(2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry


Abstract
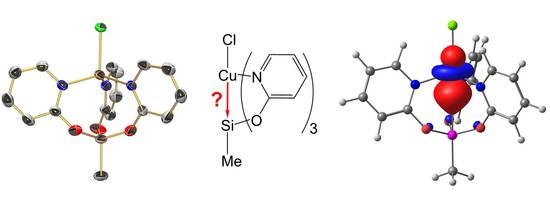
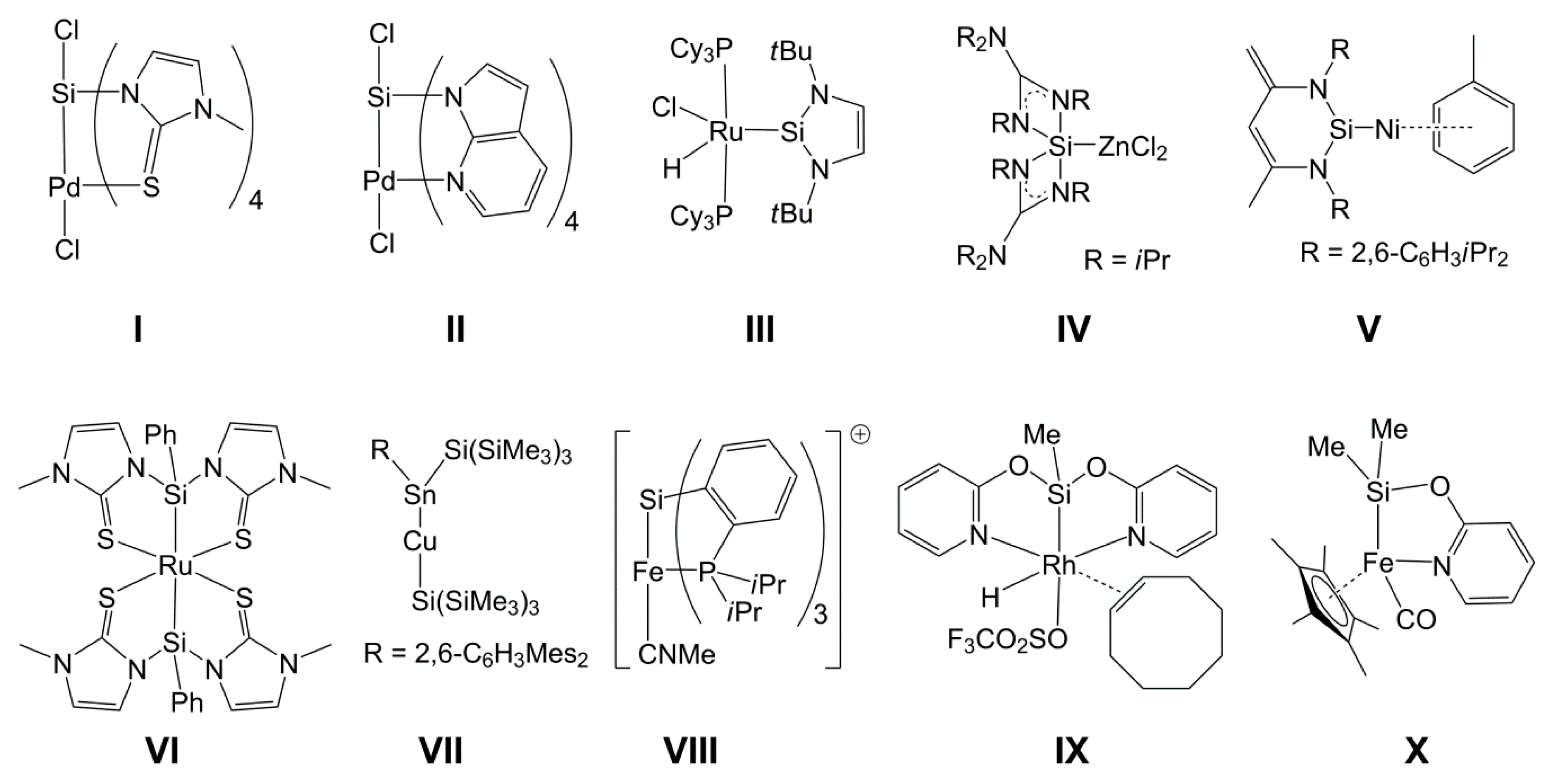
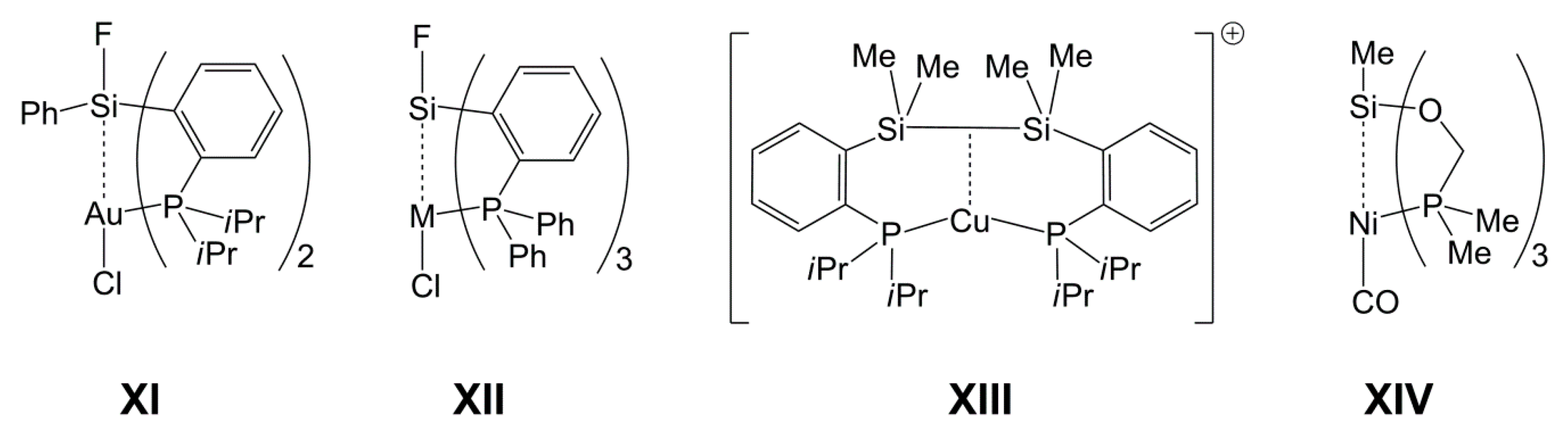
1. Introduction
2. Results and Discussion
2.1. Syntheses and Characterization of Silanes MeSi(pyO)3 and Si(pyO)4
2.2. Choice of Metals: Pd(II) and Cu(I)
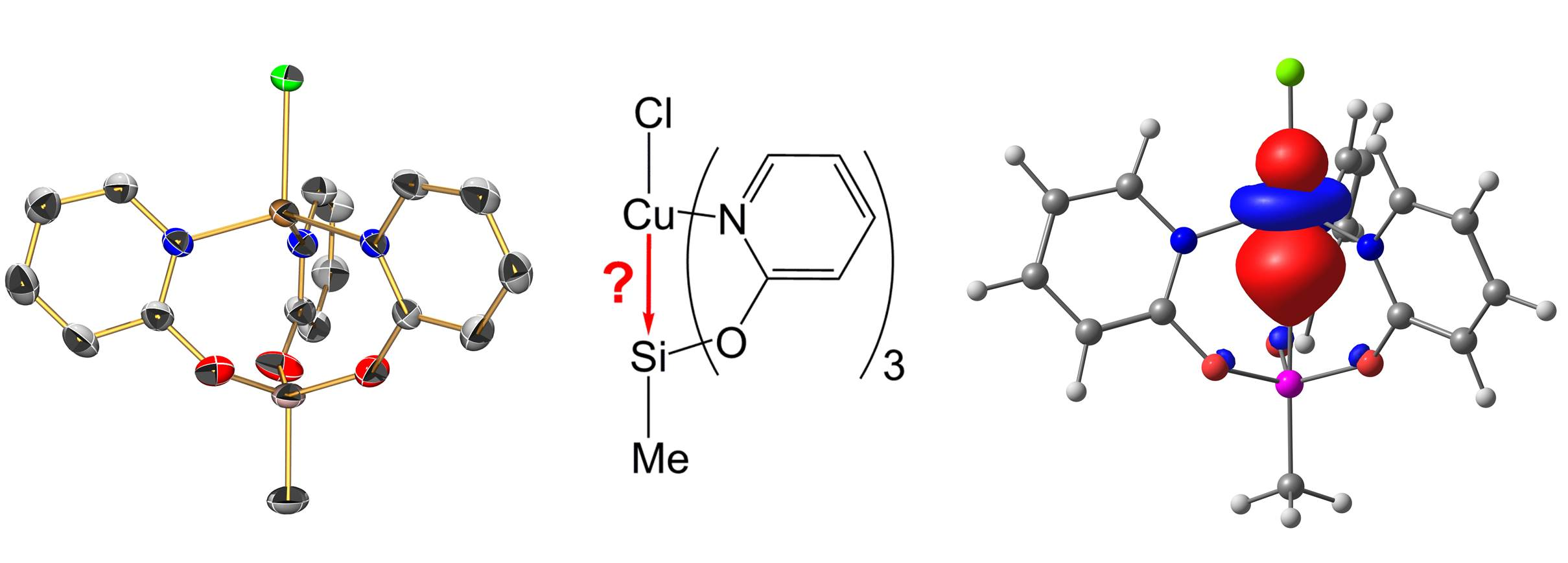
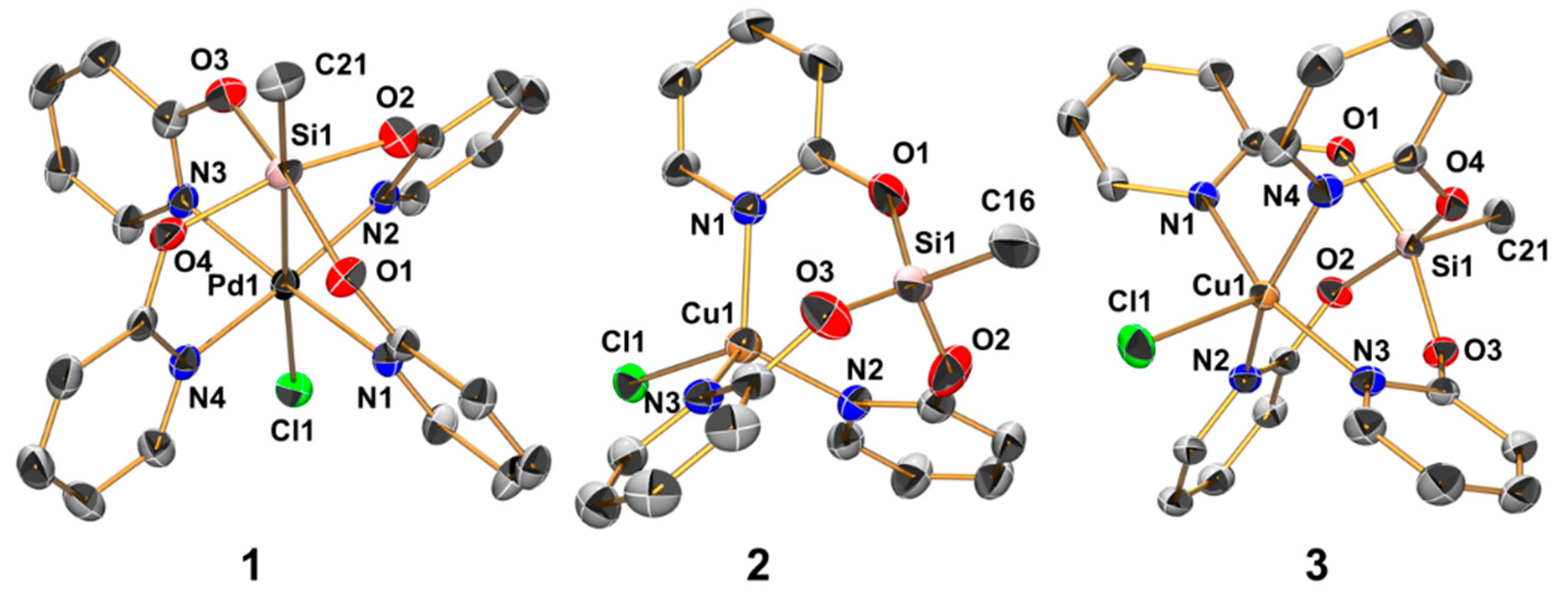
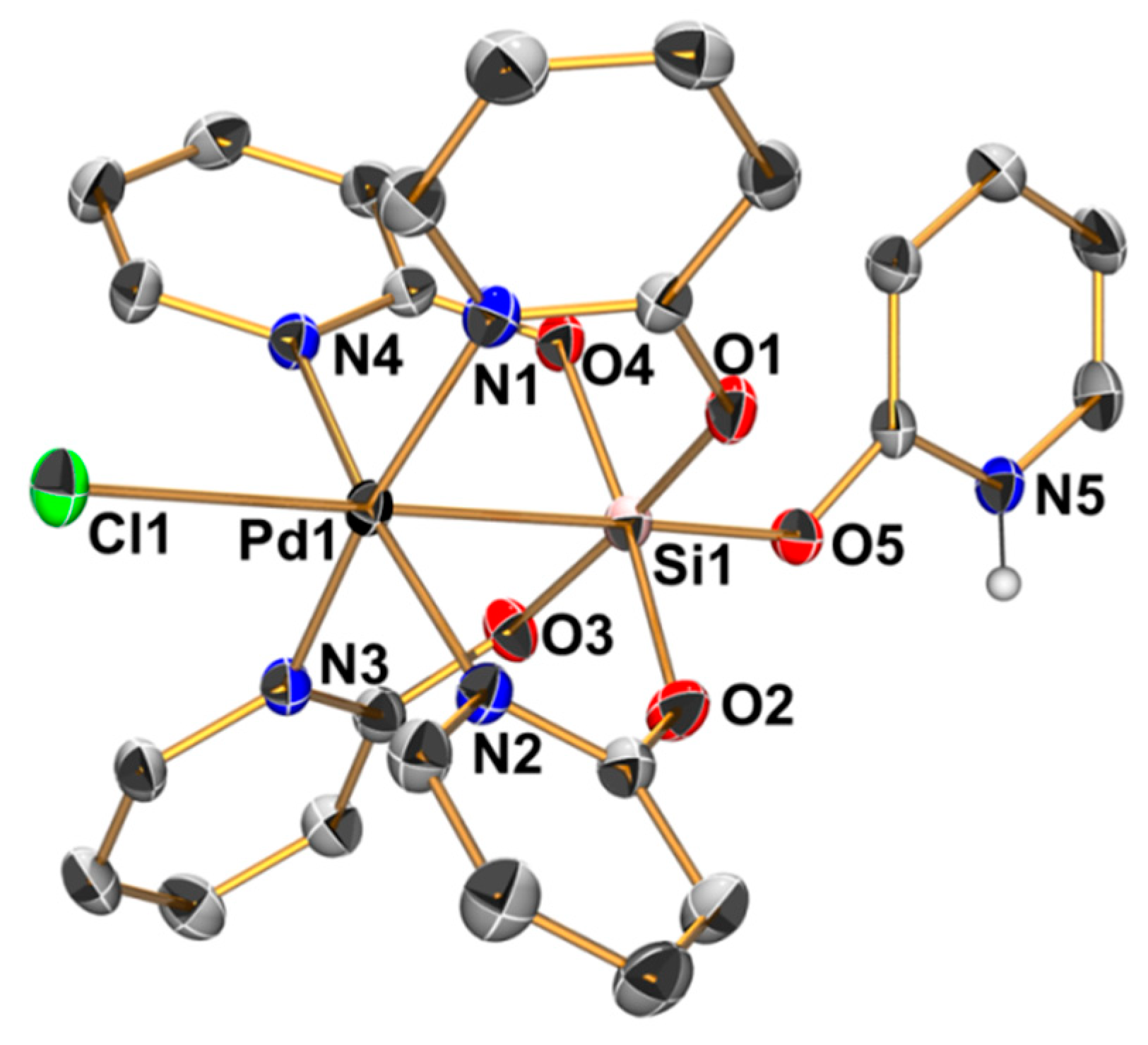
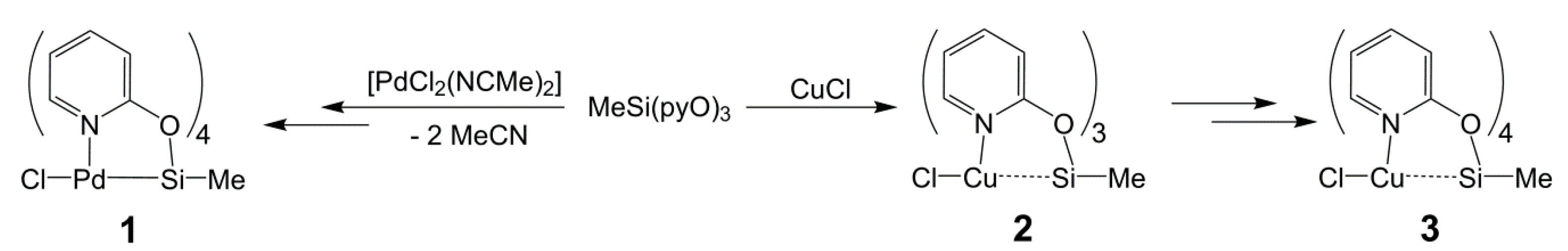
2.3. Reactions of MeSi(pyO)3 with [PdCl2(NCMe)2] and CuCl
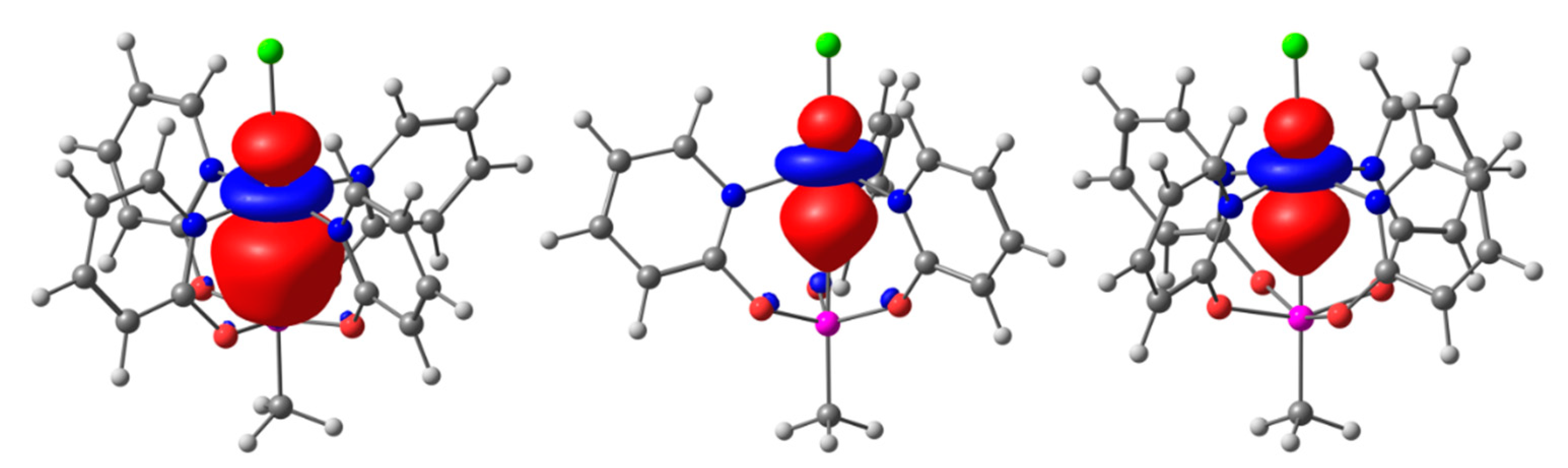
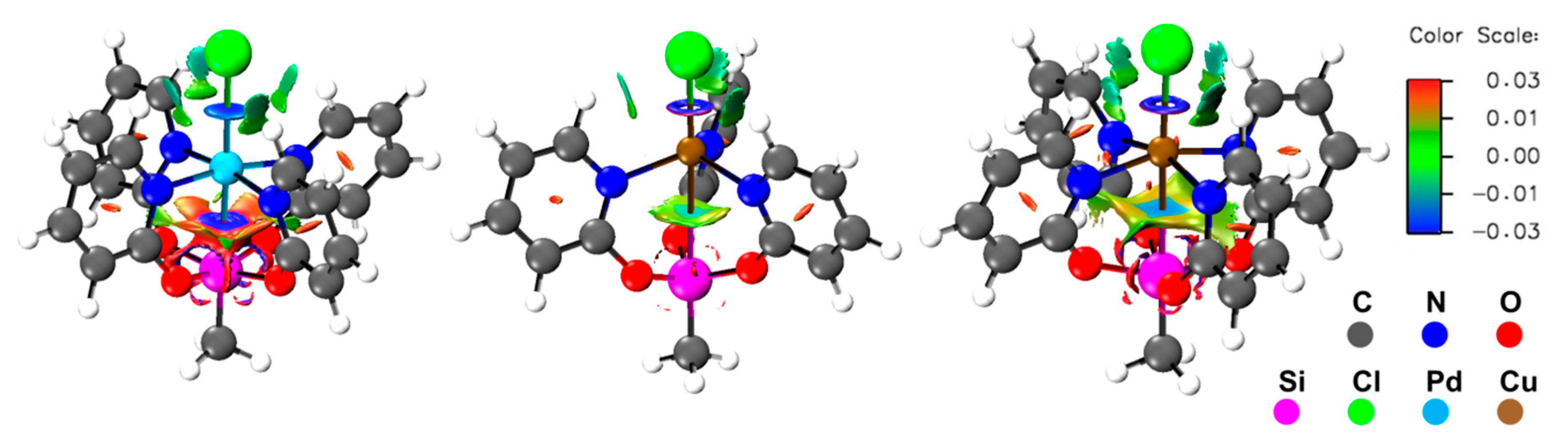
2.4. Computational Analyses of the Pd→Si and Cu→Si Interactions in Compounds 1, 2, and 3
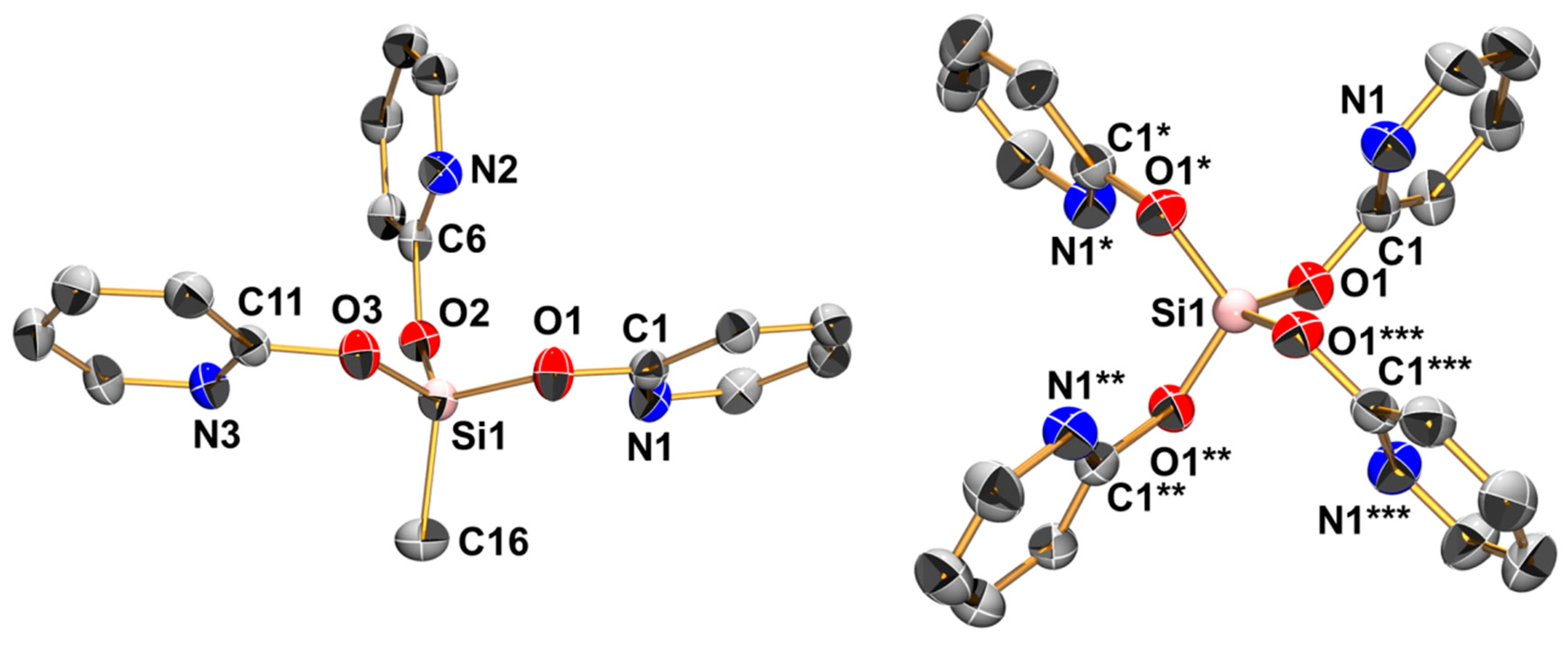
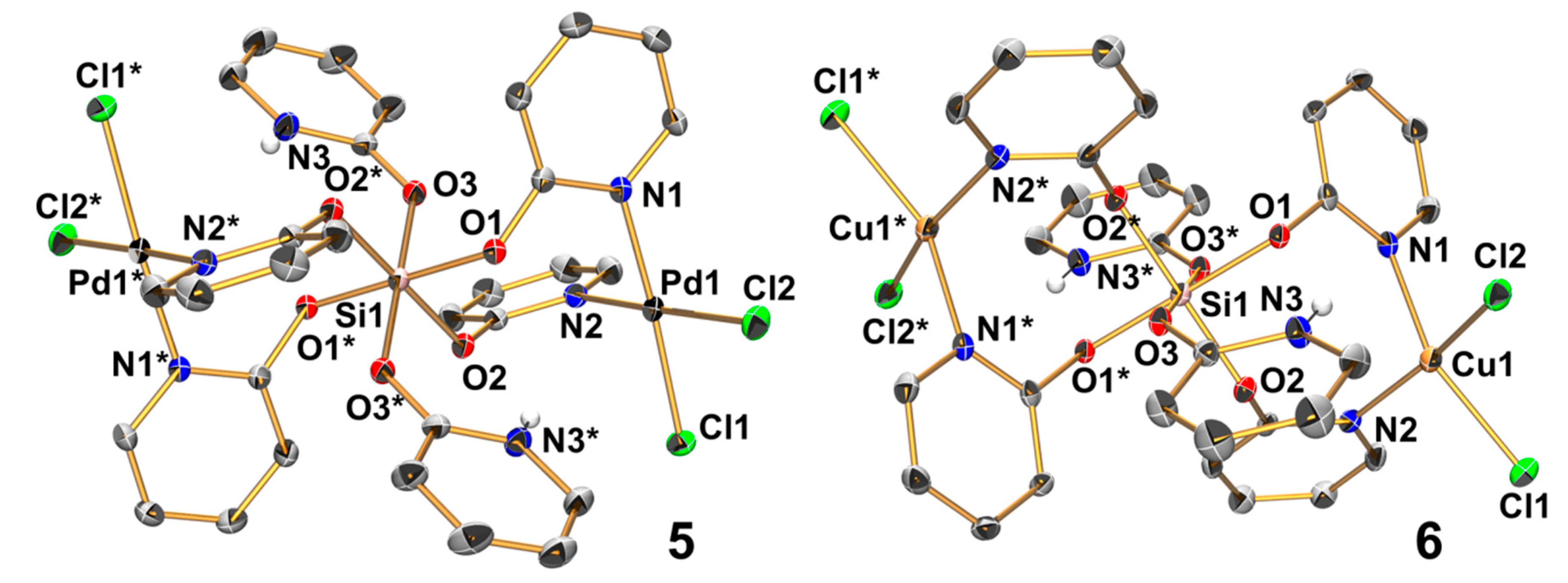
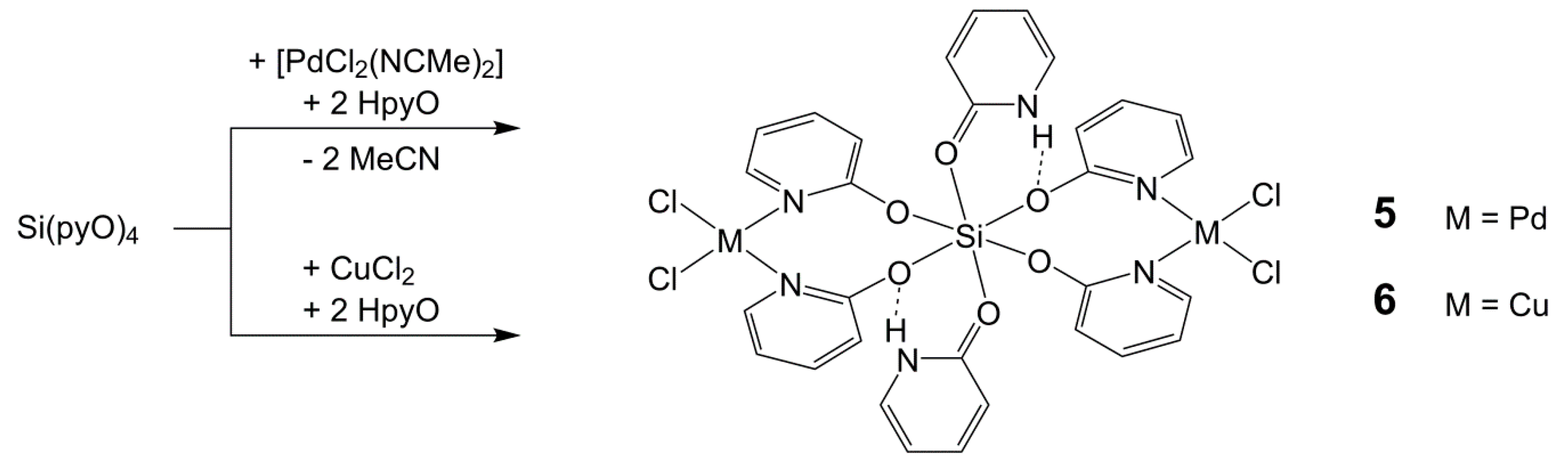
2.5. Reactions of Si(pyO)4 with [PdCl2(NCMe)2] and CuCl2
3. Experimental Section
3.1. General Considerations
3.2. Syntheses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tacke, R.; Ribbeck, T. Bis(amidinato)- and bis(guanidinato) silylenes and silylenes with one sterically demanding amidinato or guanidinato ligand: Synthesis and reactivity. Dalton Trans. 2017, 46, 13628–13659. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, D.M.; Schmedake, T.A. Recent advances in hexacoordinate silicon with pyridine-containing ligands: Chemistry and emerging applications. Coord. Chem. Rev. 2016, 323, 107–119. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Kroke, E. Higher-Coordinated Molecular Silicon Compounds. In Structure and Bonding; Scheschkewitz, D., Ed.; Springer: Berlin, Germany, 2014; Volume 155, pp. 29–105. [Google Scholar]
- Levason, W.; Reid, G.; Zhang, W. Coordination complexes of silicon and germanium halides with neutral ligands. Coord. Chem. Rev. 2011, 255, 1319–1341. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Metallasilatranes: Palladium(II) and Platinum(II) as Lone-Pair Donors to Silicon(IV). Angew. Chem. Int. Ed. 2010, 49, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Truflandier, L.A.; Brendler, E.; Wagler, J.; Autschbach, J. 29Si DFT/NMR Observation of Spin-Orbit Effect in Metallasilatrane Sheds Some Light on the Strength of the Metal → Si Interaction. Angew. Chem. Int. Ed. 2011, 50, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Autschbach, J.; Sutter, K.; Truflandier, L.A.; Brendler, E.; Wagler, J. Atomic Contributions from Spin-Orbit Coupling to 29Si NMR Chemical Shifts in Metallasilatrane Complexes. Chem. Eur. J. 2012, 18, 12803–12813. [Google Scholar] [CrossRef] [PubMed]
- Wahlicht, S.; Brendler, E.; Heine, T.; Zhechkov, L.; Wagler, J. 7-Azaindol-1-yl(organo)silanes and Their PdCl2 Complexes: Pd-Capped Tetrahedral Silicon Coordination Spheres and Paddlewheels with a Pd-Si Axis. Organometallics 2014, 33, 2479–2488. [Google Scholar] [CrossRef]
- Cade, I.A.; Hill, A.F.; Kämpfe, A.; Wagler, J. Five-Coordinate Hydrido-Ruthenium(II) Complexes Featuring N-Heterocyclic Silylene and Carbene Ligands. Organometallics 2010, 29, 4012–4017. [Google Scholar] [CrossRef]
- Baus, J.A.; Mück, F.M.; Schneider, H.; Tacke, R. Iron(II), Cobalt(II), Nickel(II), and Zinc(II) Silylene Complexes: Reaction of the Silylene [iPrNC(NiPr2)NiPr]2Si with FeBr2, CoBr2, NiBr2·MeOCH2CH2OMe, ZnCl2, and ZnBr2. Chem. Eur. J. 2017, 23, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Junold, K.; Baus, J.A.; Burschka, C.; Vent-Schmidt, T.; Riedel, S.; Tacke, R. Five-Coordinate Silicon(II) Compounds with Si−M Bonds (M = Cr, Mo, W, Fe): Bis[N,N′-diisopropylbenzamidinato(−)]silicon(II) as a Ligand in Transition-Metal Complexes. Inorg. Chem. 2013, 52, 11593–11599. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Köppe, R.; Roesky, P.W. Investigations of the Nature of ZnII–SiII Bonds. Chem. Eur. J. 2016, 22, 7127–7133. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, A.; Präsang, C.; Milsmann, C.; Driess, M. The Striking Stabilization of Ni0(η6-Arene) Complexes by an Ylide-Like Silylene Ligand. Angew. Chem. Int. Ed. 2009, 48, 3170–3173. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Neumann, H.; Wagler, J. Bis(methimazolyl)silyl Complexes of Ruthenium. Organometallics 2010, 29, 1026–1031. [Google Scholar] [CrossRef]
- Klett, J.; Klinkhammer, K.W.; Niemeyer, M. Ligand Exchange between Arylcopper Compounds and Bis(hypersilyl)tin or Bis(hypersilyl)lead: Synthesis and Characterization of Hypersilylcopper and a Stannanediyl Complex with a Cu–Sn Bond. Chem. Eur. J. 2009, 5, 2531–2536. [Google Scholar] [CrossRef]
- Rittle, J.; Peters, J.C. N−H Bond Dissociation Enthalpies and Facile H Atom Transfers for Early Intermediates of Fe−N2 and Fe−CN Reductions. J. Am. Chem. Soc. 2017, 139, 3161–3170. [Google Scholar] [CrossRef] [PubMed]
- Suess, D.L.M.; Tsay, C.; Peters, J.C. Dihydrogen Binding to Isostructural S = 1/2 and S = 0 Cobalt Complexes. J. Am. Chem. Soc. 2012, 134, 14158–14164. [Google Scholar] [CrossRef] [PubMed]
- Garcés, K.; Lalrempuia, R.; Polo, V.; Fernández-Alvarez, F.J.; García-Orduña, P.; Lahoz, F.J.; Pérez-Torrente, J.J.; Oro, L.A. Rhodium-Catalyzed Dehydrogenative Silylation of Acetophenone Derivatives: Formation of Silyl Enol Ethers versus Silyl Ethers. Chem. Eur. J. 2016, 22, 14717–14729. [Google Scholar] [CrossRef] [PubMed]
- Lalrempuia, R.; Iglesias, M.; Polo, V.; Sanz Miguel, P.J.; Fernández-Alvarez, F.J.; Pérez-Torrente, J.J.; Oro, L.A. Effective Fixation of CO2 by Iridium-Catalyzed Hydrosilylation. Angew. Chem. Int. Ed. 2012, 51, 12824–12827. [Google Scholar] [CrossRef] [PubMed]
- Julián, A.; Jaseer, E.A.; Garcés, K.; Fernández-Alvarez, F.J.; García-Orduña, P.; Lahoz, F.J.; Oro, L.A. Tuning the activity and selectivity of iridium-NSiN catalyzed CO2 hydrosilylation processes. Catal. Sci. Technol. 2016, 6, 4410–4417. [Google Scholar] [CrossRef]
- Julián, A.; Guzmán, J.; Jaseer, E.A.; Fernández-Alvarez, F.J.; Royo, R.; Polo, V.; García-Orduña, P.; Lahoz, F.J.; Oro, L.A. Mechanistic Insights on the Reduction of CO2 to Silylformates Catalyzed by Ir-NSiN Species. Chem. Eur. J. 2017, 23, 11898–11907. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ou, C.; Wang, C.; Uchiyama, M.; Deng, L. Silane-Functionalized N-Heterocyclic Carbene−Cobalt Complexes Containing a Five-Coordinate Silicon with a Covalent Co−Si Bond. Organometallics 2015, 34, 1546–1551. [Google Scholar] [CrossRef]
- Sato, T.; Okazaki, M.; Tobita, H.; Ogino, H. Synthesis, structure, and reactivity of novel iron(II) complexes with a five-membered chelate ligand κ2(Si,N)-SiMe2O(2-C5H4N). J. Organomet. Chem. 2003, 669, 189–199. [Google Scholar] [CrossRef]
- Kwok, W.-H.; Lu, G.-L.; Rickard, C.E.F.; Roper, W.R.; Wright, L.J. Tethered silyl complexes from nucleophilic substitution reactions at the Si–Cl bond of the chloro(diphenyl)silyl ligand in Ru(SiClPh2)(κ2-S2CNMe2)(CO)(PPh3)2. J. Organomet. Chem. 2004, 689, 2979–2987. [Google Scholar] [CrossRef]
- Kanno, Y.; Komuro, T.; Tobita, H. Direct Conversion of a Si−C(aryl) Bond to Si−Heteroatom Bonds in the Reactions of η3-α-Silabenzyl Molybdenum and Tungsten Complexes with 2-Substituted Pyridines. Organometallics 2015, 34, 3699–3705. [Google Scholar] [CrossRef]
- Motherwell, W.B.; Storey, L.J. Some studies on nucleophilic trifluoromethylation using the shelf-stable trifluoromethylacetophenone-N,N-dimethyltrimethylsilylamine adduct. J. Fluor. Chem. 2005, 126, 491–498. [Google Scholar] [CrossRef]
- Wächtler, E.; Gericke, R.; Kutter, S.; Brendler, E.; Wagler, J. Molecular structures of pyridinethiolato complexes of Sn(II), Sn(IV), Ge(IV), and Si(IV). Main Group Met. Chem. 2013, 36, 181–191. [Google Scholar] [CrossRef]
- Baus, J.A.; Burschka, C.; Bertermann, R.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Tacke, R. Neutral Six-Coordinate and Cationic Five-Coordinate Silicon(IV) Complexes with Two Bidentate Monoanionic N,S-Pyridine-2-thiolato(-) Ligands. Inorg. Chem. 2013, 52, 10664–10676. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.G.; Brennessel, W.W.; Kraft, B.M. Neutral and Cationic Bis-Chelate Monoorganosilicon(IV) Complexes of 1-Hydroxy-2-pyridinone. Organometallics 2017, 36, 594–604. [Google Scholar] [CrossRef]
- Kraft, B.M.; Brennessel, W.W. Chelation and Stereodynamic Equilibria in Neutral Hypercoordinate Organosilicon Complexes of 1-Hydroxy-2-pyridinone. Organometallics 2014, 33, 158–171. [Google Scholar] [CrossRef]
- Schraml, J.; Chvalovsky, V.; Magi, M.; Lippmaa, E. NMR study of organosilicon compounds. XI. The role of electronic and steric effects in silicon-29 NMR spectra of compounds with a silicon-oxygen-carbon group. Collect. Czechoslov. Chem. Commun. 1981, 46, 377–390. [Google Scholar] [CrossRef]
- Schraml, J.; Brezny, R.; Cermak, J.; Chvalovsky, V. Silicon-29 and carbon-13 NMR spectra of some substituted bis(trimethylsiloxy)benzenes. Collect. Czechoslov. Chem. Commun. 1990, 55, 2033–2037. [Google Scholar] [CrossRef]
- Wagler, J.; Heine, T.; Hill, A.F. Poly(methimazolyl)silanes: Syntheses and Molecular Structures. Organometallics 2010, 29, 5607–5613. [Google Scholar] [CrossRef]
- Gualco, P.; Mallet-Ladeira, S.; Kameo, H.; Nakazawa, H.; Mercy, M.; Maron, L.; Amgoune, A.; Bourissou, D. Coordination of a Triphosphine-Silane to Gold: Formation of a Trigonal Pyramidal Complex Featuring Au→Si Interaction. Organometallics 2015, 34, 1449–1453. [Google Scholar] [CrossRef]
- Gualco, P.; Mercy, M.; Ladeira, S.; Coppel, Y.; Maron, L.; Amgoune, A.; Bourissou, D. Hypervalent Silicon Compounds by Coordination of Diphosphine-Silanes to Gold. Chem. Eur. J. 2010, 16, 10808–10817. [Google Scholar] [CrossRef] [PubMed]
- Gualco, P.; Lin, T.-P.; Sircoglou, M.; Mercy, M.; Ladeira, S.; Bouhadir, G.; Pérez, L.M.; Amgoune, A.; Maron, L.; Gabbaï, F.P.; Bourissou, D. Gold–Silane and Gold–Stannane Complexes: Saturated Molecules as σ-Acceptor Ligands. Angew. Chem. Int. Ed. 2009, 48, 9892–9895. [Google Scholar] [CrossRef] [PubMed]
- Kameo, H.; Kawamoto, T.; Bourissou, D.; Sakaki, S.; Nakazawa, H. Evaluation of the σ-Donation from Group 11 Metals (Cu, Ag, Au) to Silane, Germane, and Stannane Based on the Experimental/Theoretical Systematic Approach. Organometallics 2015, 34, 1440–1448. [Google Scholar] [CrossRef]
- Gualco, P.; Amgoune, A.; Miqueu, K.; Ladeira, S.; Bourissou, D. A Crystalline σ Complex of Copper. J. Am. Chem. Soc. 2011, 133, 4257–4259. [Google Scholar] [CrossRef] [PubMed]
- Grobe, J.; Lütke-Brochtrup, K.; Krebs, B.; Läge, M.; Niemeyer, H.-H.; Würthwein, E.-U. Alternativ-Liganden XXXVIII. Neue Versuche zur Synthese von Pd(0)- und Pt(0)-Komplexen des Tripod-Phosphanliganden FSi(CH2CH2PMe2)3. Z. Naturforsch. 2007, 62, 55–65. [Google Scholar] [CrossRef]
- Grobe, J.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXII Neue Tetraphosphan-Nickelkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3−n (M’ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1995, 621, 583–596. [Google Scholar] [CrossRef]
- Grobe, J.; Krummen, N.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXI Nickelcarbonylkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3–n (M’ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1994, 620, 1645–1658. [Google Scholar] [CrossRef]
- Wagler, J.; Roewer, G.; Gerlach, D. Photo-Driven Si–C Bond Cleavage in Hexacoordinate Silicon Complexes. Z. Anorg. Allg. Chem. 2009, 635, 1279–1287. [Google Scholar] [CrossRef]
- Gerlach, D.; Brendler, E.; Wagler, J. Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N,N′)-Chelating Ligand. Inorganics 2016, 4, 8. [Google Scholar] [CrossRef]
- Wächtler, E.; Kämpfe, A.; Krupinski, K.; Gerlach, D.; Kroke, E.; Brendler, E.; Wagler, J. New Insights into Hexacoordinated Silicon Complexes with 8-Oxyquinolinato Ligands: 1,3-Shift of Si-Bound Hydrocarbyl Substituents and the Influence of Si-Bound Halides on the 8-Oxyquinolinate Coordination Features. Z. Naturforsch. 2014, 69, 1402–1418. [Google Scholar] [CrossRef]
- Brendler, E.; Wächtler, E.; Wagler, J. Hypercoordinate Silacycloalkanes: Step-by-Step Tuning of N→Si Interactions. Organometallics 2009, 28, 5459–5465. [Google Scholar] [CrossRef]
- Dinda, S.; Samuelson, A.G. The Nature of Bond Critical Points in Dinuclear Copper(I) Complexes. Chem. Eur. J. 2012, 18, 3032–3042. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl cation and to Bicyclobutane. Tetrahedron 1967, 24, 1083–1096. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental Electron Density Analysis of Mn2(CO)10: Metal-Metal and Metal-Ligand Bond Characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, R.; Gervasio, G.; Marabello, D. The experimental charge density in transition metal compounds. C. R. Chim. 2005, 8, 1392–1399. [Google Scholar] [CrossRef]
- Macchi, P.; Proserpio, D.M.; Sironi, A. Experimental Electron Density in a Transition Metal Dimer: Metal-Metal and Metal-Ligand Bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Lepetit, C.; Fau, P.; Fajerwerg, K.; Kahn, M.L.; Silvi, B. Topological analysis of the metal-metal bond: A tutorial review. Coord. Chem. Rev. 2017, 345, 150–181. [Google Scholar] [CrossRef]
- Seiler, O.; Burschka, C.; Fenske, T.; Troegel, D.; Tacke, R. Neutral Hexa- and Pentacoordinate Silicon(IV) Complexes with SiO6 and SiO4N Skeletons. Inorg. Chem. 2007, 46, 5419–5424. [Google Scholar] [CrossRef] [PubMed]
- Kämpfe, A.; Brendler, E.; Kroke, E.; Wagler, J. Tp*Cu(I)–CN–SiL2–NC–Cu(I)Tp*—A hexacoordinate Si-complex as connector for redox active metals via π-conjugated ligands. Dalton Trans. 2015, 44, 4744–4750. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Program for the Solution of Crystal Structures; shelxs-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; shelxl-2014/7; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- POV-RAY (Version 3.6), Trademark of Persistence of Vision Raytracer Pty. Ltd., Williamstown, Victoria (Australia). Copyright Hallam Oaks Pty. Ltd., 1994–2004. Available online: http://www.povray.org/download/ (accessed on 22 December 2011).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scurseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, A.; et al. Gaussian09; revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013; Available online: http://nbo6.chem.wisc.edu/ (accessed on 16 August 2016).
- Chemcraft ver. 1.8 (Build 164). 2016. Available online: http://www.chemcraftprog.com (accessed on 8 April 2016).
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K.J. VMD: visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 5 April 2018). [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
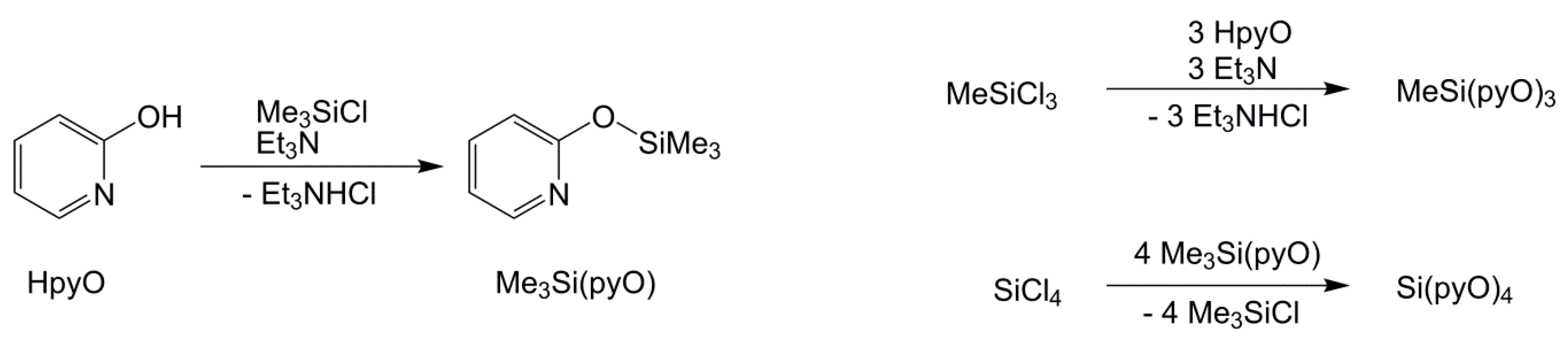{kind=link}
{kind=link}
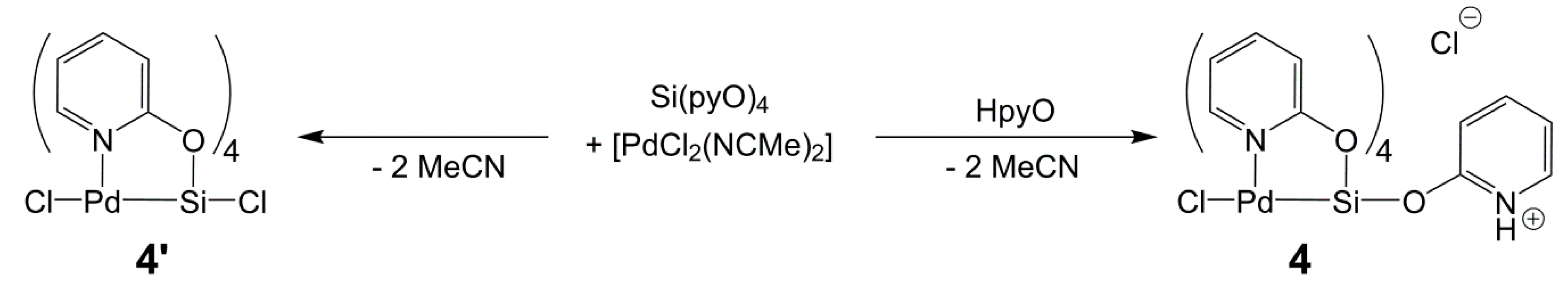{kind=link}
{kind=link}
| Parameter | MeSi(pyO)3 | Si(pyO)4 | 1 · 2 CHCl3 | 2 |
|---|---|---|---|---|
| Formula | C16H15N3O3Si | C20H16N4O4Si | C23H21Cl7N4O4PdSi | C16H15ClCuN3O3Si |
| Mr | 325.40 | 404.46 | 800.08 | 424.39 |
| T(K) | 200(2) | 200(2) | 180(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | triclinic | tetragonal | monoclinic | triclinic |
| Space group | P-1 | I41/a | C2/c | P-1 |
| a(Å) | 9.1581(7) | 9.5163(7) | 14.8719(5) | 8.7497(4) |
| b(Å) | 9.3250(7) | 9.5163(7) | 10.5112(5) | 9.2334(5) |
| c(Å) | 11.4078(9) | 21.824(2) | 39.3020(13) | 23.5781(13) |
| α(°) | 92.440(6) | 90 | 90 | 88.255(4) |
| β(°) | 109.582(6) | 90 | 95.404(3) | 89.283(4) |
| γ(°) | 116.896(6) | 90 | 90 | 68.654(4) |
| V(Å3) | 796.41(12) | 1976.4(3) | 6116.4(4) | 1773.36(17) |
| Z | 2 | 4 | 8 | 4 |
| ρcalc(g·cm−1) | 1.36 | 1.36 | 1.74 | 1.59 |
| µMo Kα (mm−1) | 0.2 | 0.2 | 1.3 | 1.5 |
| F(000) | 340 | 840 | 3184 | 864 |
| θmax(°), Rint | 28.0, 0.0263 | 25.0, 0.0238 | 25.0, 0.0355 | 28.0, 0.0310 |
| Completeness | 99.9% | 99.8% | 99.9% | 99.8% |
| Reflections collected | 12193 | 3724 | 52652 | 28693 |
| Reflns unique | 3836 | 873 | 5379 | 8555 |
| Restraints | 0 | 0 | 18 | 0 |
| Parameters | 209 | 66 | 403 | 453 |
| GoF | 1.066 | 1.137 | 1.147 | 1.073 |
| R1, wR2 [I > 2σ(I)] | 0.0343, 0.0871 | 0.0404, 0.0934 | 0.0339, 0.0765 | 0.0350, 0.0858 |
| R1, wR2 (all data) | 0.0426, 0.0925 | 0.0567, 0.1062 | 0.0423, 0.0818 | 0.0444, 0.0900 |
| Largest peak/hole (e·Å−3) | 0.22, −0.31 | 0.16, −0.28 | 0.67, −0.65 | 0.77, −0.28 |
| Parameter | 3 | 3 · CHCl3 | 4 · 4 CHCl3 |
|---|---|---|---|
| Formula | C21H19ClCuN4O4Si | C22H20Cl4CuN4O4Si | C29H25Cl14N5O5PdSi |
| Mr | 518.48 | 637.85 | 1154.33 |
| T(K) | 200(2) | 200(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | tetragonal | monoclinic | triclinic |
| Space group | I41/a | P21 | P-1 |
| a(Å) | 18.3774(5) | 9.2616(4) | 11.2079(5) |
| b(Å) | 18.3774(5) | 15.6201(7) | 13.1431(6) |
| c(Å) | 26.4847(8) | 9.2773(5) | 15.1242(7) |
| α(°) | 90 | 90 | 78.113(4) |
| β(°) | 90 | 93.049(4) | 87.403(4) |
| γ(°) | 90 | 90 | 89.899(4) |
| V(Å3) | 8944.6(6) | 1340.22(11) | 2177.81(17) |
| Z | 16 | 2 | 2 |
| ρcalc(g·cm−1) | 1.54 | 1.58 | 1.76 |
| µMo Kα (mm−1) | 1.2 | 1.3 | 1.4 |
| F(000) | 4240 | 646 | 1144 |
| θmax(°), Rint | 28.0, 0.0425 | 28.0, 0.0302 | 27.0, 0.0271 |
| Completeness | 99.9% | 99.9% | 99.9% |
| Reflns collected | 71,378 | 22,760 | 34,910 |
| Reflns unique | 5409 | 6449 | 9502 |
| Restraints | 0 | 1 | 12 |
| Parameters | 290 | 326 | 562 |
| GoF | 1.081 | 1.060 | 1.059 |
| χFlack | −0.008(4) | ||
| R1, wR2 [I > 2σ(I)] | 0.0278, 0.0679 | 0.0269, 0.0624 | 0.0230, 0.0557 |
| R1, wR2 (all data) | 0.0355, 0.0712 | 0.0302, 0.0639 | 0.0275, 0.0577 |
| Largest peak/hole (e·Å−3) | 0.45, −0.25 | 0.44, −0.31 | 0.52, −0.48 |
| Feature | 1 | 2 | 3 1 |
|---|---|---|---|
| % contribution TM | 90.7 | 97.9 | 98.6 |
| Hybrid (TM) | 97.6% 4d, 2.2% 5s | 99.6% 3d | 99.5% 3d |
| % contribution Si | 8.3 | 0.8 | 0.8 |
| Hybrid (Si) | 37.7% 3s, 61.7% 3p | 18.2% 3s, 79.5% 3p | 18.0% 3s, 79.7% 3p |
| Feature 1 | 1 | 2 | 3 |
|---|---|---|---|
| ρ(rb) | 0.04461 | 0.01229 | 0.01742 |
| ∇2ρ(rb) | −0.00127 | 0.02835 | 0.02896 |
| G(rb) | 0.01721 | 0.00721 | 0.00958 |
| V(rb) | −0.03475 | −0.00734 | −0.01192 |
| |V(rb)|/G(rb) | 2.018 | 1.017 | 1.244 |
| G(rb)/ρ(rb) | 0.386 | 0.587 | 0.550 |
| H(rb) | −0.01753 | −0.00012 | −0.00234 |
| H(rb)/ρ(rb) | −0.393 | −0.010 | −0.134 |
| Eint | −10.9 | −2.3 | −3.7 |
| WBO | 0.270 | 0.057 | 0.037 |
| Parameter | 5 · 2 CHCl3 | 5 · 6 CHCl3 | 5 · 8 CHCl3 | 6 |
|---|---|---|---|---|
| Formula | C32H28Cl10N6O6Pd2Si | C36H32Cl22N6O6Pd2Si | C38H34Cl28N6O6Pd2Si | C30H26Cl4Cu2N6O6Si |
| Mr | 1187.99 | 1665.46 | 1904.20 | 863.54 |
| T(K) | 200(2) | 200(2) | 200(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic |
| Space group | C2/c | P-1 | P21/n | P21/n |
| a(Å) | 22.458(2) | 12.1698(6) | 12.3681(5) | 9.1063(8) |
| b(Å) | 11.8102(7) | 12.1951(7) | 13.1356(4) | 11.0475(6) |
| c(Å) | 17.4818(18) | 13.1643(8) | 22.2337(9) | 17.1744(17) |
| α(°) | 90 | 107.904(4) | 90 | 90 |
| β(°) | 111.330(7) | 103.147(4) | 95.990(3) | 104.705(7) |
| γ(°) | 90 | 114.671(4) | 90 | 90 |
| V(Å3) | 4319.1(7) | 1539.81(19) | 3592.4(2) | 1671.2(2) |
| Z | 4 | 1 | 2 | 2 |
| ρcalc(g·cm−1) | 1.83 | 1.80 | 1.76 | 1.72 |
| µMo Kα (mm−1) | 1.5 | 1.6 | 1.6 | 1.7 |
| F(000) | 2344 | 818 | 1868 | 872 |
| θmax(°), Rint | 25.0, / | 27.0, 0.0264 | 27.0, 0.0396 | 25.0, 0.0983 |
| Completeness | 99.8% | 99.9% | 99.9% | 99.7% |
| Reflns collected | 16169 | 18872 | 46707 | 16933 |
| Reflns unique | 3789 | 6728 | 7828 | 2933 |
| Restraints | 9 | 66 | 166 | 0 |
| Parameters | 289 | 410 | 539 | 226 |
| GoF | 1.034 | 1.055 | 1.033 | 0.924 |
| R1, wR2 [I > 2σ(I)] | 0.0474, 0.1009 | 0.0331, 0.0720 | 0.0299, 0.0663 | 0.0441, 0.0776 |
| R1, wR2 (all data) | 0.0958, 0.1166 | 0.0448, 0.0776 | 0.0399, 0.0699 | 0.1057, 0.0907 |
| Largest peak/hole (e·Å−3) | 0.76, −0.85 | 0.56, −0.58 | 0.46, −0.35 | 0.46, −0.72 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics 2018, 6, 119. https://doi.org/10.3390/inorganics6040119
Ehrlich L, Gericke R, Brendler E, Wagler J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics. 2018; 6(4):119. https://doi.org/10.3390/inorganics6040119
Chicago/Turabian StyleEhrlich, Lisa, Robert Gericke, Erica Brendler, and Jörg Wagler. 2018. "(2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry" Inorganics 6, no. 4: 119. https://doi.org/10.3390/inorganics6040119
APA StyleEhrlich, L., Gericke, R., Brendler, E., & Wagler, J. (2018). (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics, 6(4), 119. https://doi.org/10.3390/inorganics6040119