2.2.5. Oxidation of Compounds 2 and 3 over LDH Catalysts
In order to determine the efficacy of these catalysts with electron-rich substrates, model compound
2 was used as a starting material. Catalysts that returned a yield of >80% in the conversion of
1 to
1a were screened for catalytic activity in the oxidation of
2. Additionally, Ni-Al-LDH-1 was also used in the reaction for comparison purposes [
21]. Oxidation of
2 with the aforementioned catalysts afforded two products, the expected ketone,
2a, as well as the alcohol elimination product,
2b. As can be seen from
Table 5, catalysts containing nickel yielded higher amounts of the dehydration product
2a. Dehydration was found to be most prominent for Ni-Al-LDH-2. On the other hand, copper-containing catalysts tended to be more selective towards the ketone product,
2a. In an attempt to elucidate the relationship between catalyst functionality and alcohol dehydration, acidity and basicity measurements were compared to catalyst performance. No absolute trend between acidity or basicity and alkene yield was elucidated, although the highest yields of
2b and
3b (see
Table 5 and
Table 6) were obtained for the LDH catalysts possessing the highest numbers of acid and base sites (Ni-Al-LDH-1, Ni-Al-LDH-2, and Ni-Cr-LDH). Mechanistically this may occur via an E2-type mechanism in which the metal alkoxide is formed on the catalyst surface, followed by deprotonation of the β-carbon and subsequent elimination of the metal oxide to form the alkene. Although both Ni-Al-LDH catalysts both performed well, overall, oxidation with Ni-Cr-LDH resulted in the highest yield of
2a. In the case of Cu-Cr-LDH, near quantitative conversion of
2 was observed. However, the selectivity to
2a (ca. 50%), which was the only identifiable compound by GC/MS, was significantly lower than other LDH catalysts (entries 2–6,
Table 5), presumably due to the conversion of
2 to unidentifiable products, evidenced by the darkening of the reaction mixture.
In order to further increase lignin-like functionality on the substrate and explore functional group sensitivity, the phenolic model compound
3 was chosen. As shown in
Table 6, the use of Ni-containing catalysts for the oxidation of
3 favored the formation of the dehydration product
3b. Unfortunately, poor mass balances were obtained for the oxidation of compound
3 due to suspected polymerization (chromatographically immobile material). This suggests that phenolic groups may need to be protected, e.g., by benzylation, prior to benzylic oxidation. Unexpectedly, a small amount of benzaldehyde
3c was also formed during the oxidation of
3 as a result of C
α–C
β bond cleavage. Aldehyde formation was most prevalent when the Ni-Cu-Cr-LDH and Ni-Al-LDH-1 were used as catalysts. Moreover, Cu-Cr-LDH was active in the oxidation of
3 but did not yield identifiable products. The production of
3b likely results in phenolic or styrenic coupling reactions leading to high molecular weight polymers.
2.2.6. Catalyst Reusability Study
Catalyst reusability was studied using Ni-Al-LDH-1 and Ni-Cr-LDH in the oxidation of
1 (
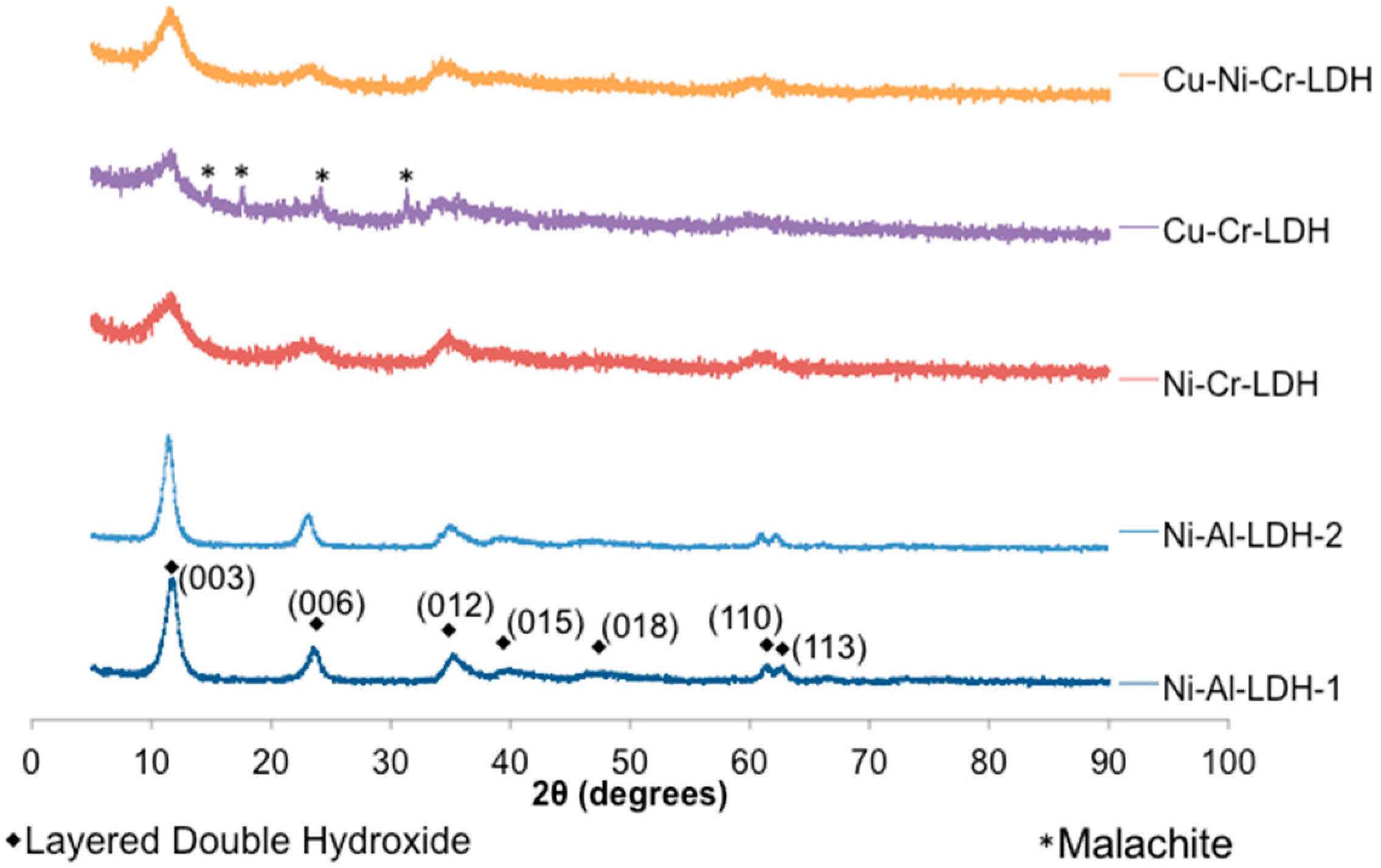
Table 7). After the reaction, the catalysts were filtered and washed with THF and hexanes, and then dried in a vacuum oven prior to re-use. Re-usability tests for Ni-Al-LDH-1 and Ni-Cr-LDH demonstrated a significant decrease in activity upon successive use. The X-ray diffractogram of the spent Ni-Cr-LDH (
Figure S10) displayed similar peaks to the fresh catalyst with the exception of a new highly crystalline peak corresponding to chromium (III) oxide, while N
2 physisorption analysis revealed a significant decrease in surface area (27.8 m
2·g
−1 post reaction), which is believed to be the result of phase segregation in the LDH, in addition to adsorbed organics blocking pores. Similarly, Ni-Al-LDH-1 displayed the characteristic LDH diffraction pattern but also a highly crystalline peak corresponding to Al(OH)
3 (
Figure S11). From these observations, it is evident that the LDH structure of the catalysts was largely retained after use, although limited segregation of the M(OH)
3 phase occurs (which decomposes, in the case of Cr, to Cr
2O
3). Similar results were obtained for LDHs tested with other substrates (see, for example,
Figure S12).
Other researchers have reported that full activity of LDH catalysts for the oxidation of benzyl alcohol is regained upon washing LDH catalysts with aqueous sodium carbonate [
29,
30]. After washing Ni-Cr-LDH with Na
2CO
3, a small amount of activity was regained. We believe that inability to completely regenerate the Ni-Cr-LDH catalyst may be related to the phase segregation observed by X-ray diffraction (
Figure S10). The effect of carbonate washing was more pronounced in the case of Ni-Al-LDH-1. Indeed, Ni-Al-LDH-1 showed no activity for the oxidation of compound
1 after the first use. However, after washing with carbonate solution, activity was completely regained. In fact, conversion increased from 72% to 100%, possibly due to an increase in the number of defect sites after reconstitution with Na
2CO
3.
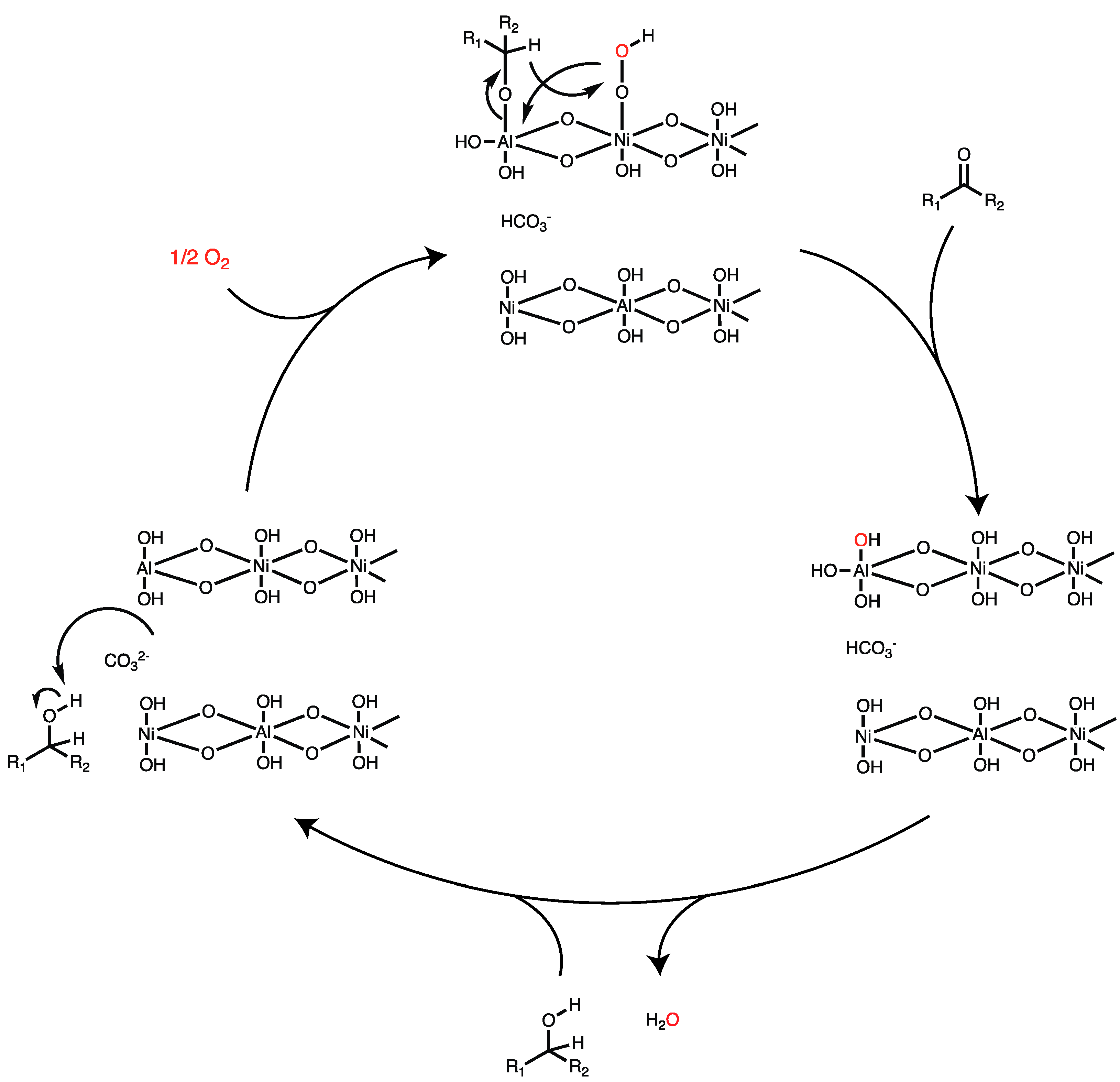
Other workers have reported that LDH anions may play an integral role in alcohol oxidation as evidenced by the reduced catalyst activity when anions are absent or substituted by another anion in the LDH [
21,
25]. In order to ascertain whether carbonate acts as a stoichiometric base, the amount of CO
32− present in each LDH was calculated based on the idealized LDH formula [M
2+1−χM
3+χ(OH)
2]
χ+(A
n−)
χ/n, where χ is the trivalent metal ratio, A is the anionic species, and
n is the charge of the anionic species. The water content was purposefully ignored as this can vary between LDHs [
26]. As can be seen in
Table 8, CO
32− was present in a sub-stoichiometric quantity compared to the substrate
1; hence, carbonate did not act as a stoichiometric base in these oxidation reactions.
2.2.8. Oxidation of Lignin Model Dimer Compounds over LDH Catalysts
Lignin is an amorphous biopolymer synthesized
in planta in the secondary cell walls via oxidative radical condensation of three monolignols (sinapyl, coniferyl, and
p-coumaryl alcohol) [
31]. As such, it is composed of a variety of linkages, the most abundant of which is the β-O-4 linkage, which can compose as much as 60% of the linkages in hardwood species [
31]. Moreover, several recent reports on the depolymerization of lignin have focused on the benzylic oxidation of the β-O-4 linkage, followed by a secondary cleavage step [
14,
16,
17,
32,
33].
While promising results were obtained when benchtop reactions were performed on non-phenolic compounds
1 and
2, only the dehydration product (
4c, 100% yield) was observed when lignin model dimer
4 was subjected to optimized reaction conditions (100% O
2, 0.5 g Ni-Al-LDH-1, 150 °C, in DPE). Thus, in order to determine if higher temperatures would enhance the rate of oxidation rather than dehydration, a pressurized reaction system was used. Indeed, when lignin model dimer compounds were reacted under slightly elevated temperatures (i.e., 180 °C) using 8% O
2/N
2 (50 bar) significant conversion was observed (
Table 9,
Table 10 and
Table 11). It should be noted that the use of pressurized oxygen significantly increases safety concerns for reactions in organic media. Indeed, Stahl and coworkers [
34] recently reported limiting oxygen concentrations (LOC) for nine organic solvents, finding that ca. 8% O
2 counter-balanced with inert gas was generally non-combustible. Thus, in this study 8% oxygen counter-balanced with nitrogen was used, which provides a nearly stoichiometric amount of oxygen (ca. 3 equiv.) for the oxidation of lignin model dimers. In addition to addressing safety concerns, the use of near stoichiometric amounts of oxygen limits over-oxidation to dicarboxylic acids, which are common products of aromatic ring over-oxidation [
35].
Although modest amounts of the ketone resulting from benzylic oxidation were detected, small molecules resulting from cleavage of the model linkages were observed in more substantial yields (
Table 9). Wang et al. [
36] recently reported that lignin β-O-4 models oxidized at the benzylic position are more easily fragmented than the benzylic alcohol analogue, due to the weakening of the C
β–O
4 bond by approximately 87 kJ/mol. Thus, it follows that the modest yields of
4d are explained by the ready cleavage of the C
β–O
4 bond, as indicated by the observation of the phenol
4b. Product
4a, which likely also results from oxidative cleavage of
4d, was generally present in higher yield than
4b presumably due to phenol polymerization; while the mechanism for oxidative cleavage of
4d is unclear, it is presumed to undergo a similar route as that observed by Mottweiler et al. [
37]. Indeed, Mottweiler et al. noted that the use of a Cu-V-LDH in the presence of O
2 resulted in a large amount of the A-ring acid and aldehyde. In addition, enol ether product
4c was observed, resulting from benzylic alcohol dehydration. Product
4c is particularly interesting because enol ethers are known to undergo hydrolysis under acidic conditions [
38]. This production of enol ethers in lignin would result in an easily hydrolysable linkage. Additionally, product
4e, that likely results from non-oxidative cleavage of
4d, was observed as a minor co-product. Products such as
4e are commonly observed in heterogeneous oxidation reactions of lignin model compounds [
39,
40,
41,
42]. Given the tendency of enol ethers to undergo metal-catalyzed cleavage reactions under oxidizing conditions, one can speculate that benzoic acid
4a is produced from the enol ether (
4c and
5c) [
43]. Indeed, when
4c was used as the feedstock in a control experiment (using Cu-Cr-LDH as catalyst),
4a was produced in low yield (7%) after 24 h.
Surprisingly, the Ni-Cr-LDH catalyst, which successfully oxidized compounds
1–
3, produced modest conversions in the cases of dimer models
4–
6. This may be due, in part at least, to the catalyst’s small average pore diameter (2.9 nm). Unlike compounds
1–
3, which are relatively small, lignin dimer model compounds
4–
6 (ca. 1.5 nm) [
44] approach the pore diameter of Ni-Cr-LDH (
Table 1). Moreover, other catalysts with larger pore diameters showed increased conversion of compounds
4–
5. Indeed, Ni-Al-LDH-1, with a pore diameter of 7.3 nm, was found to be the most active catalyst for conversion of the lignin model compounds used in this study, resulting in >99% conversion of models
4 and
5 (
Table 9 and
Table 10). Unfortunately, yields of individual products were low (<20%).
In these experiments phenols resulting from β-aryl ether cleavage were recovered in low yields. As commonly reported for oxidation of lignin model compounds, phenols are often converted into unidentifiable products [
16,
17,
36]. In order to investigate the stability of phenols under the reaction conditions, guaiacol (
4b) was subjected to the same conditions in the absence of catalyst. After 16 h at 180 °C under 8% O
2/N
2 (720 psi), guaiacol was converted (76%) to unidentifiable products and evident darkening of the reaction solution was observed. This leads us to the conclusion that polymerization of phenols in the presence of oxygen was responsible for their low yields.
While models
4 and
5 serve as sufficiently complex models to establish reactivity trends, they do not accurately represent lignin linkages. Consequently, to better reflect native and technical lignins, model complexity was increased by the addition of a
γ-carbinol group (compound
6;
Table 11). The addition of a
γ-carbinol provides another alcohol that can be oxidized. Once oxidized at the
α or
γ-position,
6 can undergo retro-aldol reactions further complicating the product mixture. Indeed, a retro-aldol reaction at the oxidized γ-position produces
4 via loss of formaldehyde, while oxidation at the α-position produces a ketone that can also undergo further oxidation.
The oxidative fragmentation of model
6 was investigated using Ni-Al-LDH-1, Cu-Cr-LDH, and Ni-Cr-LDH. Unexpectedly, Ni-Al-LDH-1 catalyzed oxidation resulted in only 34% conversion (
Table 11), whereas Cu-Cr-LDH afforded similar conversion as for models
4 and
5 (80–90%). In contrast, Ni-Cr-LDH, which showed similar reactivity trends for models
4 and
5, showed relatively lower conversion for
6 (23%). The benzoic acid (
4a) resulting from cleavage of the C
α–C
β bond was observed in low yield (<7%) for all three catalysts. Furthermore, compounds similar to those produced after the oxidation of
4 and
5 (e.g.,
6c) were not observed when
6 was subjected to the same reaction conditions.
The difference in the reactivity of 6 compared to 4 and 5 may have been due to diverging reaction pathways. It is hypothesized that reaction intermediates included both benzylic ketones formed via oxidation and enol ethers via dehydration. Basic sites were likely responsible for the deprotonation of the benzylic carbinol for catalysts such as Ni-Al-LDH-1. After the substrate was coordinated to the catalyst surface, base sites likely deprotonated the β-carbon. Thus, it stands to reason that substitution at the β-carbon by a γ-carbinol likely made deprotonation of the β-carbon more difficult, as suggested by the absence of 6c in the product mixture.






{kind=link}
{kind=link}
{kind=link}
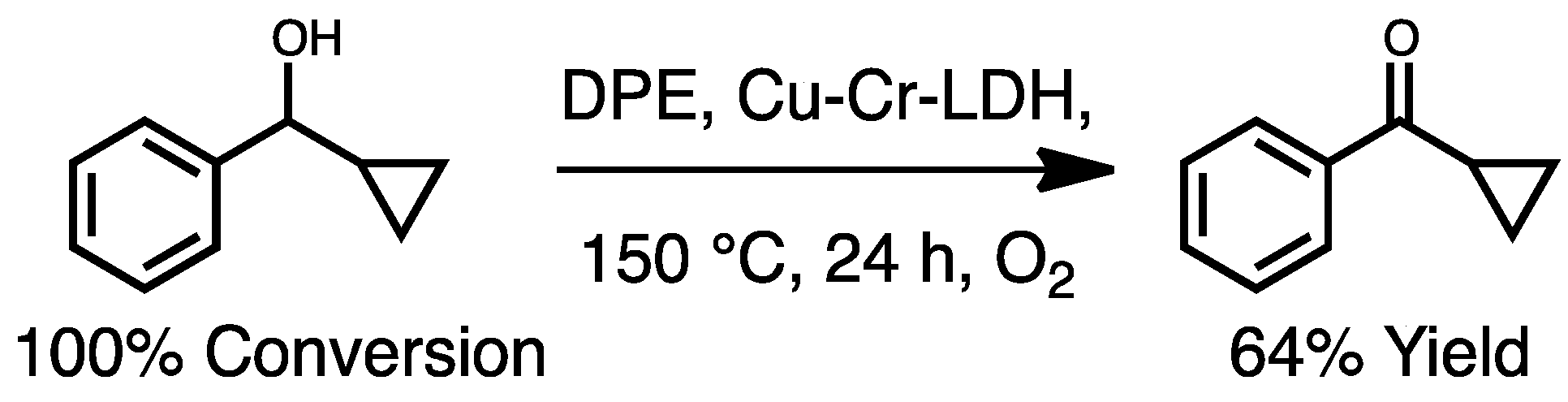{kind=link}







