Mononuclear Dysprosium(III) Complexes with Triphenylphosphine Oxide Ligands: Controlling the Coordination Environment and Magnetic Anisotropy

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussions
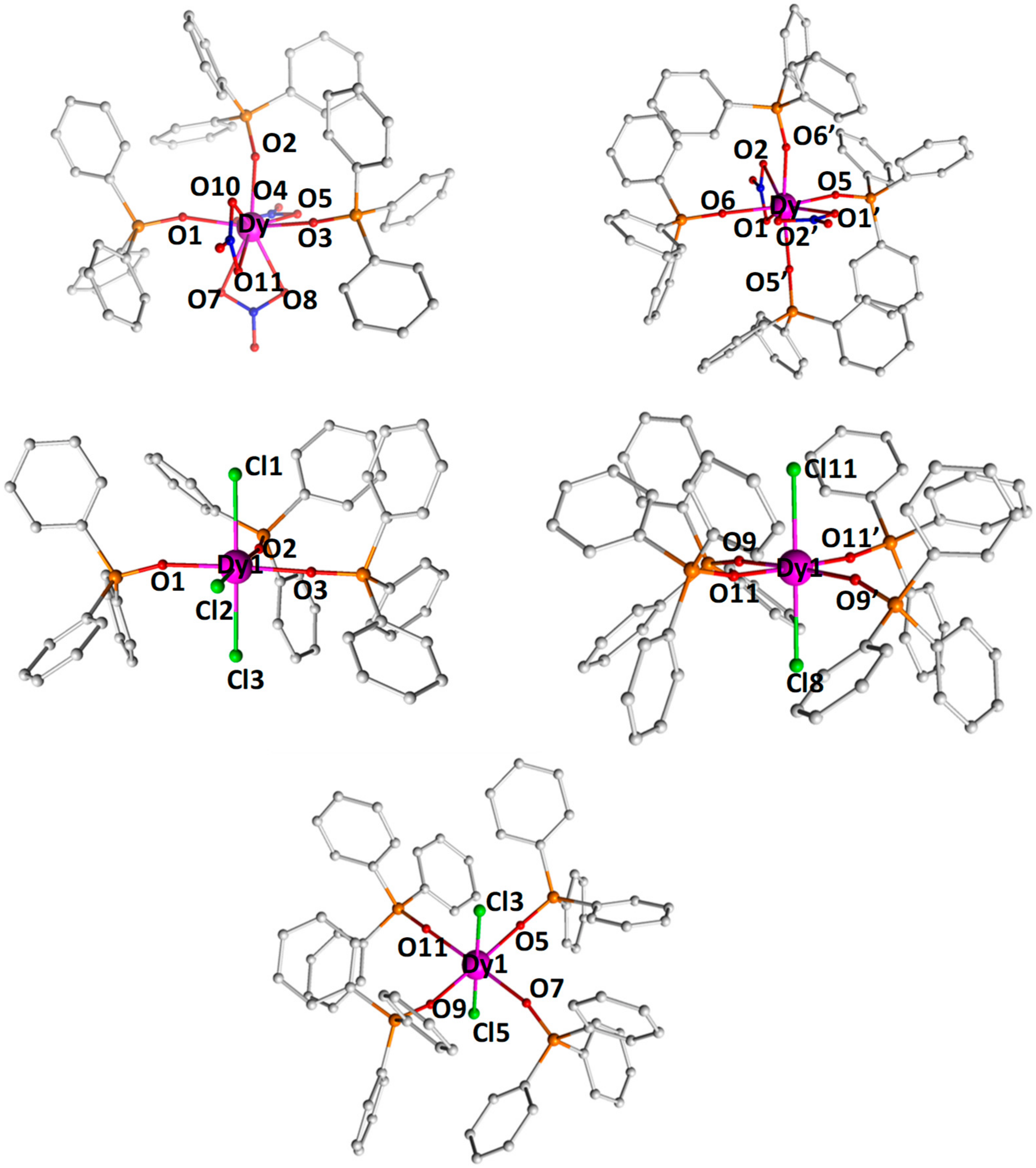
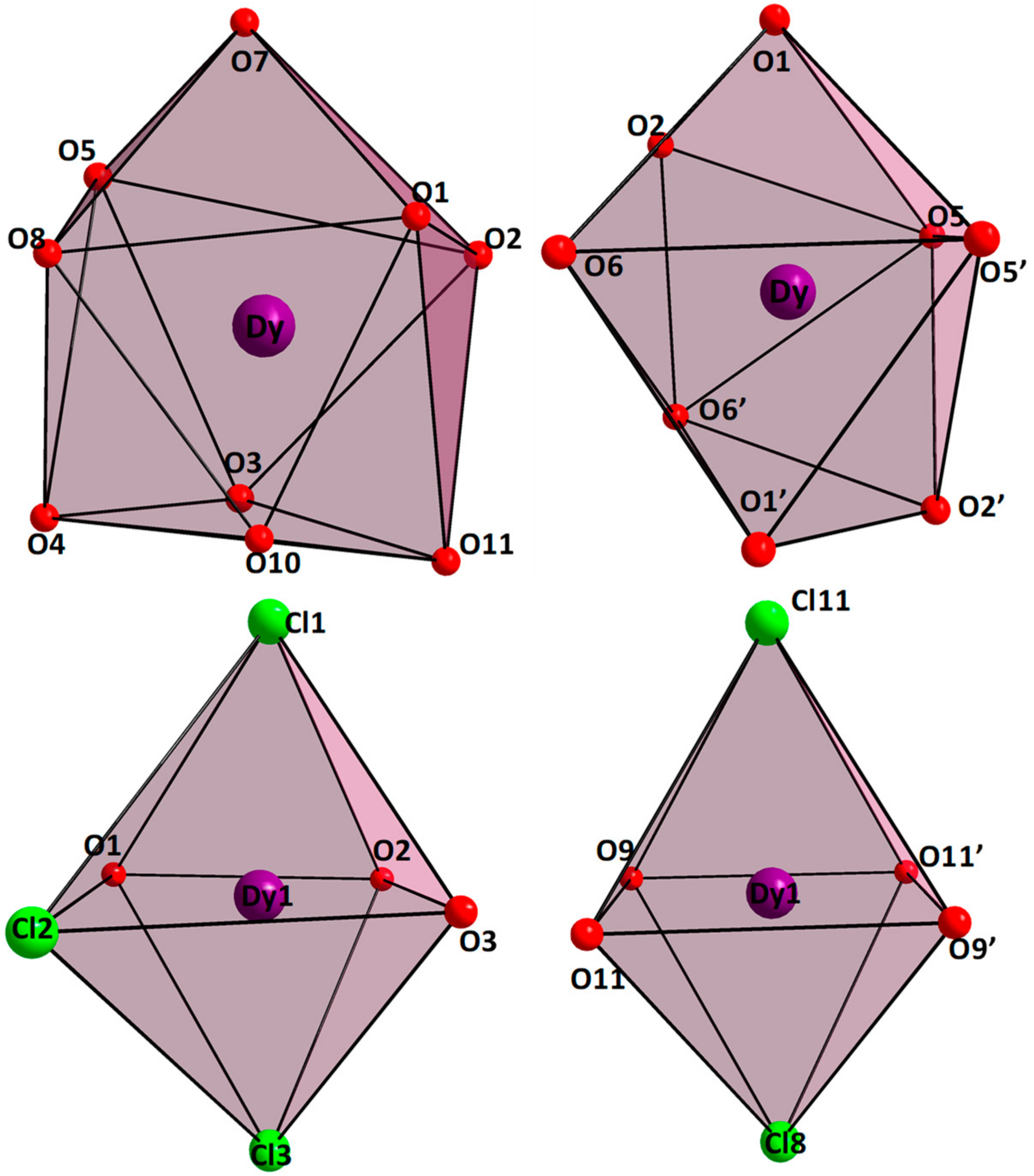
2.1. Synthesis and Molecular Structure
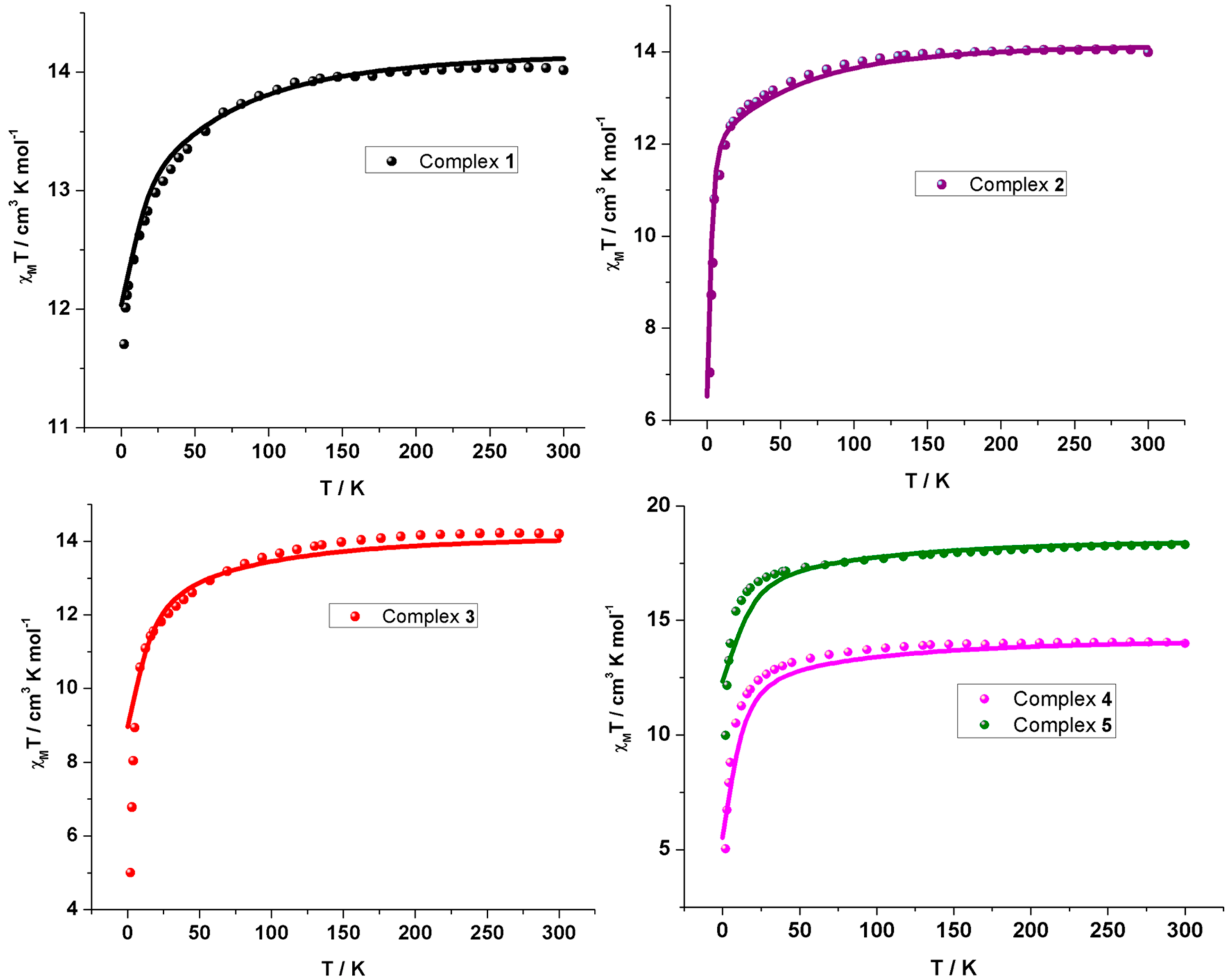
2.2. Magnetic Properties
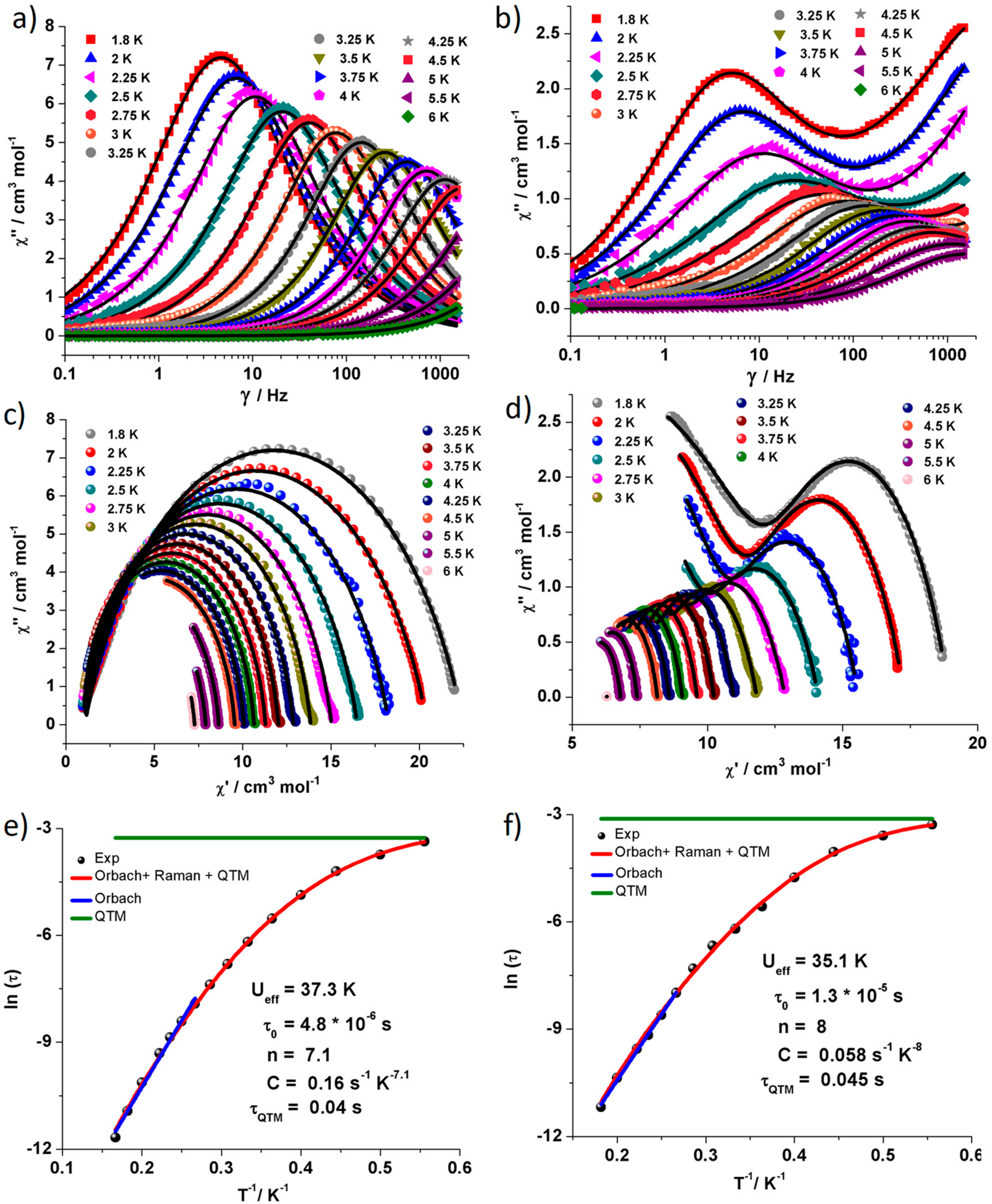
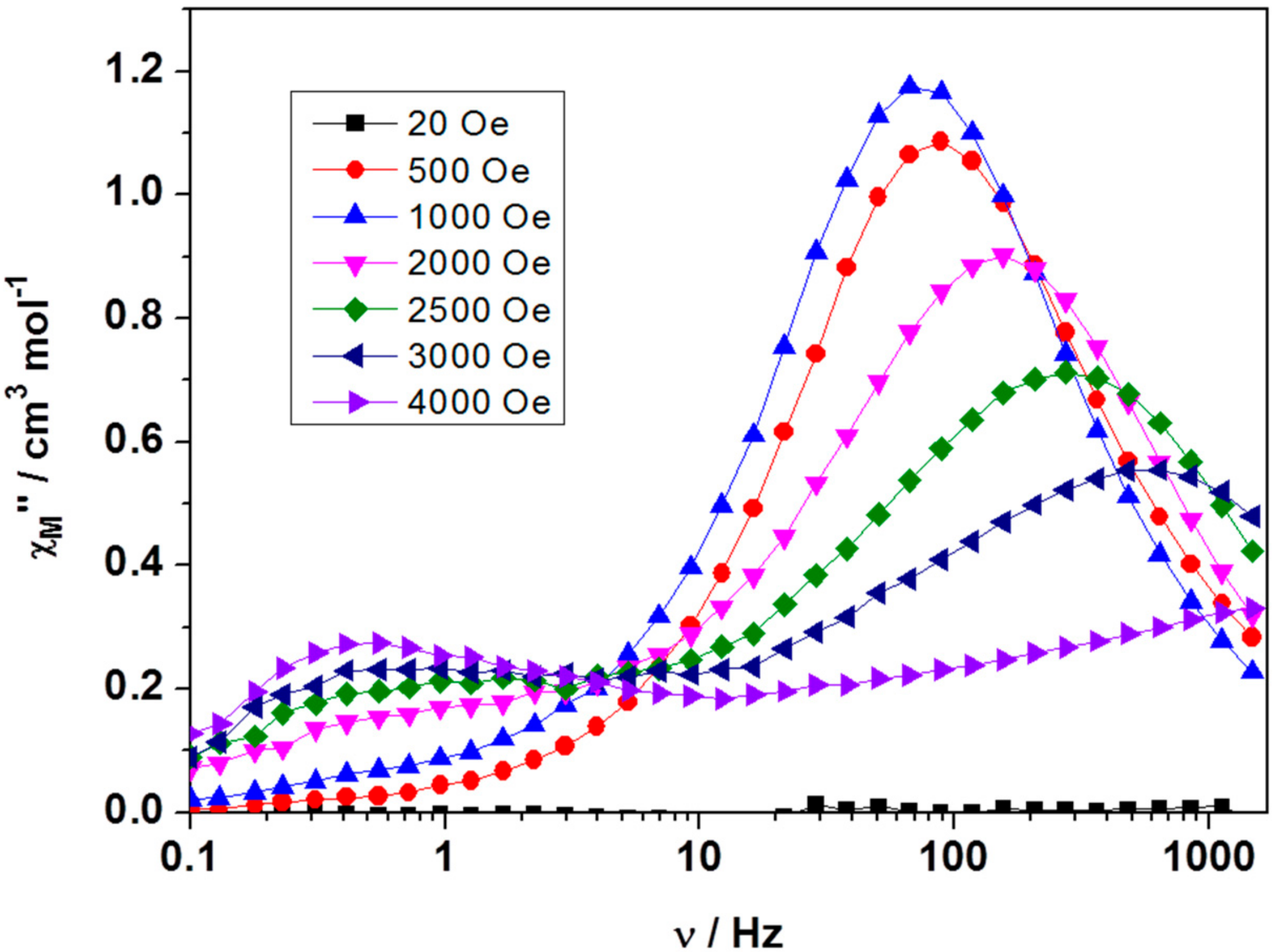
2.3. Dynamic Magnetic Properties
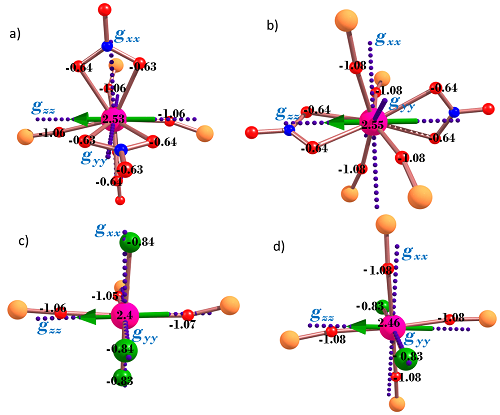
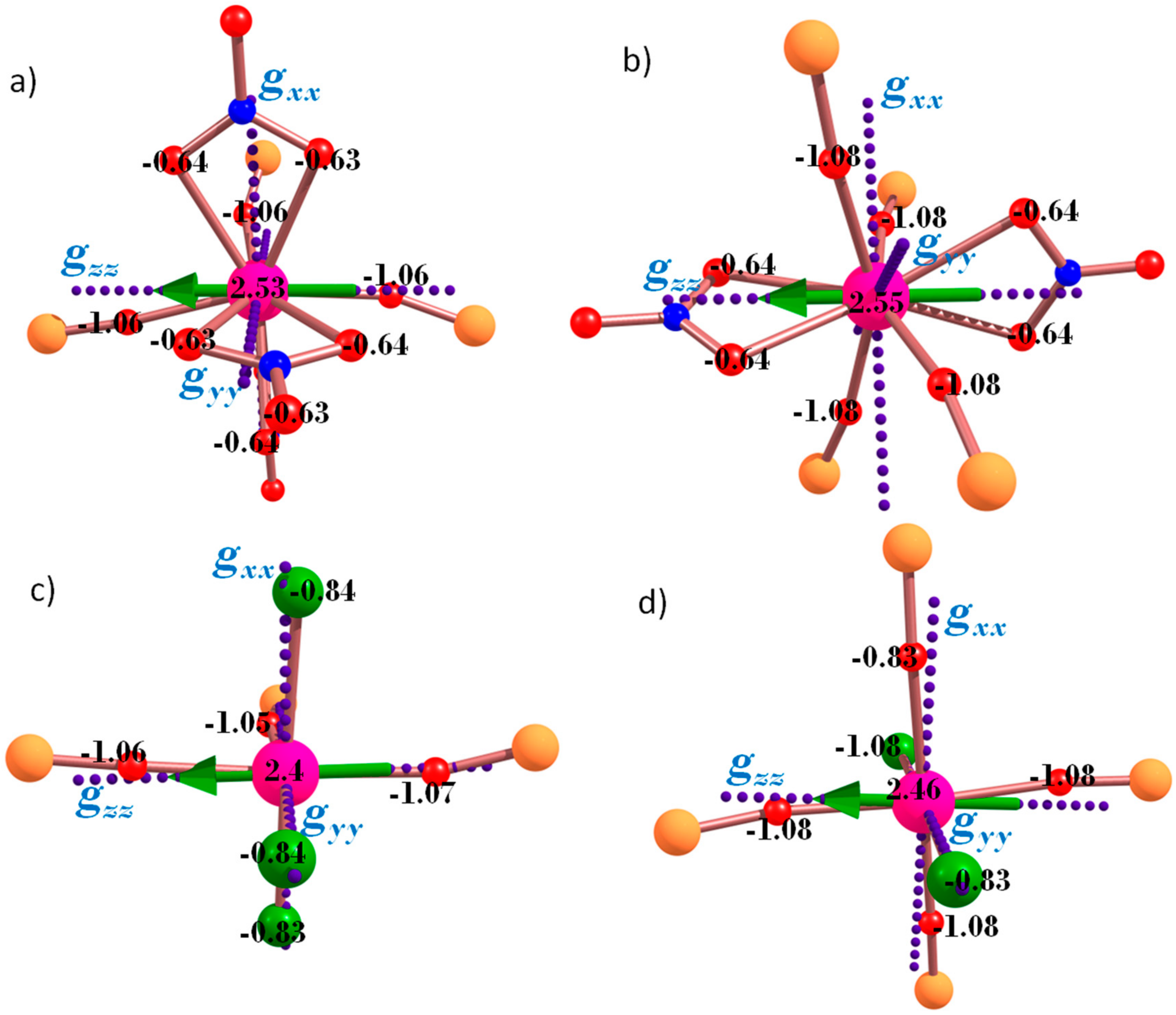
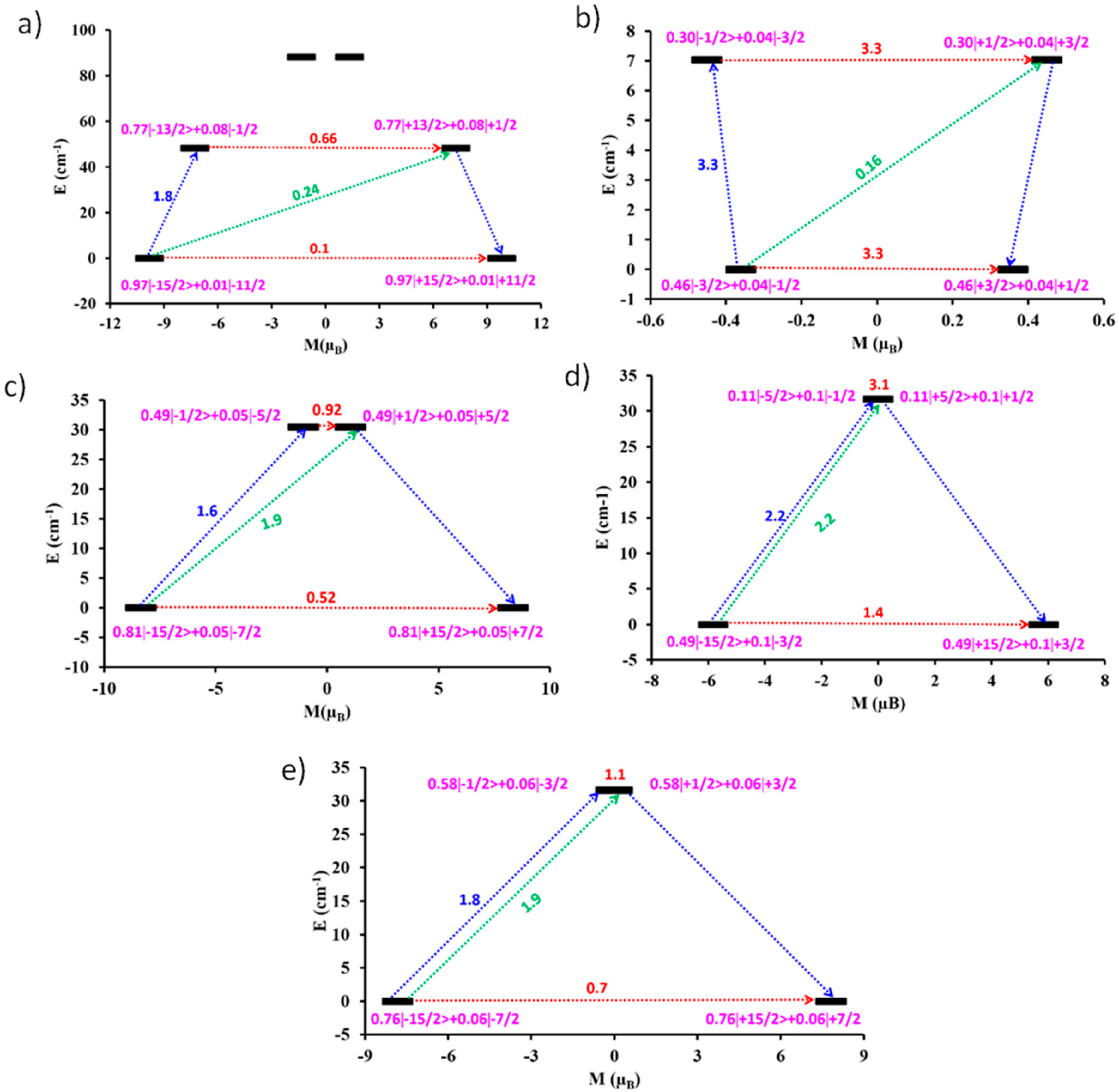
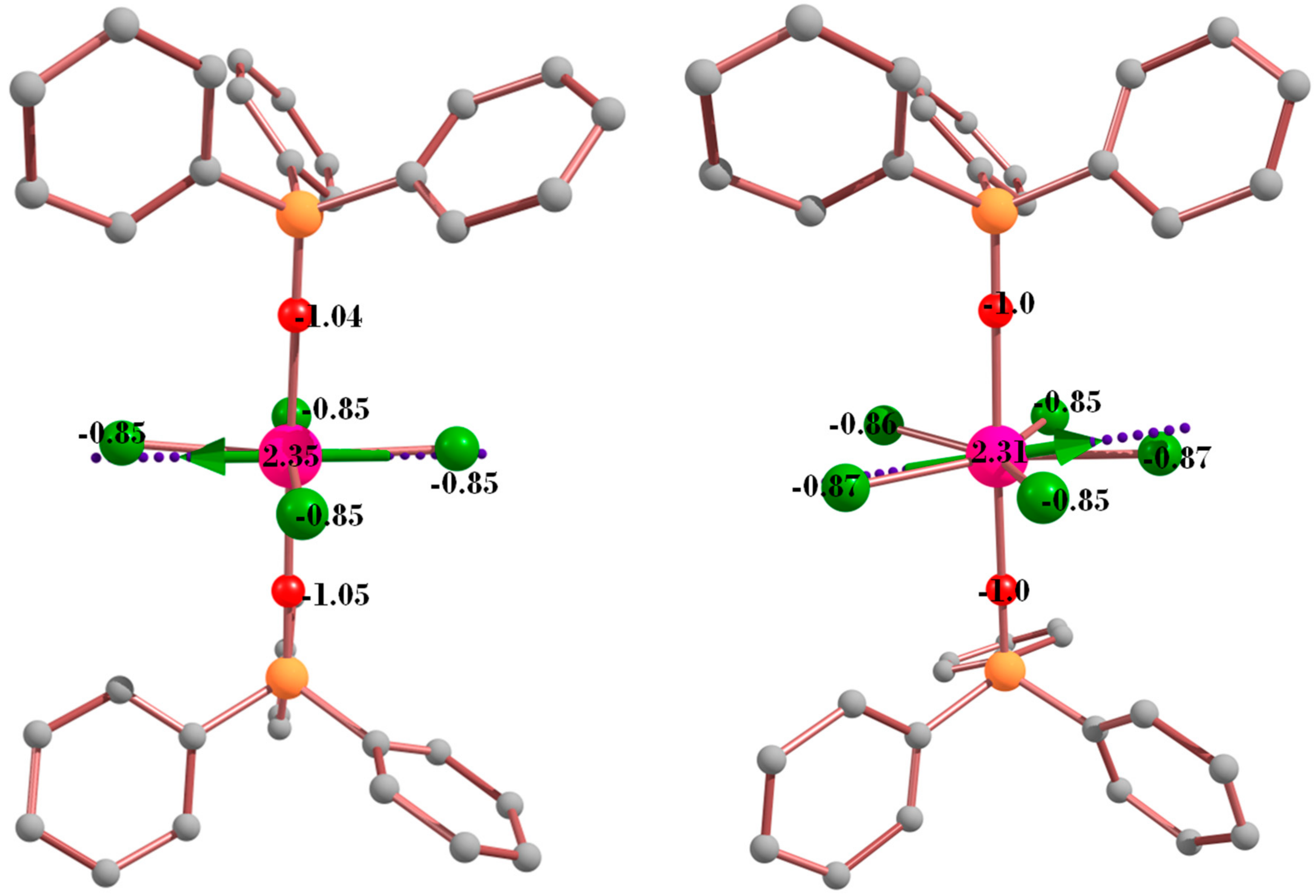
3. Theoretical Studies
4. Materials and Methods
4.1. Experimental section
4.1.1. Synthesis of [DyIII(OPPh3)3(NO3)3]·2(CH3)2CO (1)
4.1.2. Synthesis of [DyIII(OPPh3)4(NO3)2](NO3) (2)
4.1.3. Synthesis of [DyIII(OPPh3)3Cl3]·0.5(CH3)2CO (3)
4.1.4. Synthesis of [DyIII(OPPh3)4Cl2]Cl·2H2O·2EtOH (4)
4.1.5. Synthesis of [DyIII(OPPh3)4(Cl)2](FeCl4) (5)
4.2. X-ray Crystallography
4.3. Magnetic Measurements
4.4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Mononuclear lanthanide complexes with a long magnetization relaxation time at high temperatures: A new category of magnets at the single-molecular level. J. Phys. Chem. B 2004, 108, 11265–11271. [Google Scholar] [CrossRef]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Lanthanide double-decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef] [PubMed]
- Sessoli, R.; Powell, A.K. Strategies towards single molecule magnets based on lanthanide ions. Coord. Chem. Rev. 2009, 253, 2328–2341. [Google Scholar] [CrossRef]
- Meng, Y.-S.; Jiang, S.-D.; Wang, B.-W.; Gao, S. Understanding the Magnetic Anisotropy toward Single-Ion Magnets. Acc. Chem. Res. 2016, 49, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Guo, Y.-N.; Tang, J. Recent advances in dysprosium-based single molecule magnets: Structural overview and synthetic strategies. Coord. Chem. Rev. 2013, 257, 1728–1763. [Google Scholar] [CrossRef]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [Google Scholar] [CrossRef]
- Chilton, N.F.; Collison, D.; McInnes, E.J.L.; Winpenny, R.E.P.; Soncini, A. An electrostatic model for the determination of magnetic anisotropy in dysprosium complexes. Nat. Commun. 2013, 4, 2551. [Google Scholar] [CrossRef] [Green Version]
- Rinehart, J.D.; Long, J.R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2011, 2, 2078–2085. [Google Scholar] [CrossRef]
- Aravena, D.; Ruiz, E. Shedding Light on the Single-Molecule Magnet Behavior of Mononuclear Dy(III) Complexes. Inorg. Chem. 2013, 52, 13770–13778. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 2017, 548, 439–442. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem. Int. Ed. 2017, 56, 11445–11449. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Rajeshkumar, T.; Rajaraman, G.; Murugavel, R. An air-stable Dy(III) single-ion magnet with high anisotropy barrier and blocking temperature. Chem. Sci. 2016, 7, 5181–5191. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.-C.; Liu, J.-L.; Vieru, V.; Ungur, L.; Jia, J.-H.; Chibotaru, L.F.; Lan, Y.; Wernsdorfer, W.; Gao, S.; et al. A Stable Pentagonal Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization Reversal Barrier over 1000 K. J. Am. Chem. Soc. 2016, 138, 5441–5450. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Liu, J.-L.; Wernsdorfer, W.; Liu, D.; Chibotaru, L.F.; Chen, X.-M.; Tong, M.-L. Hyperfine-Interaction-Driven Suppression of Quantum Tunneling at Zero Field in a Holmium(III) Single-Ion Magnet. Angew. Chem. Int. Ed. 2017, 56, 4996–5000. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Liu, J.-L.; Ungur, L.; Liu, J.; Li, Q.-W.; Wang, L.-F.; Ni, Z.-P.; Chibotaru, L.F.; Chen, X.-M.; Tong, M.-L. Symmetry-Supported Magnetic Blocking at 20 K in Pentagonal Bipyramidal Dy(III) Single-Ion Magnets. J. Am. Chem. Soc. 2016, 138, 2829–2837. [Google Scholar] [CrossRef] [PubMed]
- Bannister, R.D.; Levason, W.; Reid, G.; Zhang, W. Diphosphine dioxide complexes of lanthanum and lutetium—The effects of ligand architecture and counter-anion. Polyhedron 2017, 133, 264–269. [Google Scholar] [CrossRef]
- Burt, J.; Levason, W.; Reid, G. Coordination chemistry of the main group elements with phosphine, arsine and stibine ligands. Coord. Chem. Rev. 2014, 260, 65–115. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-K.; Zhang, M.-J.; Jing, X.-Y.; Wang, J.-T.; Wang, R.-J.; Wang, H.-G. Pentakis(triphenylphosphine oxide)chlorolanthanide bis(feric tetrachloride) complexes. Inorg. Chim.Acta 1989, 163, 19–23. [Google Scholar]
- Pinsky, M.; Avnir, D. Continuous Symmetry Measures. 5. The Classical Polyhedra. Inorg. Chem. 1998, 37, 5575–5582. [Google Scholar] [CrossRef]
- Chilton, N.F. CC-Fit Model; The Univesity of Manchester: Manchester, UK, 2014. [Google Scholar]
- Sertphon, D.; Murray, K.S.; Phonsri, W.; Jover, J.; Ruiz, E.; Telfer, S.G.; Alkas, A.; Harding, P.; Harding, D.J. Slow relaxation of magnetization in a bis-mer-tridentate octahedral Co(II) complex. Dalton Trans. 2018, 47, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Vignesh, K.R.; Langley, S.K.; Murray, K.S.; Rajaraman, G. Role of Diamagnetic Ions on the Mechanism of Magnetization Relaxation in “Butterfly” {CoIII2LnIII2} (Ln = Dy, Tb, Ho) Complexes. Inorg. Chem. 2017, 56, 2518–2532. [Google Scholar] [CrossRef] [PubMed]
- Das, C.; Upadhyay, A.; Vaidya, S.; Singh, S.K.; Rajaraman, G.; Shanmugam, M. Origin of SMM behaviour in an asymmetric Er(III) Schiff base complex: A combined experimental and theoretical study. Chem. Commun. 2015, 51, 6137–6140. [Google Scholar] [CrossRef] [PubMed]
- Hallmen, P.P.; Köppl, C.; Rauhut, G.; Stoll, H.; van Slageren, J. Fast and reliable ab initio calculation of crystal field splittings in lanthanide complexes. J. Chem. Phys. 2017, 147, 164101. [Google Scholar] [CrossRef] [PubMed]
- Blagg, R.J.; Ungur, L.; Tuna, F.; Speak, J.; Comar, P.; Collison, D.; Wernsdorfer, W.; McInnes, E.J.L.; Chibotaru, L.F.; Winpenny, R.E.P. Magnetic relaxation pathways in lanthanide single-molecule magnets. Nat. Chem. 2013, 5, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Meihaus, K.R.; Long, J.R. Magnetic blocking at 10 K and a Dipolar-mediated avalanche in salts of the bis(η8-cyclooctatetraenide) complex [Er(COT)2]. J. Am. Chem. Soc. 2013, 135, 17952–17957. [Google Scholar] [CrossRef]
- Ungur, L.; Lin, S.-Y.; Tang, J.; Chibotaru, L.F. Single-molecule toroics in Ising-type lanthanide molecular clusters. Chem. Soc. Rev. 2014, 43, 6894–6905. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.C.; Reid, J.S. CrysAlis Pro. Agilent Technologies, Yarnton, England. Acta Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL-97, Programs for X-Ray Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Spek, A. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef]
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; Vico, L.D.; Galván, I.F.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. MOLCAS 8: New Capabilities for Multiconfigurational Quantum Chemical Calculations across the Periodic Table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef]
- Hess, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. [Google Scholar] [CrossRef]
- Roos, B.O.; Malmqvist, P.-A. Relativistic quantum chemistry: The multiconfigurational approach. Phys. Chem. Chem. Phys. 2004, 6, 2919–2927. [Google Scholar] [CrossRef]
- Roos, B.O.; Lindh, R.; Malmqvist, P.-A.; Veryazov, V.; Widmark, P.-O.; Borin, A.C. New Relativistic Atomic Natural Orbital Basis Sets for Lanthanide Atoms with Applications to the Ce Diatom and LuF3. J. Phys. Chem. A 2008, 112, 11431–11435. [Google Scholar] [CrossRef] [PubMed]
- Malmqvist, P.A.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin-orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- Chibotaru, L.F.; Ungur, L. Ab initio calculation of anisotropic magnetic properties of complexes. I. Unique definition of pseudospin Hamiltonians and their derivation. J. Chem. Phys. 2012, 137, 064112. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, L.; Lindh, R.; Karlström, G. Local properties of quantum chemical systems: The LoProp approach. J. Chem. Phys. 2004, 121, 4494–4500. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Vignesh, K.R.; Das, C.; Singh, S.K.; Rajaraman, G.; Shanmugam, M. Influence of the Ligand Field on the Slow Relaxation of Magnetization of Unsymmetrical Monomeric Lanthanide Complexes: Synthesis and Theoretical Studies. Inorg. Chem. 2017, 56, 14260–14276. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, S.L.; Truhlar, D.G. Modeling the Partial Atomic Charges in Inorganometallic Molecules and Solids and Charge Redistribution in Lithium-Ion Cathodes. J. Chem. Theory Comput. 2014, 10, 5640–5650. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KDs | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 2 | 48.3 | 7.0 | 30.5 | 31.7 | 31.6 |
| 3 | 88.2 | 125.8 | 64.2 | 53.2 | 52.9 |
| 4 | 148.4 | 148.1 | 145.9 | 148.3 | 153.5 |
| 5 | 185.0 | 185.4 | 276.9 | 294.8 | 302.0 |
| 6 | 212.8 | 231.4 | 307.4 | 322.3 | 331.0 |
| 7 | 265.9 | 274.6 | 343.4 | 364.7 | 374.9 |
| 8 | 333.1 | 312.1 | 368.0 | 394.8 | 396.7 |
| gx | 0.2287 | 11.9707 | 0.1352 | 2.1939 | 1.2982 |
| gy | 0.3597 | 8.0477 | 2.9545 | 5.9830 | 2.8906 |
| gz | 19.6158 | 0.7205 | 16.6803 | 11.6635 | 15.6449 |
| KDs | Model a | Model b |
|---|---|---|
| 1 | 0.0 | 0.0 |
| 2 | 49.1 | 61.4 |
| 3 | 99.6 | 162.0 |
| 4 | 158.3 | 307.7 |
| 5 | 296.1 | 463.4 |
| 6 | 306.0 | 609.8 |
| 7 | 354.9 | 691.4 |
| 8 | 373.7 | 749.1 |
| gx | 0.6575 | 1.0122 |
| gy | 3.5133 | 4.8452 |
| gz | 16.8965 | 15.7556 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langley, S.K.; Vignesh, K.R.; Holton, K.; Benjamin, S.; Hix, G.B.; Phonsri, W.; Moubaraki, B.; Murray, K.S.; Rajaraman, G. Mononuclear Dysprosium(III) Complexes with Triphenylphosphine Oxide Ligands: Controlling the Coordination Environment and Magnetic Anisotropy. Inorganics 2018, 6, 61. https://doi.org/10.3390/inorganics6020061
Langley SK, Vignesh KR, Holton K, Benjamin S, Hix GB, Phonsri W, Moubaraki B, Murray KS, Rajaraman G. Mononuclear Dysprosium(III) Complexes with Triphenylphosphine Oxide Ligands: Controlling the Coordination Environment and Magnetic Anisotropy. Inorganics. 2018; 6(2):61. https://doi.org/10.3390/inorganics6020061
Chicago/Turabian StyleLangley, Stuart K., Kuduva R. Vignesh, Kerey Holton, Sophie Benjamin, Gary B. Hix, Wasinee Phonsri, Boujemaa Moubaraki, Keith S. Murray, and Gopalan Rajaraman. 2018. "Mononuclear Dysprosium(III) Complexes with Triphenylphosphine Oxide Ligands: Controlling the Coordination Environment and Magnetic Anisotropy" Inorganics 6, no. 2: 61. https://doi.org/10.3390/inorganics6020061
APA StyleLangley, S. K., Vignesh, K. R., Holton, K., Benjamin, S., Hix, G. B., Phonsri, W., Moubaraki, B., Murray, K. S., & Rajaraman, G. (2018). Mononuclear Dysprosium(III) Complexes with Triphenylphosphine Oxide Ligands: Controlling the Coordination Environment and Magnetic Anisotropy. Inorganics, 6(2), 61. https://doi.org/10.3390/inorganics6020061