The Mechanism of Rh-Catalyzed Transformation of Fatty Acids to Linear Alpha olefins


Abstract
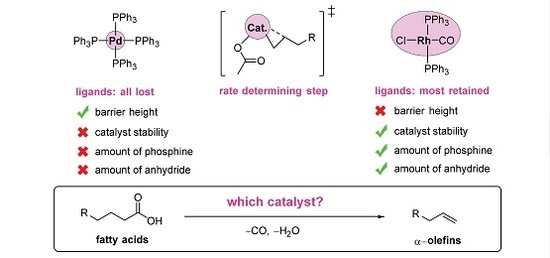
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
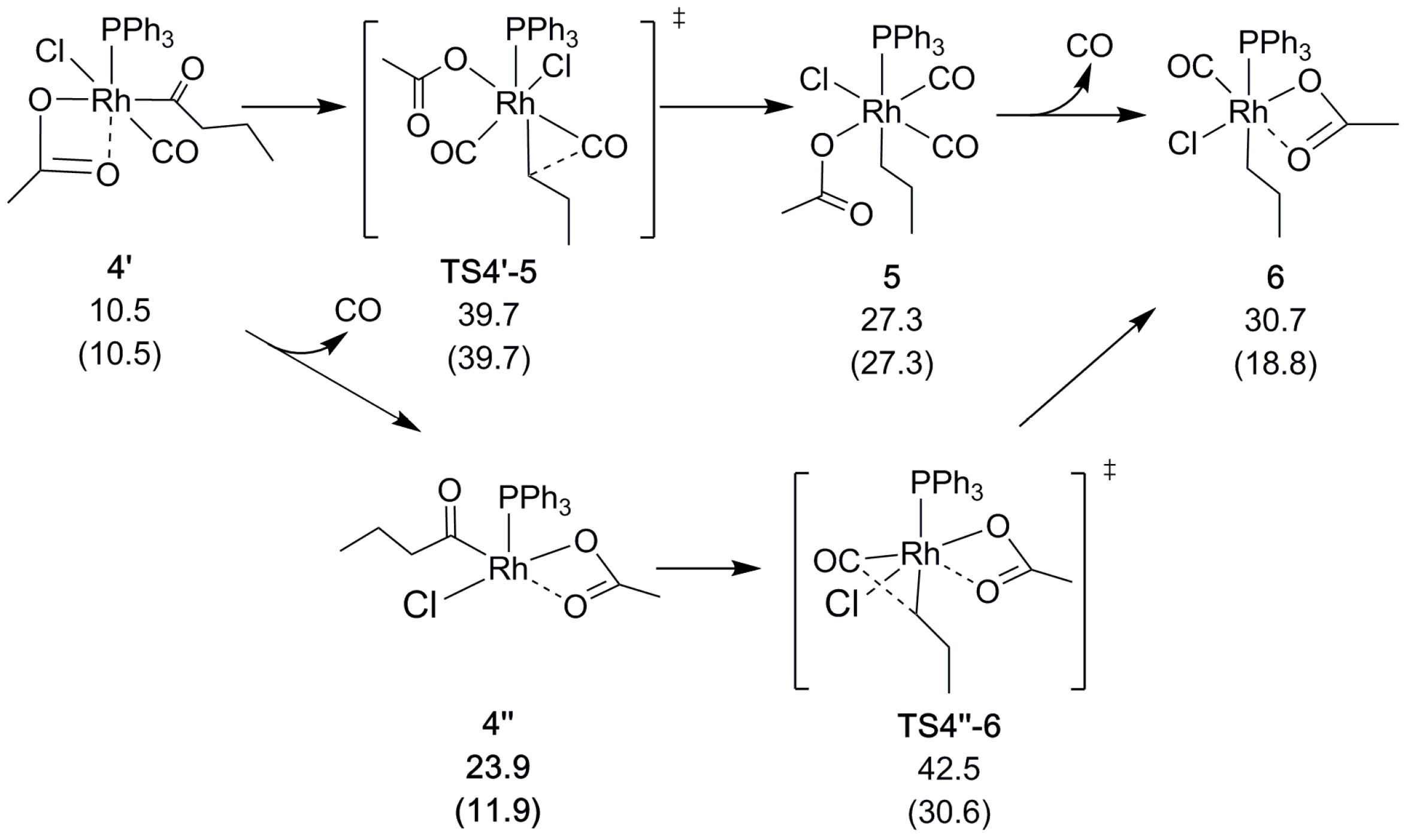{kind=link}
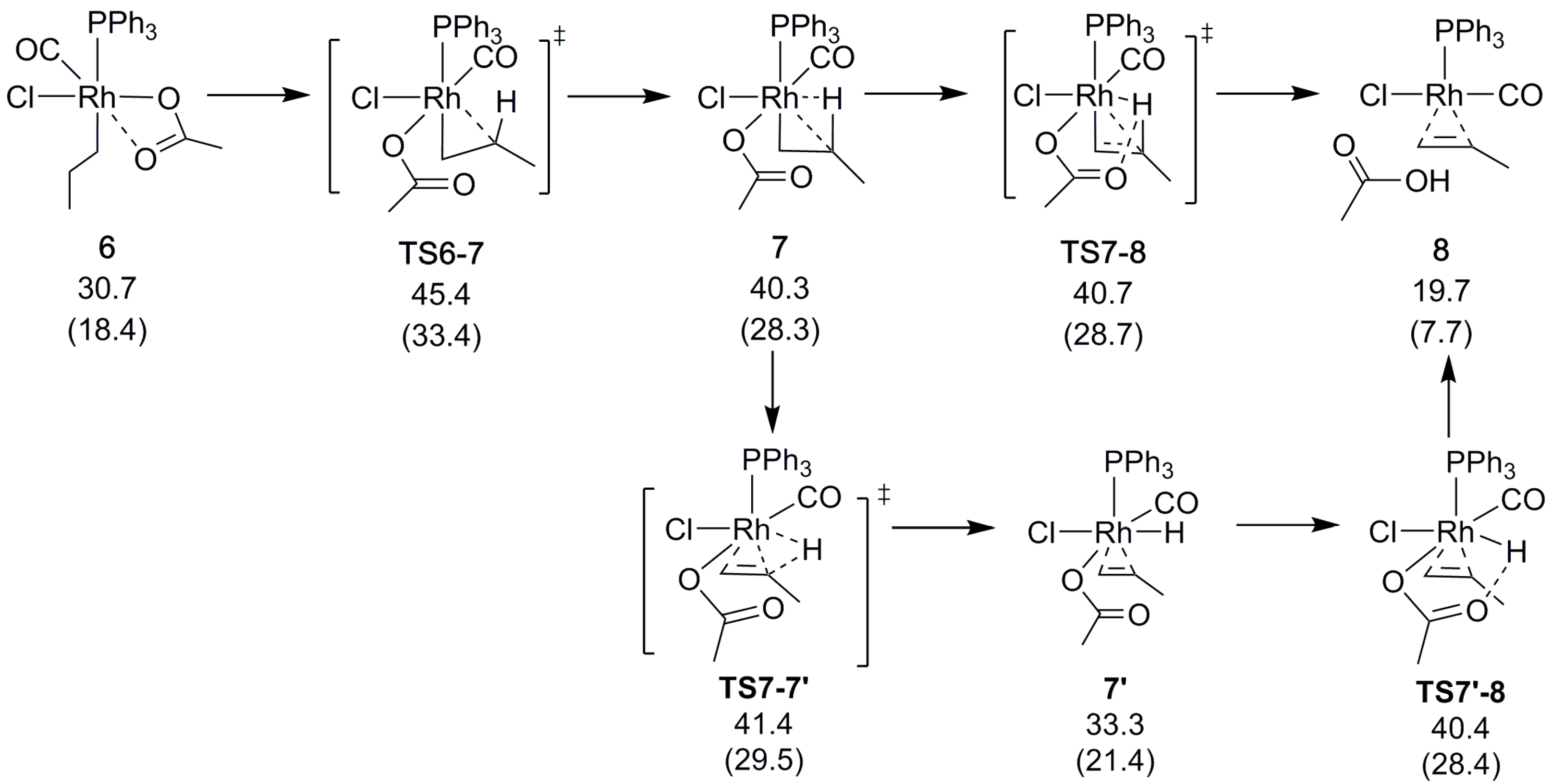{kind=link}
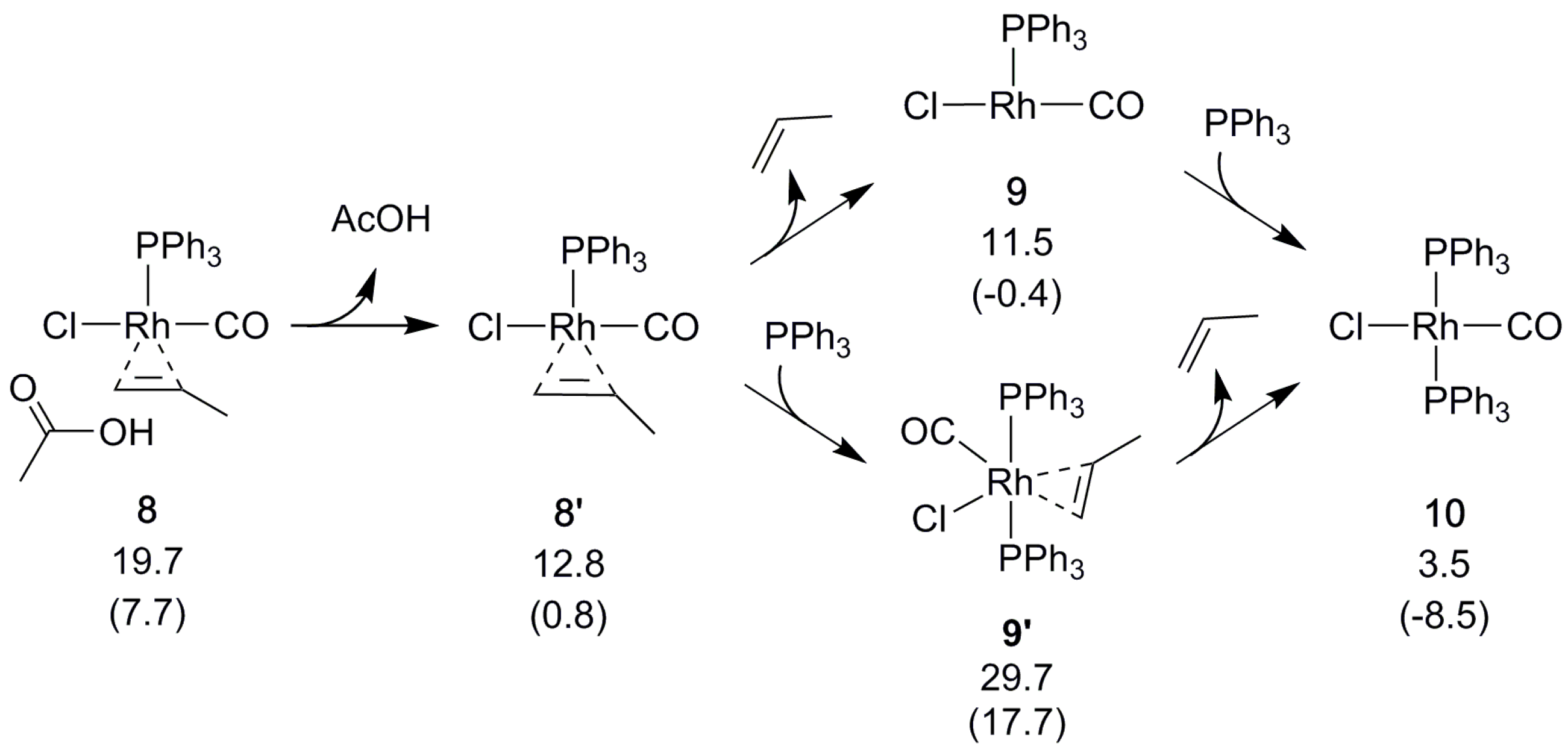{kind=link}
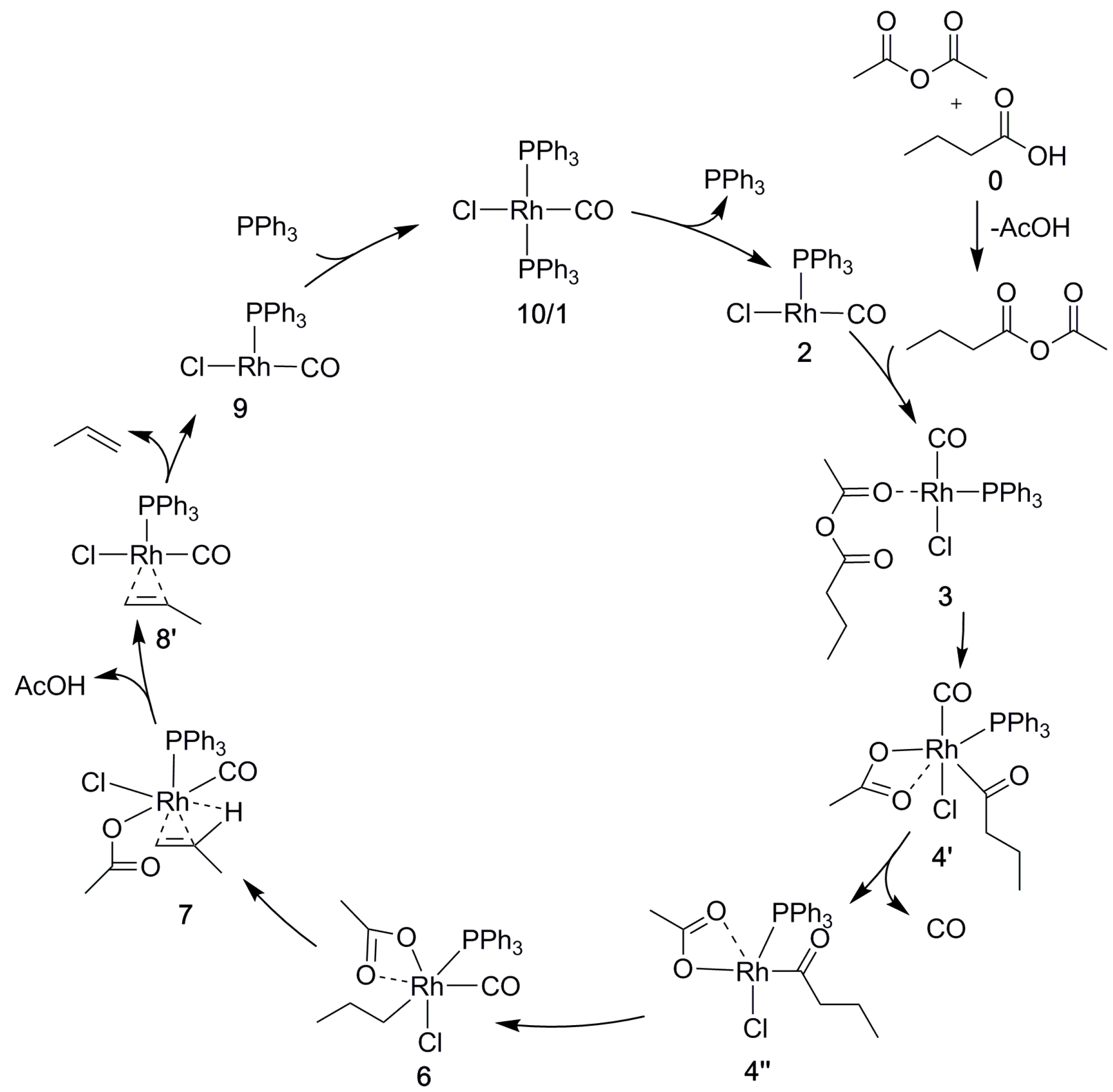{kind=link}
{kind=link}
1. Introduction
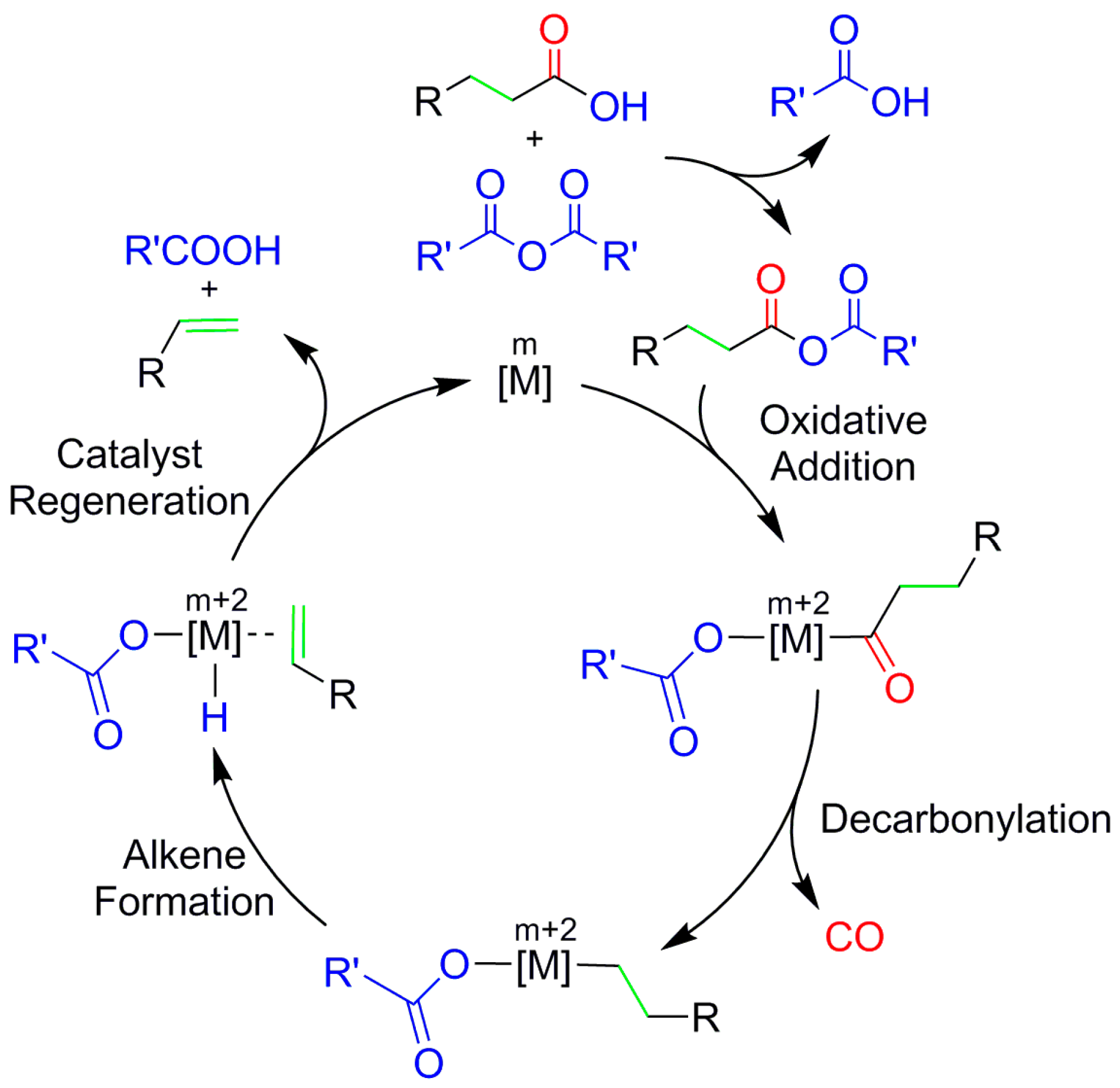
2. Results and Discussion
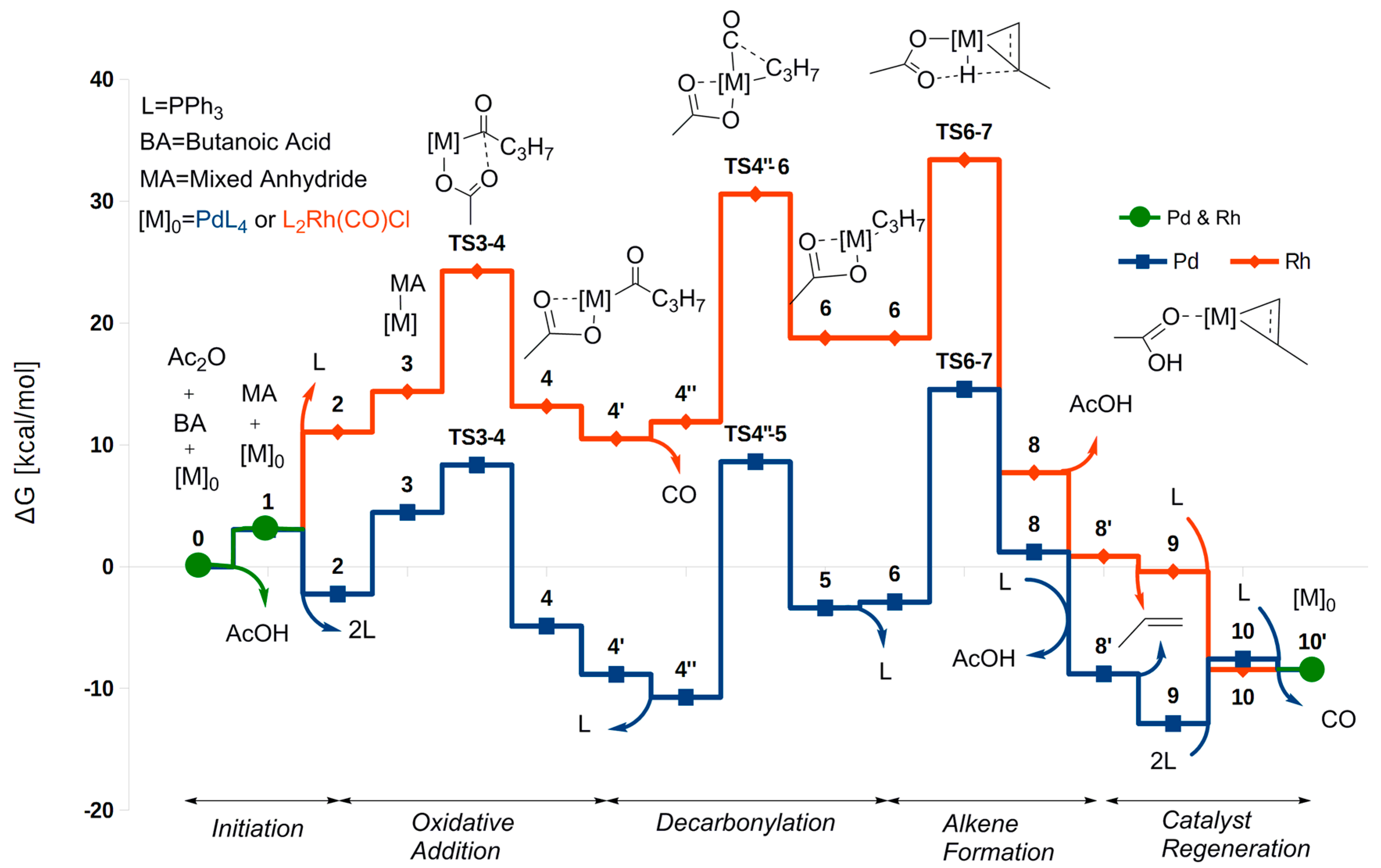
2.1. Oxidative Addition
2.2. Decarbonylation
2.3. Alkene Formation
2.4. Catalyst Regeneration
2.5. The Overall Reaction Mechanism and Comparison to the Pd-Catalyzed Reaction
3. Computational Method
3.1. Geometry Optimization
3.2. Thermochemistry
3.3. Single-Point Calculations (SP)
3.4. Free Energies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Franke, R.; Selent, D.; Börner, A. Applied hydroformylation. Chem. Rev. 2012, 112, 5675–5732. [Google Scholar] [CrossRef] [PubMed]
- Dawes, G.J.S.; Scott, E.L.; Le Notre, J.; Sanders, J.P.M.; Bitter, J.H. Deoxygenation of biobased molecules by decarboxylation and decarbonylation—A review on the role of heterogeneous, homogeneous and bio-catalysis. Green Chem. 2015, 17, 3231–3250. [Google Scholar] [CrossRef]
- Arpe, H.J.; Hawkins, S. Industrial Organic Chemistry, 5th ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2010; ISBN 978-3-527-32002-8. [Google Scholar]
- Agapie, T. Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Coord. Chem. Rev. 2011, 255, 861–880. [Google Scholar] [CrossRef]
- Skupinska, J. Oligomerization of α-olefins to higher oligomers. Chem. Rev. 1991, 91, 613–648. [Google Scholar] [CrossRef]
- Dodds, D.R.; Gross, R.A. Chemicals from biomass. Science 2007, 318, 1250–1251. [Google Scholar] [CrossRef] [PubMed]
- Dapsens, P.Y.; Mondelli, C.; Pérez-Ramírez, J. Biobased chemicals from conception toward industrial reality: Lessons learned and to be learned. ACS Catal. 2012, 2, 1487–1499. [Google Scholar] [CrossRef]
- Vennestrøm, P.N.R.; Osmundsen, C.M.; Christensen, C.H.; Taarning, E. Beyond petrochemicals: The renewable chemicals industry. Angew. Chem. Int. Ed. 2011, 50, 10502–10509. [Google Scholar] [CrossRef] [PubMed]
- Santillan-Jimenez, E.; Crocker, M. Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation. J. Chem. Technol. Biotechnol. 2012, 87, 1041–1050. [Google Scholar] [CrossRef]
- Gosselink, R.W.; Hollak, S.A.W.; Chang, S.-W.; van Haveren, J.; de Jong, K.P.; Bitter, J.H.; van Es, D.S. Reaction pathways for the deoxygenation of vegetable oils and related model compounds. ChemSusChem 2013, 6, 1576–1594. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, L.J.; Rodríguez, N.; Gooßen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Hopen Eliasson, S.H.; Törnroos, K.W.; Jensen, V.R. Palladium precatalysts for decarbonylative dehydration of fatty acids to linear alpha olefins. ACS Catal. 2016, 6, 7784–7789. [Google Scholar] [CrossRef]
- Goossen, L.J.; Rodriguez, N. A mild and efficient protocol for the conversion of carboxylic acids to olefins by a catalytic decarbonylative elimination reaction. Chem. Commun. 2004, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Nelson, J.A.; Byrne, M.P. A highly catalytic and selective conversion of carboxylic-acids to 1-alkenes of one less carbon-atom. J. Org. Chem. 1993, 58, 18–20. [Google Scholar] [CrossRef]
- Liu, Y.; Kim, K.E.; Herbert, M.B.; Fedorov, A.; Grubbs, R.H.; Stoltz, B.M. Palladium-catalyzed decarbonylative dehydration of fatty acids for the production of linear alpha olefins. Adv. Synth. Catal. 2014, 356, 130–136. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Hogan, L.T.; Hillmyer, M.A.; Tolman, W.B. Olefins from biomass feedstocks: Catalytic ester decarbonylation and tandem heck-type coupling. Chem. Commun. 2015, 51, 2731–2733. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.E.; Walter, E.L.; Doll, K.M. Tandem isomerization-decarboxylation for converting alkenoic fatty acids into alkenes. ACS Catal. 2014, 4, 3517–3520. [Google Scholar] [CrossRef]
- Chatterjee, A.; Jensen, V.R. A heterogeneous catalyst for the transformation of fatty acids to α-olefins. ACS Catal. 2017, 7, 2543–2547. [Google Scholar] [CrossRef]
- Dennig, A.; Kuhn, M.; Tassoti, S.; Thiessenhusen, A.; Gilch, S.; Bülter, T.; Haas, T.; Hall, M.; Faber, K. Oxidative decarboxylation of short-chain fatty acids to 1-alkenes. Angew. Chem. Int. Ed. 2015, 54, 8819–8822. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-B.; Lonsdale, R.; Reetz, M.T. Exploring substrate scope and stereoselectivity of p450 peroxygenase OleTJE in olefin-forming oxidative decarboxylation. Chem. Commun. 2016, 52, 8131–8133. [Google Scholar] [CrossRef] [PubMed]
- Herman, N.A.; Zhang, W. Enzymes for fatty acid-based hydrocarbon biosynthesis. Curr. Opin. Chem. Biol. 2016, 35, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kraus, G.A.; Riley, S. A large-scale synthesis of α-olefins and α,ω-dienes. Synthesis 2012, 44, 3003–3005. [Google Scholar] [CrossRef]
- Foglia, T.A.; Barr, P.A. Decarbonylation dehydration of fatty acids to alkenes in the presence of transition metal complexes. J. Am. Oil Chem. Soc. 1976, 53, 737–741. [Google Scholar] [CrossRef]
- John, A.; Miranda, M.O.; Ding, K.; Dereli, B.; Ortuño, M.A.; LaPointe, A.M.; Coates, G.W.; Cramer, C.J.; Tolman, W.B. Nickel catalysts for the dehydrative decarbonylation of carboxylic acids to alkenes. Organometallics 2016, 35, 2391–2400. [Google Scholar] [CrossRef]
- Le Nôtre, J.; Scott, E.L.; Franssen, M.C.R.; Sanders, J.P.M. Selective preparation of terminal alkenes from aliphatic carboxylic acids by a palladium-catalysed decarbonylation–Elimination reaction. Tetrahedron Lett. 2010, 51, 3712–3715. [Google Scholar] [CrossRef]
- Le Notre, J.; Scott, E.L.; Franssen, M.C.R.; Sanders, J.P.M. Biobased synthesis of acrylonitrile from glutamic acid. Green Chem. 2011, 13, 807–809. [Google Scholar] [CrossRef]
- Maetani, S.; Fukuyama, T.; Suzuki, N.; Ishihara, D.; Ryu, I. Efficient iridium-catalyzed decarbonylation reaction of aliphatic carboxylic acids leading to internal or terminal alkenes. Organometallics 2011, 30, 1389–1394. [Google Scholar] [CrossRef]
- Maetani, S.; Fukuyama, T.; Suzuki, N.; Ishihara, D.; Ryu, I. Iron-catalyzed decarbonylation reaction of aliphatic carboxylic acids leading to α-olefins. Chem. Commun. 2012, 48, 2552–2554. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.O.; Pietrangelo, A.; Hillmyer, M.A.; Tolman, W.B. Catalytic decarbonylation of biomass-derived carboxylic acids as efficient route to commodity monomers. Green Chem. 2012, 14, 490–494. [Google Scholar] [CrossRef]
- John, A.; Dereli, B.; Ortuño, M.A.; Johnson, H.E.; Hillmyer, M.A.; Cramer, C.J.; Tolman, W.B. Selective decarbonylation of fatty acid esters to linear α-olefins. Organometallics 2017, 36, 2956–2964. [Google Scholar] [CrossRef]
- Liu, Y.; Virgil, S.C.; Grubbs, R.H.; Stoltz, B.M. Palladium-catalyzed decarbonylative dehydration for the synthesis of α-vinyl carbonyl compounds and total synthesis of (−)-aspewentins A, B, and C. Angew. Chem. Int. Ed. 2015, 54, 11800–11803. [Google Scholar] [CrossRef] [PubMed]
- Ortuño, M.A.; Dereli, B.; Cramer, C.J. Mechanism of Pd-catalyzed decarbonylation of biomass-derived hydrocinnamic acid to styrene following activation as an anhydride. Inorg. Chem. 2016, 55, 4124–4131. [Google Scholar] [CrossRef] [PubMed]
- Ternel, J.; Lebarbé, T.; Monflier, E.; Hapiot, F. Catalytic decarbonylation of biosourced substrates. ChemSusChem 2015, 8, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Hillmyer, M.A.; Tolman, W.B. Anhydride-additive-free nickel-catalyzed deoxygenation of carboxylic acids to olefins. Organometallics 2017, 36, 506–509. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis: A Practical Approach, 2nd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2006; ISBN 978-3-527-31144-6. [Google Scholar]
- Miller, J.A.; Nelson, J.A. Oxidative addition of carboxylic acid anhydrides to rhodium(I) phosphine complexes to produce novel rhodium(III) acyl derivatives. Organometallics 1991, 10, 2958–2961. [Google Scholar] [CrossRef]
- Fristrup, P.; Kreis, M.; Palmelund, A.; Norrby, P.-O.; Madsen, R. The mechanism for the rhodium-catalyzed decarbonylation of aldehydes: A combined experimental and theoretical study. J. Am. Chem. Soc. 2008, 130, 5206–5215. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, W. Empirical correction to density functional theory for van der Waals interactions. J. Chem. Phys. 2002, 116, 515–524. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, A.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef] [PubMed]
- Spartan ’08, Wavefunction Inc.: Irvine, CA, USA, 2008.
- Allen, F.H. The Cambridge structural database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Dolg, M.; Stoll, H. Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y–Pd. J. Chem. Phys. 2007, 126, 124101. [Google Scholar] [CrossRef] [PubMed]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Ribeiro, R.F.; Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem. B 2011, 115, 14556–14562. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Applications and validations of the Minnesota density functionals. Chem. Phys. Lett. 2011, 502, 1–13. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Burns, L.A.; Patkowski, K.; Sherrill, C.D. Revised damping parameters for the D3 dispersion correction to density functional theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Minenkov, Y.; Occhipinti, G.; Jensen, V.R. Complete reaction pathway of ruthenium-catalyzed olefin metathesis of ethyl vinyl ether: Kinetics and mechanistic insight from DFT. Organometallics 2013, 32, 2099–2111. [Google Scholar] [CrossRef]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eliasson, S.H.H.; Chatterjee, A.; Occhipinti, G.; Jensen, V.R. The Mechanism of Rh-Catalyzed Transformation of Fatty Acids to Linear Alpha olefins. Inorganics 2017, 5, 87. https://doi.org/10.3390/inorganics5040087
Eliasson SHH, Chatterjee A, Occhipinti G, Jensen VR. The Mechanism of Rh-Catalyzed Transformation of Fatty Acids to Linear Alpha olefins. Inorganics. 2017; 5(4):87. https://doi.org/10.3390/inorganics5040087
Chicago/Turabian StyleEliasson, Sondre H. Hopen, Anamitra Chatterjee, Giovanni Occhipinti, and Vidar R. Jensen. 2017. "The Mechanism of Rh-Catalyzed Transformation of Fatty Acids to Linear Alpha olefins" Inorganics 5, no. 4: 87. https://doi.org/10.3390/inorganics5040087
APA StyleEliasson, S. H. H., Chatterjee, A., Occhipinti, G., & Jensen, V. R. (2017). The Mechanism of Rh-Catalyzed Transformation of Fatty Acids to Linear Alpha olefins. Inorganics, 5(4), 87. https://doi.org/10.3390/inorganics5040087