N-Heterocyclic Carbene Coinage Metal Complexes Containing Naphthalimide Chromophore: Design, Structure, and Photophysical Properties



Abstract
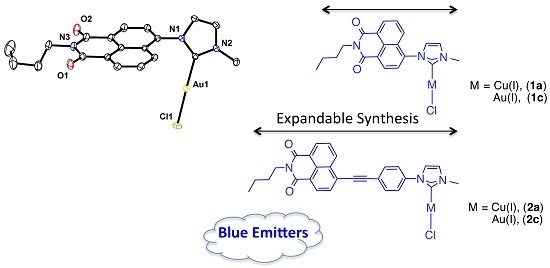
:
1. Introduction
2. Results and Discussions
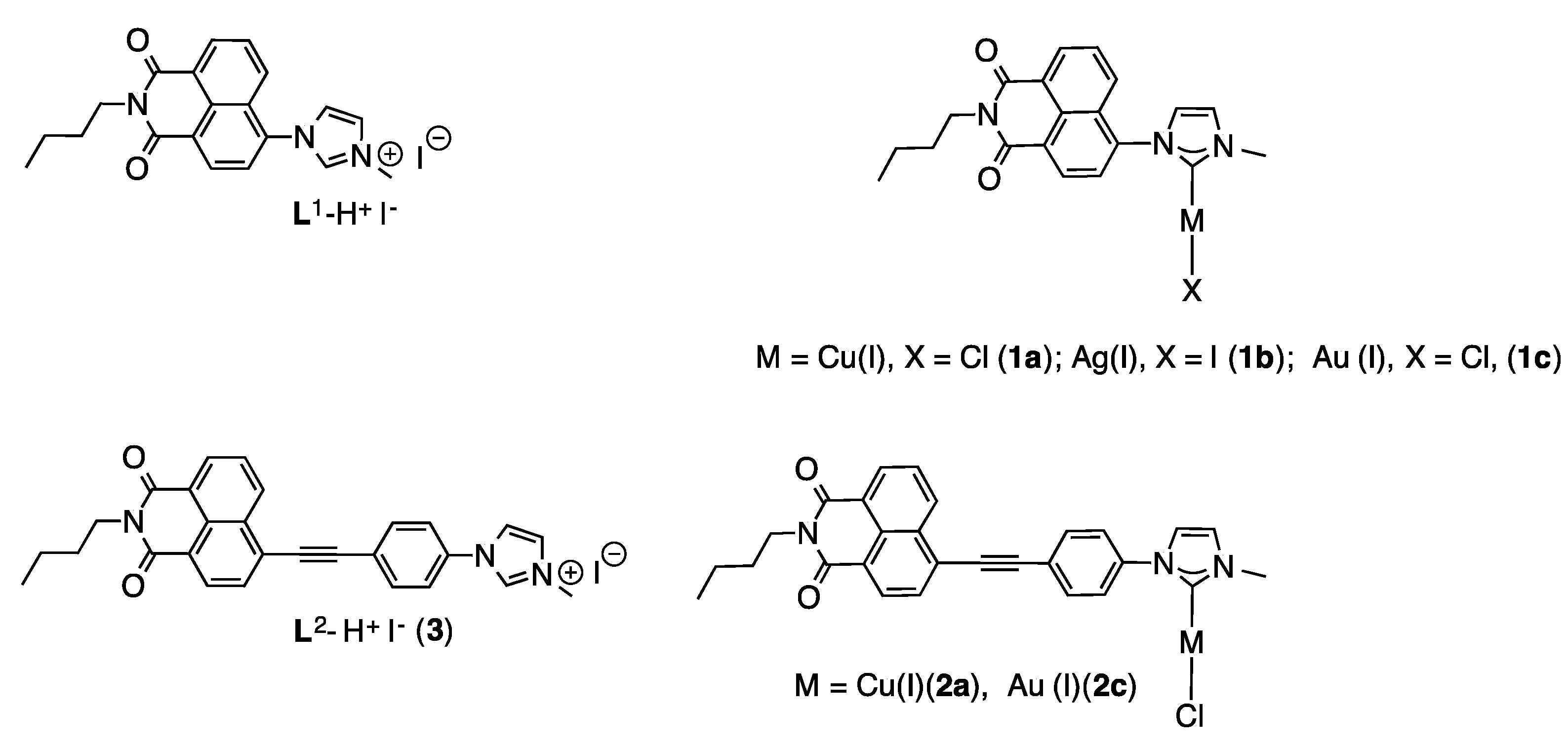
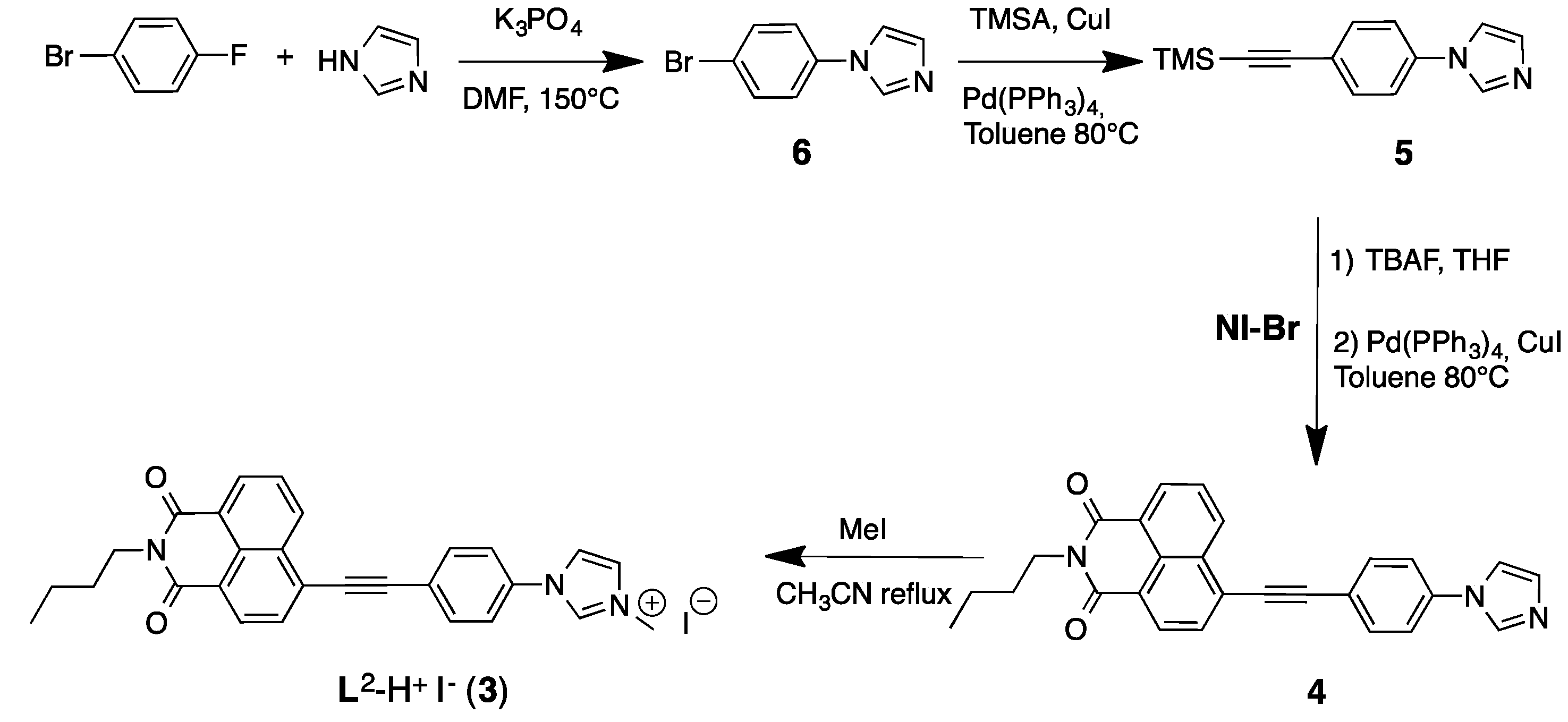
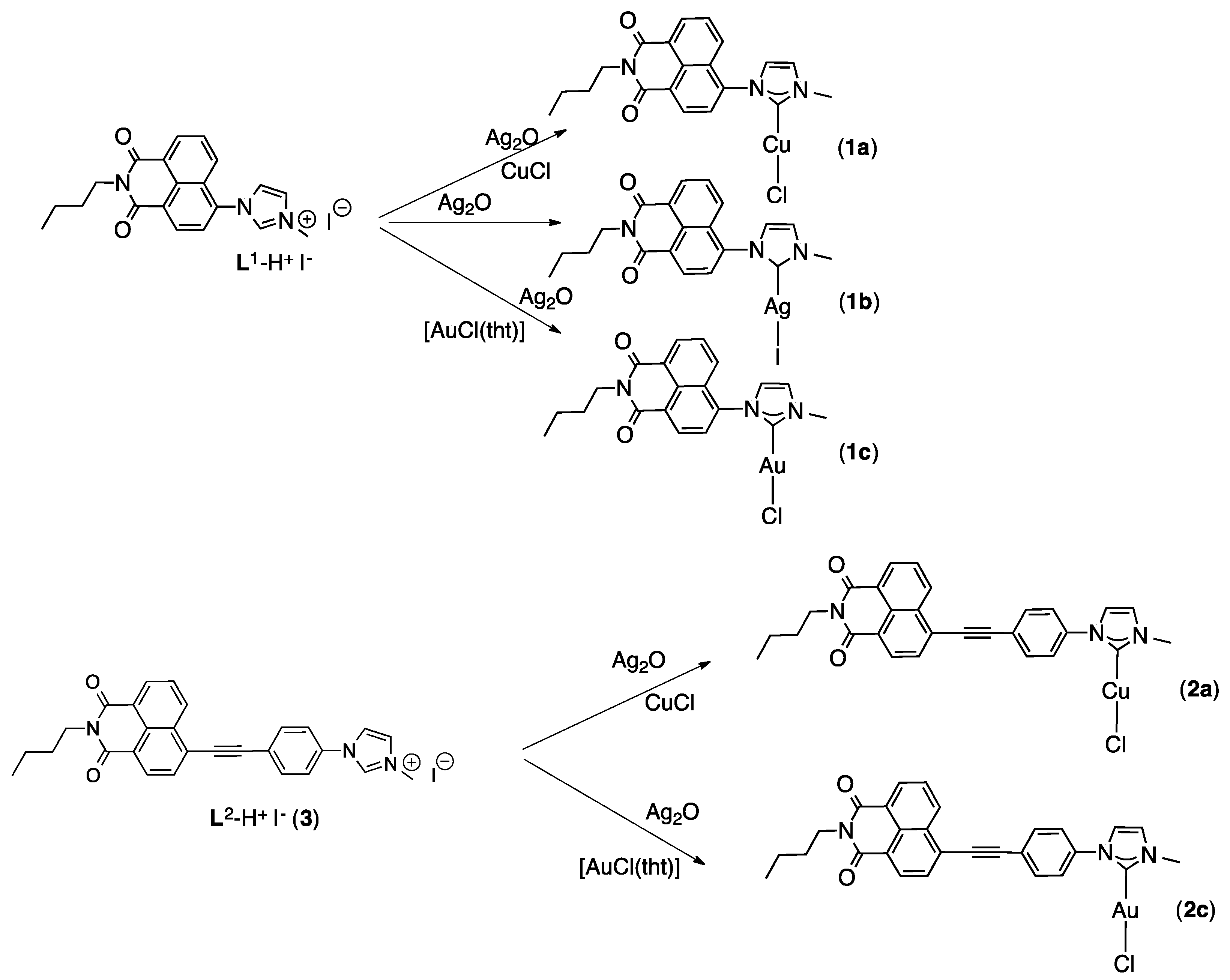
2.1. Synthesis and Characterization of the Imidazolium Salts
2.2. Synthesis and Characterization of the NHC Coinage Metal Complexes
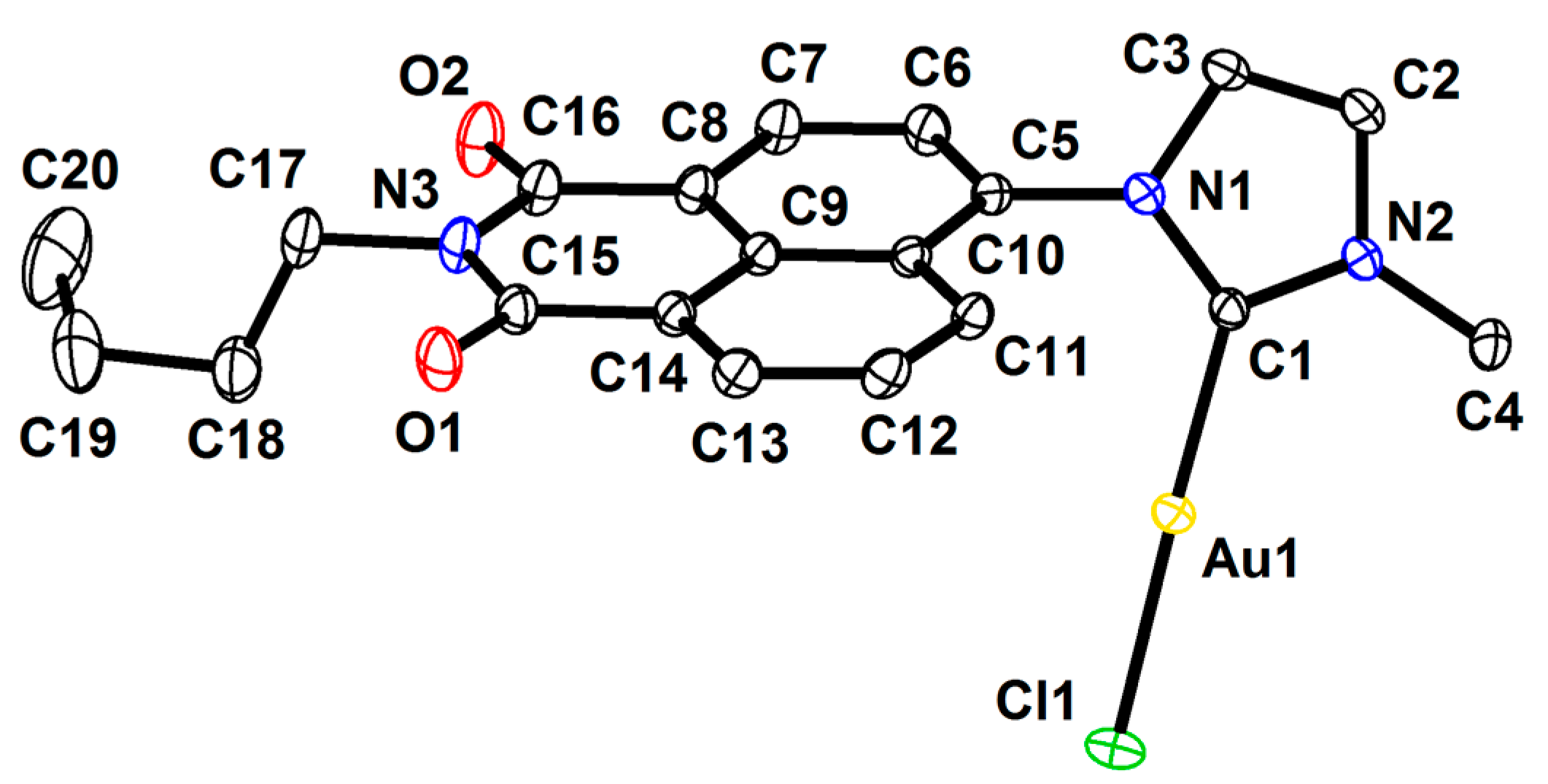
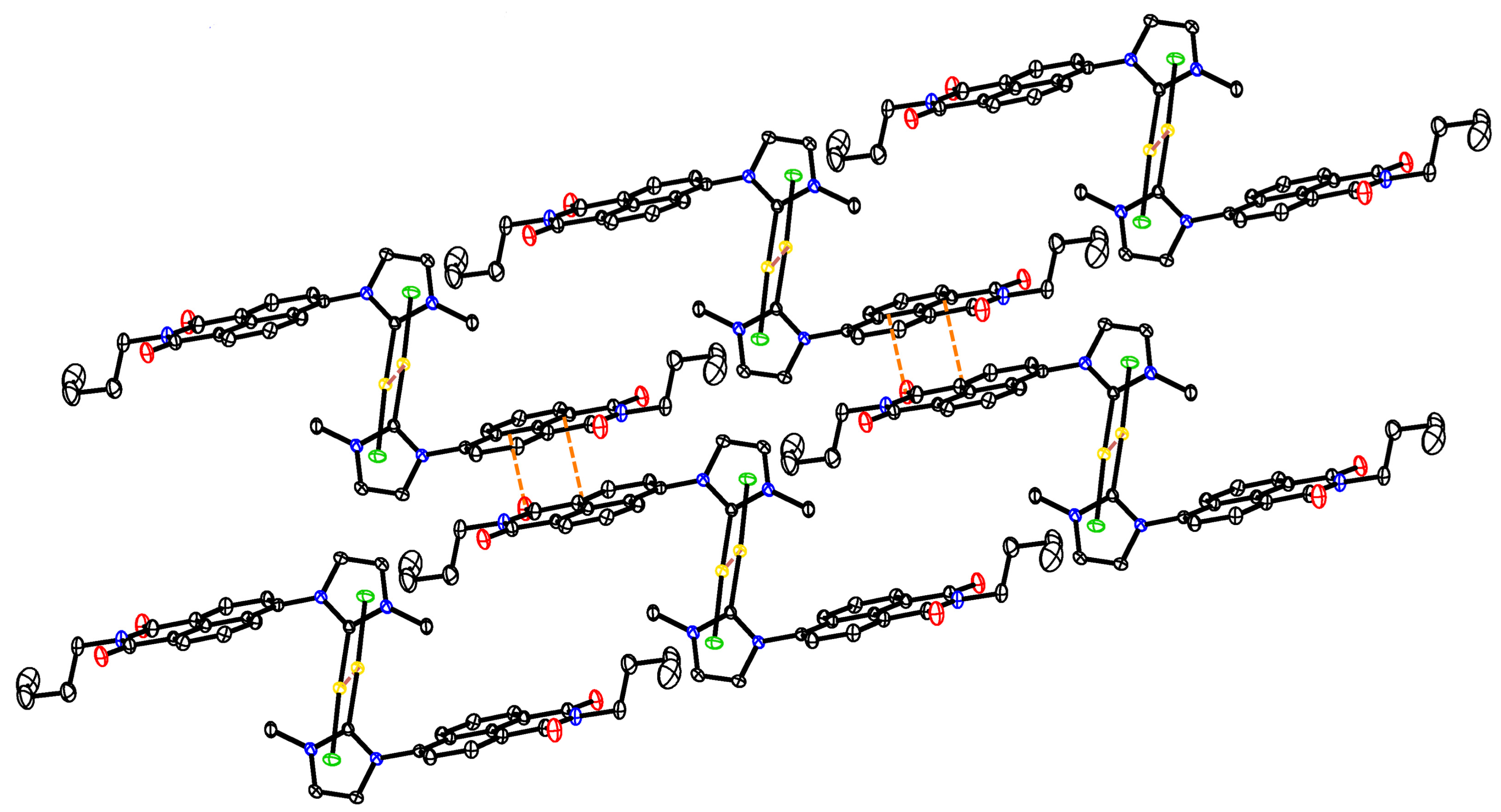
2.3. X-ray Molecular Structure of the N-heterocyclic Gold Carbene Complex [(L1)–Au–Cl] (1c)
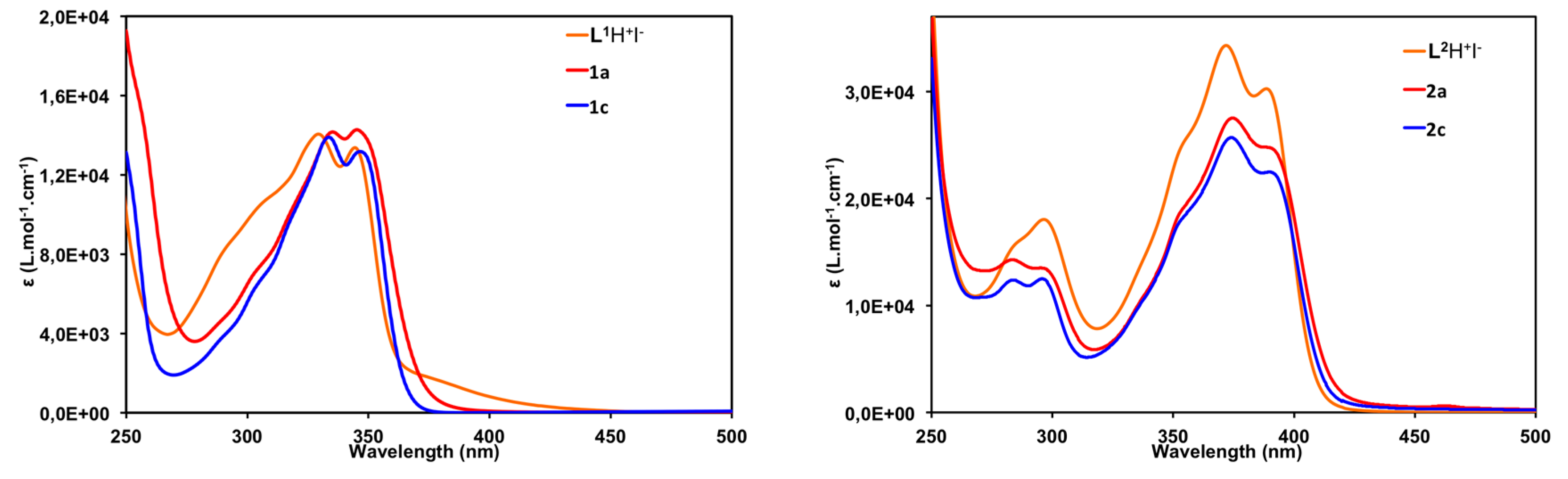
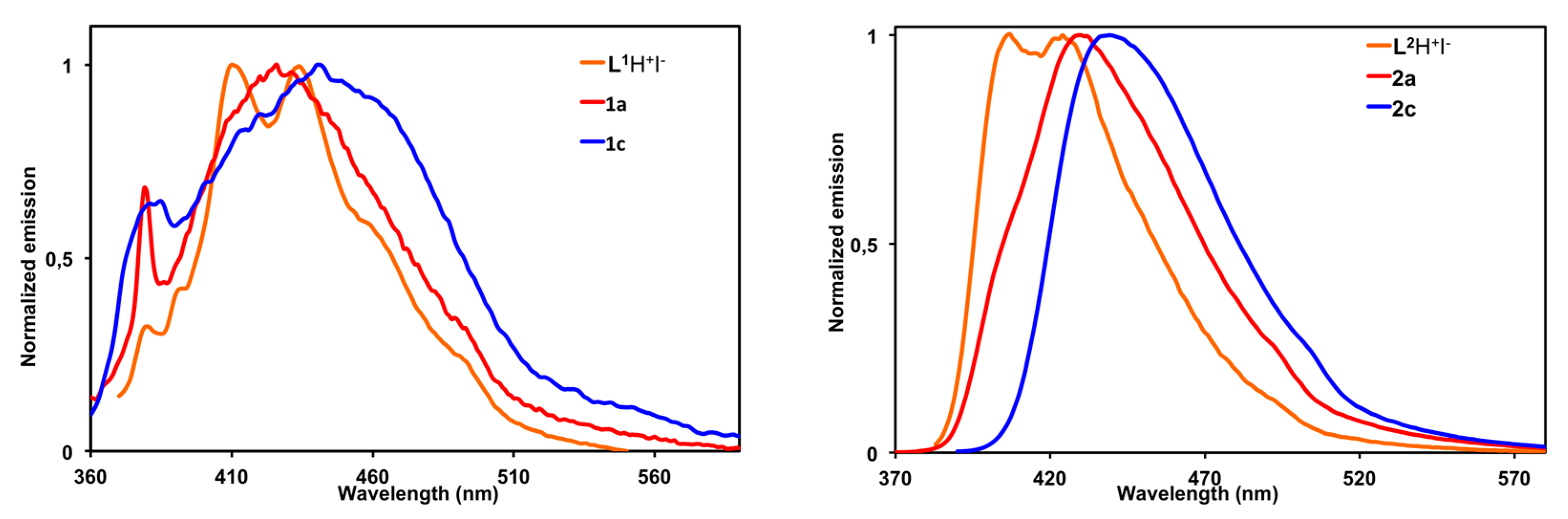
2.4. Photophysical Properties
2.5. Concluding Remarks
3. Experimental Section
3.1. General Experimental Methods
3.2. Synthesis of Compound 5
3.3. Synthesis of Compound 4
3.4. Synthesis of L2–H+I− (3)
3.5. Synthesis of Complex 1b
3.6. General Procedure for the Synthesis of 1a and 1c
3.7. For Complex 1a
3.8. For Complex 1c
3.9. General Procedure for the Synthesis of 2a and 2c
3.10. For Complex 2a
3.11. For Complex 2c
3.12. X-ray Crystal Structure Determination
3.13. Crystal Data for (1c)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arduengo, A.J.; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Ofele, K. A New Transition-Metal Carbon Complexe 1,3-Dimethyl-4-Imidazolinylid-2-Ene Pentacarbonylchromium. J. Organomet. Chem. 1968, 12, 42–43. [Google Scholar] [CrossRef]
- Wanzlick, H.W.; Schonher, H.J. Direct Synthesis of a Mercury Salt-Carbene Complex. Angew. Chem. Inter. Ed. 1968, 7, 141–142. [Google Scholar] [CrossRef]
- Huynh, H.V. The Organometallic Chemistry of N-Heterocyclic Carbenes; Wiley: Chichester, UK, 2017. [Google Scholar]
- Mercs, L.; Albrecht, M. Beyond catalysis: N-heterocyclic carbene complexes as components for medicinal, luminescent, and functional materials applications. Chem. Soc. Rev. 2010, 39, 1903–1912. [Google Scholar] [CrossRef] [PubMed]
- De Fremont, P.; Marion, N.; Nolan, S.P. Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev. 2009, 253, 862–892. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Inter. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Hindi, K.M.; Panzner, M.J.; Tessier, C.A.; Cannon, C.L.; Youngs, W.J. The Medicinal Applications of Imidazolium Carbene–Metal Complexes. Chem. Rev. 2009, 109, 3859–3884. [Google Scholar] [CrossRef] [PubMed]
- Visbal, R.; Gimeno, M.C. N-Heterocyclic carbene metal complexes: Photoluminescence and applications. Chem. Soc. Rev. 2014, 43, 3551–3574. [Google Scholar] [CrossRef] [PubMed]
- Moussa, J.; Freeman, G.R.; Williams, J.A.G.; Chamoreau, L.M.; Herson, P.; Amouri, H. Synthesis and Luminescence Properties of Cycloplatinated Complexes with a Chelating N^C Pyridine-Derived N-Heterocyclic Carbene—Influence of 2,4,6-Triphenylphosphinine versus Triphenylphosphine. Eur. J. Inorg. Chem. 2016, 2016, 761–767. [Google Scholar] [CrossRef]
- Moussa, J.; Haddouche, K.; Chamoreau, L.M.; Amouri, H.; Williams, J.A.G. New N^C^ N-coordinated Pd(II) and Pt(II) complexes of a tridentate N-heterocyclic carbene ligand featuring a 6-membered central ring: synthesis, structures and luminescence. Dalton Trans. 2016, 45, 12644–12648. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.L.F.; So, M.H.; Lu, W.; Zhu, N.Y.; Che, C.M. Synthesis, Photophysical Properties, and Molecular Aggregation of Gold(I) Complexes Containing Carbon-Donor Ligands. Chem. Asian J. 2011, 6, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.Y.; Huang, R.T.W.; Lee, C.S.; Bhattacharyya, A.; Hwang, W.S.; Lin, I.J.B. Coinage Metal–N-Heterocyclic Carbene Complexes. Chem. Rev. 2009, 109, 3561–3598. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.J.B.; Vasam, C.S. Review of gold(I) N-heterocyclic carbenes. Can. J. Chem. Rev. Can. 2005, 83, 812–825. [Google Scholar] [CrossRef]
- Kriechbaum, M.; Winterleitner, G.; Gerisch, A.; List, M.; Monkowius, U. Synthesis, Characterization and Luminescence of Gold Complexes Bearing an NHC Ligand Based on the Imidazo[1,5-a]quinolinol Scaffold. Eur. J. Inorg. Chem. 2013, 2013, 5567–5575. [Google Scholar] [CrossRef]
- Hong, E.Y.-H.; Wong, H.-L.; Yam, V.W.-W. From Spherical to Leaf-Like Morphologies: Tunable Supramolecular Assembly of Alkynylgold(I) Complexes through Variations of the Alkyl Chain Length. Chem. Eur. J. 2015, 21, 5732–5735. [Google Scholar] [CrossRef] [PubMed]
- Lanoe, P.H.; Chan, J.; Gontard, G.; Monti, F.; Armaroli, N.; Barbieri, A.; Amouri, H. Deep-Red Phosphorescent Iridium(III) Complexes with Chromophoric N-Heterocyclic Carbene Ligands: Design, Photophysical Properties, and DFT Calculations. Eur. J. Inorg. Chem. 2016, 1631–1634. [Google Scholar] [CrossRef]
- Nitsch, J.; Lacemon, F.; Lorbach, A.; Eichhorn, A.; Cisnetti, F.; Steffen, A. Cuprophilic interactions in highly luminescent dicopper(I)–NHC–picolyl complexes—Fast phosphorescence or TADF? Chem. Commun. 2016, 52, 2932–2935. [Google Scholar] [CrossRef] [PubMed]
- Penney, A.A.; Sizov, V.V.; Grachova, E.V.; Krupenya, D.V.; Gurzhiy, V.V.; Starova, G.L.; Tunik, S.P. Aurophilicity in Action: Fine-Tuning the Gold(I)–Gold(I) Distance in the Excited State to Modulate the Emission in a Series of Dinuclear Homoleptic Gold(I)–NHC Complexes. Inorg. Chem. 2016, 55, 4720–4732. [Google Scholar] [CrossRef] [PubMed]
- Diness, F.; Fairlie, D.P. Catalyst-Free N-Arylation Using Unactivated Fluorobenzenes. Angew. Chem. Inter. Ed. 2012, 51, 8012–8016. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.J.; Lin, I.J.B. Facile synthesis of silver(I)–carbene complexes. Useful carbene transfer agents. Organometallics 1998, 17, 972–975. [Google Scholar] [CrossRef]
- Visbal, R.; Laguna, A.; Gimeno, M.C. Simple and efficient synthesis of [MCI(NHC)] (M = Au, Ag) complexes. Chem. Commun. 2013, 49, 5642–5644. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.C.; Laguna, A.; Visbal, R. N-Heterocyclic Carbene Coinage Metal Complexes as Intense Blue-Green Emitters. Organometallics 2012, 31, 7146–7157. [Google Scholar] [CrossRef]
- Wang, H.A.J.; Vasam, C.S.; Tsai, T.Y.R.; Chen, S.H.; Chang, A.H.H.; Lin, I.J.B. Gold(I) N-heterocyclic carbene and carbazolate complexes. Organometallics 2005, 24, 486–493. [Google Scholar] [CrossRef]
- Alexiou, M.S.; Tychopoulos, V.; Ghorbanian, S.; Tyman, J.H.P.; Brown, R.G.; Brittain, P.I. The UV-Visible Absorption and Fluorescence of Some Substituted 1,8-Naphthalimides and Naphthalic Anhydrides. J. Chem. Soc. Perkin Trans. 1990, 837–842. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Singh, A.; Mishra, L. Tuning of Aggregation Enhanced Emission and Solid State Emission from 1,8-Naphthalimide Derivatives: Nanoaggregates, Spectra, and DFT Calculations. J. Phys. Chem. A 2016, 120, 4490–4504. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Elsevier Science: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs/nm (L·mol−1·cm−1 × 103) | λem/nm (φfluo a) |
|---|---|---|
| L1–H+ I− | 330 (14.0); 345 (13.3) | 410 b, 430 (0.21) c |
| [(L1)–Cu–Cl] (1a) | 332 (13.8); 346 (14.2) | 426 (0.07) c |
| [(L1)–Au–Cl] (1c) | 335 (13.7); 349 (12.9) | 451 (0.01) c |
| L2–H+ I− (3) | 285 (15.8); 296 (18.0); 372 (34.3); 389 (30.2) | 406, 424 b (0.73) d |
| [(L2)–Cu–Cl] (2a) | 284 (14.2); 299 (13.2); 376 (13.2); 3393 (24.2) | 428 (0.20) d |
| [(L1)–Au–Cl] (2c) | 284 (12.4); 298 (12.1); 375 (25.6); 393 (21.9) | 429 (0.29) d |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanoë, P.-H.; Najjari, B.; Hallez, F.; Gontard, G.; Amouri, H. N-Heterocyclic Carbene Coinage Metal Complexes Containing Naphthalimide Chromophore: Design, Structure, and Photophysical Properties. Inorganics 2017, 5, 58. https://doi.org/10.3390/inorganics5030058
Lanoë P-H, Najjari B, Hallez F, Gontard G, Amouri H. N-Heterocyclic Carbene Coinage Metal Complexes Containing Naphthalimide Chromophore: Design, Structure, and Photophysical Properties. Inorganics. 2017; 5(3):58. https://doi.org/10.3390/inorganics5030058
Chicago/Turabian StyleLanoë, Pierre-Henri, Btissam Najjari, Florine Hallez, Geoffrey Gontard, and Hani Amouri. 2017. "N-Heterocyclic Carbene Coinage Metal Complexes Containing Naphthalimide Chromophore: Design, Structure, and Photophysical Properties" Inorganics 5, no. 3: 58. https://doi.org/10.3390/inorganics5030058
APA StyleLanoë, P.-H., Najjari, B., Hallez, F., Gontard, G., & Amouri, H. (2017). N-Heterocyclic Carbene Coinage Metal Complexes Containing Naphthalimide Chromophore: Design, Structure, and Photophysical Properties. Inorganics, 5(3), 58. https://doi.org/10.3390/inorganics5030058