubovich, A.A.; Shcheglova, L.V.; Petrovsky, P.V.; Chekulaeva, L.A. NMR spectra of a hydrocarbon-soluble organosodium compound and its lithium analogs. Rus. Chem. Bull. 1996, 45, 838–840. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Khvostov, A.V.; Lappert, M.F. Synthesis and structures of crystalline bis(trimethylsilyl)methanide complexes of potassium, calcium and ytterbium. J. Organomet. Chem. 2002, 663, 263–268. [Google Scholar] [CrossRef]
- Boesveld, W.M.; Hitchcock, P.B.; Lappert, M.F.; Liu, D.-S.; Tian, S. Synthesis and structures of the crystalline heavier alkali metal alkyls: X-ray structures of [K(μ-R){O(Me)But}]∞, [(pmdeta)K(μ-R)K(μ-R)2K(μ-R)K(pmdeta)], and [Cs(μ-R)(tmeda)]∞ (R = CH(SiMe3)2). Organometallics 2000, 19, 4030–4035. [Google Scholar] [CrossRef]
- Lewis, H.L.; Brown, T.B. Association of alkyllithium compounds in hydrocarbon media. Alkyllithium-base interactions. J. Am. Chem. Soc. 1970, 92, 4664–4670. [Google Scholar] [CrossRef]
- Lappert, M.F.; Engelhardt, L.M.; Raston, C.L.; White, A.H. Synthesis of [Li(CH2SiMe3)(pmdeta)] and the crystalline monomeric bulky alkyl-lithium complexes[LiR(tmeda)]and [LiR(pmdeta)][R = CH(SiMe3)2]; X-ray crystal structure of [Li{CH(SiMe3)2}(pmdeta)]{tmeda = Me2NCH2CH2NMe2, pmdeta = Me2N[CH2]2N(Me)[CH2]2NMe2}. J. Chem. Soc. Chem. Commun. 1982, 1323–1324. [Google Scholar] [CrossRef]
- Lochmann, L.; Janata, M. 50 years of superbases made from organolithium compounds and heavier alkali metal alkoxides. Cent. Eur. J. Chem. 2014, 12, 537–548. [Google Scholar] [CrossRef]
- Unkelbach, C.; O’Shea, D.F.; Strohmann, C. Insights into the metalation of benzene and toluene by Schlosser’s base: A superbasic cluster comprising PhK, PhLi, and tBuOLi. Angew. Chem. Int. Ed. 2014, 53, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Benrath, P.; Kaiser, M.; Limbach, T.; Mondeshki, M.; Klett, J. Combining neopentyllithium with potassium tert-butoxide: Formation of an alkane-soluble lochmann-schlosser superbase. Angew. Chem. Int. Ed. 2016, 55, 10886–10889. [Google Scholar] [CrossRef] [PubMed]
- Stokes, R.H.; Tomlins, R.P. Thermodynamic functions of melting for cyclohexane. J. Chem. Thermodyn. 1974, 6, 379–386. [Google Scholar] [CrossRef]
- Armstrong, D.R.; Barr, D.; Clegg, W.; Mulvey, R.E.; Reed, D.; Snaith, D.; Wade, K. The laddering principle in lithium amide chemistry: the crystal and molecular structure of the pyrrolididolithium adduct [H2C(CH2)3NLi]3MeN(CH2CH2NMe2)2. J. Chem. Soc. Chem. Commun. 1986, 11, 869–870. [Google Scholar] [CrossRef]
- Bond, A.D. Ring-laddering and ring-stacking: Unifying concepts in the structural chemistry of organic ammonium halides. Cryst. Growth Des. 2005, 5, 755–771. [Google Scholar] [CrossRef]
- Hevia, E.; Kennedy, A.R.; Mulvey, R.E.; Ramsay, D.L.; Robertson, S.D. Concealed cyclotrimeric polymorph of lithium 2,2,6,6-tetramethylpiperidide unconcealed: X-Ray crystallographic and NMR spectroscopic studies. Chem. Eur. J. 2013, 19, 1521–3765. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, G.; Chow, A.; Winchester, W.R. Structure and dynamic behavior of solvated neopentyllithium monomers, dimers, and tetramers: Proton, carbon-13 and lithium-6 NMR. J. Am. Chem. Soc. 1990, 112, 6190–6198. [Google Scholar] [CrossRef]
- Neufeld, R.; John, M.; Stalke, D. The donor-base-free aggregation of lithium diisopropyl amide in hydrocarbons revealed by a DOSY method. Angew. Chem. Int. Ed. 2015, 54, 6994–6998. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Lagow, R.J. Isolation and structure analysis of neohexyllithium generated by ether cleavage: Primary and secondary coordination in alkyllithium aggregates. Organometallics 1997, 16, 789–792. [Google Scholar] [CrossRef]
- Kottke, T.; Stalke, D. Crystal handling at low temperatures. J. Appl. Cryst. 1993, 26, 615–619. [Google Scholar] [CrossRef]
- The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 8 May 2017).
- Engelhardt, L.M.; Jolly, B.S.; Junk, P.C.; Raston, C.L.; Skelton, B.W.; White, A.H. Highly hindered amido-lithium and amido-magnesium complexes. Crystal-structures of [Li(Mu-N(SiMe3)2)(tetrahydrofuran)]2 and [MgBus(Mu-N(SiMe3)2)]2. Aust. J. Chem. 1986, 39, 1337–1345. [Google Scholar] [CrossRef]
- Teclé, B.; Ilsley, W.H.; Oliver, J.P. Metal-silicon bonded compounds. 16. The structure of (LiSiMe3)2·(Me2NCH2CH2NMe2)3, a highly reactive silylating agent. Organometallics 1982, 1, 875–877. [Google Scholar] [CrossRef]
- Wiberg, N.; Wagner, G. Auf dem Wege zu einem stabilen Silaethen: Sterisch überladene Trisilylmethane tBu2SiX–CY(SiMe3)2 (X, Y = H, Hal, Li). Eur. J. Inorg. Chem. 1986, 119, 1455–1466. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Herbst-Irmer, R.; Spek, A.L.; Schneider, T.R.; Sawaya, M.R. Crystal Structure Refinement—A Crystallographer’s Guide to SHELXL; Oxford University Press: Oxford, UK, 2006. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Concentration (mol/L) | ∆T (K) | M(Exp) (g/mol) | M(Oligomer) (g/mol) | ∆M |
|---|---|---|---|---|---|
| Li-1 | 0.040 | −0.50 | 345 | 332.68 (1-dimer) | +3.7% |
| Na-1 | 0.021 | −0.12 | 804 | 729.52 (2-tetramer) | +10.2% |
| Na-2 | 0.041 | −0.29 | 663 | 729.52 (2-tetramer) | −9.1% |
| Na-3 | 0.087 | −0.35 | 1175 | 1094.28 (2-hexamer) | +7.4% |
| Na-4 | 0.087 | −0.37 | 1098 | 1094.28 (2-hexamer) | +0.3% |
| Compound in Solvent | 1H | 13C | 29Si | 7Li | ||
|---|---|---|---|---|---|---|
| SiMe3 | CH2 | SiMe3 | CH2 | SiMe3 | ||
| 1 in C6D6 | 0.15 | −2.52 | 5.1 | 2.4 | −6.6 | 2.2 |
| 1 in [D8]THF | −0.14 | −2.26 | 6.6 | 0.4 | −8.3 | 1.0 |
| 1 in C6D12 | 0.05 | −2.29 | 4.8 | 3.4 | −7.9 | 3.6 |
| 2 in C6D6 [11] | 0.20 | −2.04 | 7.0 | 0.4 | 12.4 | – |
| 2 in C6D6 | 0.22 | −2.01 | 7.0 | 0.0 | −11.8 | – |
| 2 in [D8]THF | −0.16 | −2.09 | 6.9 | -0.4 | −11.3 | – |
| 2 in C6D12 | 0.04 | −2.08 | 7.1 | -0.1 | −12.1 | – |
| Compound | Conc (mol/L) | D (10−10 m2/s) | rH (nm) | D [Si(SiMe3)4] (10−10 m2/s) | rH [Si(SiMe3)4] (nm) |
|---|---|---|---|---|---|
| 1 | 0.08 | 6.258 | 0.33 | 5.828 | 0.35 |
| 1 | 0.19 | 6.020 | 0.34 | 5.781 | 0.36 |
| 1 | <0.3 1 | 5.243 | 0.39 | 5.998 | 0.34 |
| 2 | 0.1 | 2.877 | 0.72 | 6.295 | 0.33 |
| 2 | 0.2 | 2.355 | 0.88 | 5.959 | 0.35 |
| 2 | <0.3 1 | 1.920 | 1.10 | 5.454 | 0.38 |
| Compound | 1H | 13C | 29Si | 7Li | ||||
|---|---|---|---|---|---|---|---|---|
| SiMe3 | CH2 | Ligand | SiMe3 | CH2 | Ligand | SiMe3 | ||
| 1 | 0.05 | −2.29 | – | 4.8 | 3.4 | – | −6.6 | 3.6 |
| 1a | −0.02 | −2.39 | 1.89 (β-CH2) | 5.7 | 2.0 | 26.1 (β-CH2) | −6.0 | 2.9 |
| 3.88 (α-CH2) | 69.2 (α-CH2) | |||||||
| 1b | −0.10 | −2.05 | 2.30 (Me) | 6.4 | 2.3 | 45.9 (Me) | −7.9 | 3.1 |
| 2.37 (CH2) | 57.3 (CH2) | |||||||
| 2 | 0.04 | −2.08 | – | 7.1 | −0.1 | – | −12.1 | – |
| 2a | 0.00 | −2.28 | 1.83 (β-CH2) | 6.7 | 1.1 | 27.0 (β-CH2) | −10.1 | – |
| 3.76 (α-CH2) | 68.7 (α-CH2) | |||||||
| 2b | −0.08 | −2.04 | 2.25 (Me) | 6.7 | 1.0 | 46.2 (Me) | −8.5 | – |
| 2.34 (CH2) | 58.0 (CH2) | |||||||
| Compound | 1a | 1b | 2a | 2b |
|---|---|---|---|---|
| Formula | C11H27LiOSi2 | C13H35LiN2Si2 | C11H27NaOSi2 | C16H43NaN3Si2 |
| Mr(g·mol−1) | 238.44 | 282.55 | 254.49 | 713.39 |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group | P21/n | P21/c | P21/n | P21/n |
| a (Å) | 9.4930(9) | 18.7636(8) | 11.3470(19) | 10.450(4) |
| b (Å) | 9.9165(9) | 13.2303(5) | 9.7379(17) | 17.414(6) |
| c (Å) | 16.7191(14) | 17.7299(7) | 14.622(2) | 14.258(5) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 92.527(2) | 112.040(2) | 90.876(5) | 100.824(9) |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 1572.4(3) | 4079.8(3) | 1615.5(5) | 2548.5(16) |
| Z | 4 | 8 | 4 | 6 |
| ρcalcd (g·cm−3) | 1.007 | 0.920 | 1.046 | 0.930 |
| μ(Mo Kα) (mm−1) | 0.203 | 0.163 | 0.226 | 0.158 |
| T (K) | 173 | 173 | 173 | 173 |
| measured refl. [b] | 51,345 | 51,744 | 17,850 | 37,556 |
| independent refl. | 3766 | 9687 | 3904 | 6057 |
| refined parameters | 192 | 183 | 141 | 236 |
| R1 [c] | 0.0320 | 0.0449 | 0.0690 | 0.0441 |
| R1, all data | 0.0428 | 0.1013 | 0.1539 | 0.0932 |
| wR2 [d] | 0.0898 | 0.0964 | 0.1572 | 0.1034 |
| wR2, all data | 0.0964 | 0.1112 | 0.1894 | 0.1196 |
| max, min peaks (eÅ−3) | 0.369, −0.161 | 0.270, −0.187 | 0.910, −0.510 | 0.265, −0.203 |
| CCDC numbers [28] | 1,548,189 | 1,548,191 | 1,548,190 | 15,481,892 |
| Compound | 1a (M = Li) | 1b (M = Li) | 2a (M = Na) | 2b (M = Na) |
|---|---|---|---|---|
| M1–C1 | 2.204(2) | 2.070(3)/2.083(3) | 2.778(4) | 2.520(2) |
| M1–C1A | 2.274(3) | – | 2.657(4) | – |
| M1–O1 | 1.953(8) | – | 2.375(3) | – |
| M1–N21 | – | – | – | 2.559(2) |
| M1–N22 | – | – | – | 2.569(2) |
| M1–N31 | – | 2.054(6)/2.133(7) | – | 2.635(2) |
| M1–N32 | – | 2.071(9)/2.061(9) | – | – |
| C1–Si11 | 1.835(2) | 1.809(2)/1.813(2) | 1.809(5) | 1.808(2) |
| C1–Si12 | 1.838(2) | 1.807(2)/1.803(2) | 1.800(5) | 1.808(2) |
| M1–M1A | 2.395(4) | – | – | – |
| M1–H1 | 2.81 | 2.30/2.43 | 2.68/2.70 | 2.71 |
| M1–C1–M1A | 64.64(10) | – | 159.30(18) | – |
| C1–M1–C1A | 115.36(10) | – | 130.74(6) | – |
| C1–M1–O1 | 137.4(7) | – | 129.93(13) | – |
| C1A–M1–O1 | 110.5(6) | – | 99.33(13) | – |
| Si11–C1–Si12 | 117.06(7) | 123.25(10)/122.48(11) | 127.9(3) | 120.91 |
| ƩCHSi2 | 327.2 | 341.1/341.0 | 359.3 | 336.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Von Pilgrim, M.; Mondeshki, M.; Klett, J. [Bis(Trimethylsilyl)Methyl]Lithium and -Sodium: Solubility in Alkanes and Complexes with O- and N- Donor Ligands. Inorganics 2017, 5, 39. https://doi.org/10.3390/inorganics5020039
Von Pilgrim M, Mondeshki M, Klett J. [Bis(Trimethylsilyl)Methyl]Lithium and -Sodium: Solubility in Alkanes and Complexes with O- and N- Donor Ligands. Inorganics. 2017; 5(2):39. https://doi.org/10.3390/inorganics5020039
Chicago/Turabian StyleVon Pilgrim, Markus, Mihail Mondeshki, and Jan Klett. 2017. "[Bis(Trimethylsilyl)Methyl]Lithium and -Sodium: Solubility in Alkanes and Complexes with O- and N- Donor Ligands" Inorganics 5, no. 2: 39. https://doi.org/10.3390/inorganics5020039
APA StyleVon Pilgrim, M., Mondeshki, M., & Klett, J. (2017). [Bis(Trimethylsilyl)Methyl]Lithium and -Sodium: Solubility in Alkanes and Complexes with O- and N- Donor Ligands. Inorganics, 5(2), 39. https://doi.org/10.3390/inorganics5020039