A Computational Study of AlF3 and ACF Surfaces

Abstract
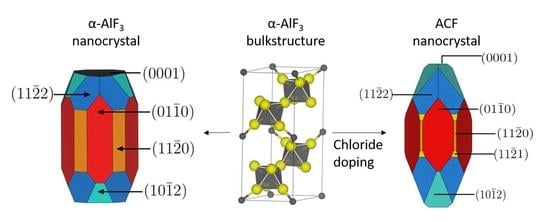
:
1. Introduction
2. Methodology and Models
2.1. Calculation of Surface Energy
2.2. Slab Construction
2.3. Computational Details
2.4. Effect of DFT-D3 Correction
3. Results and Discussion
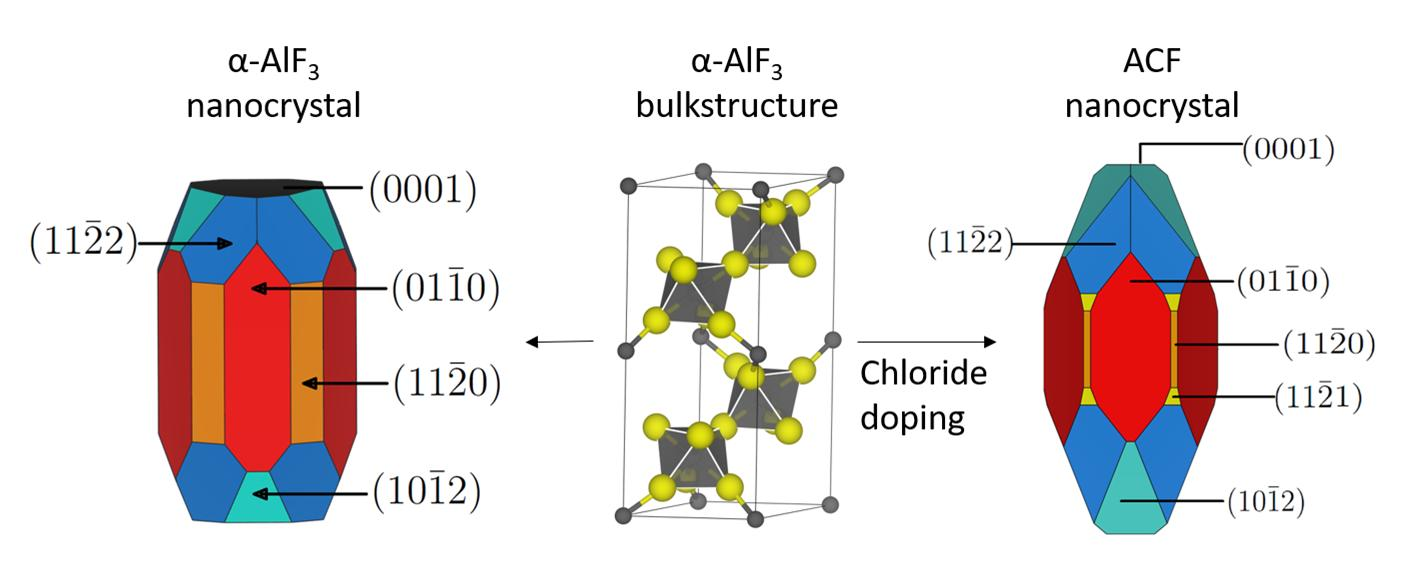
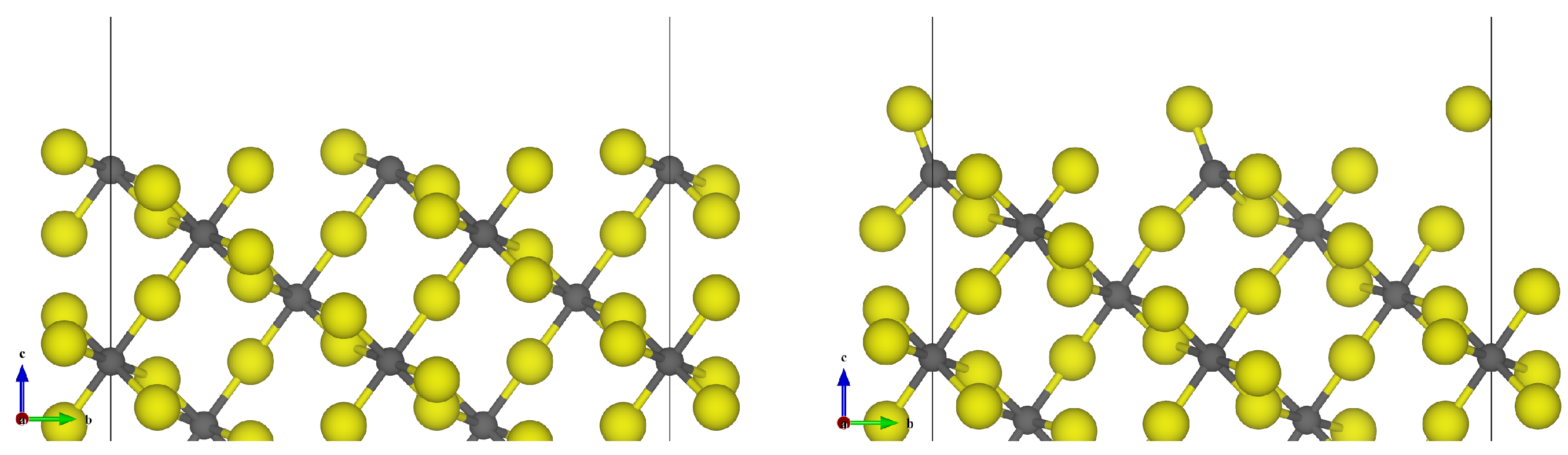
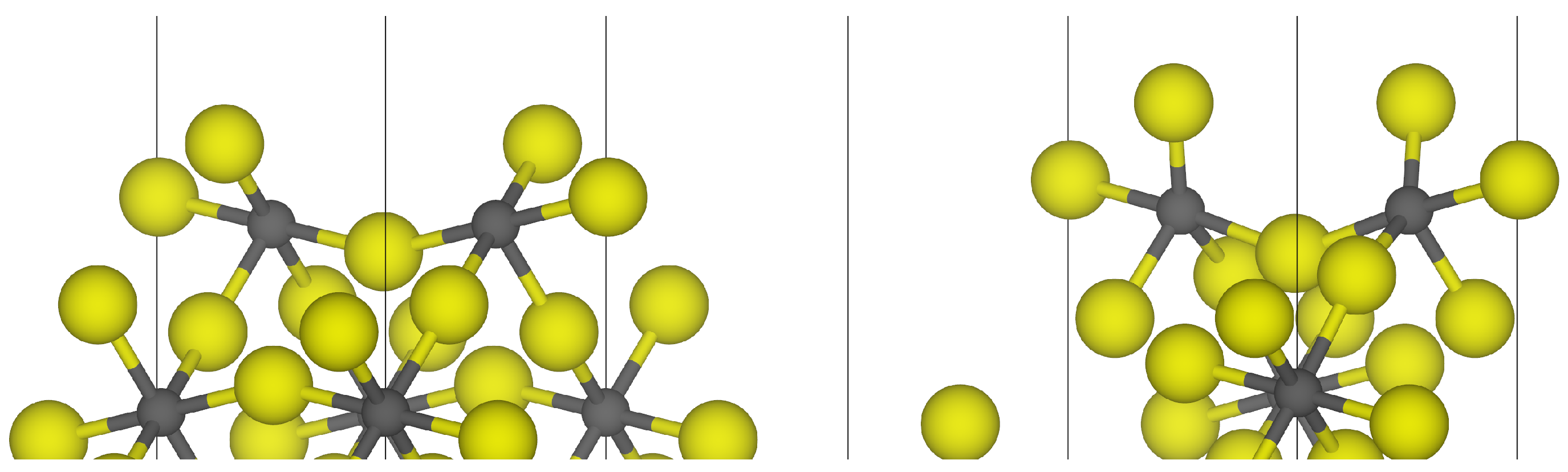
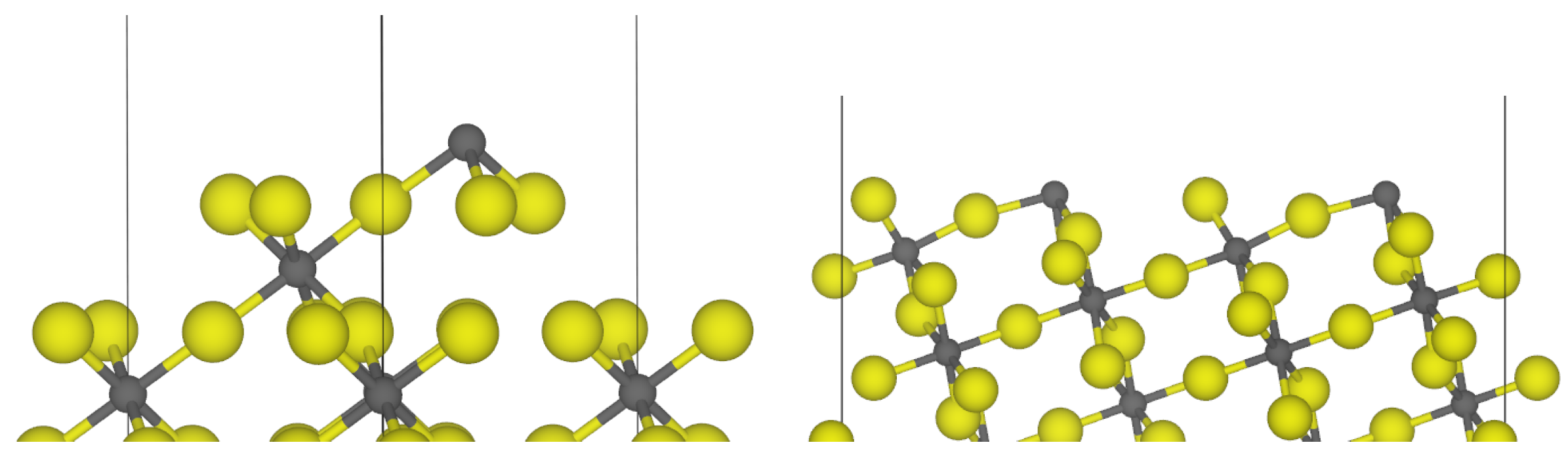
3.1. -AlF3
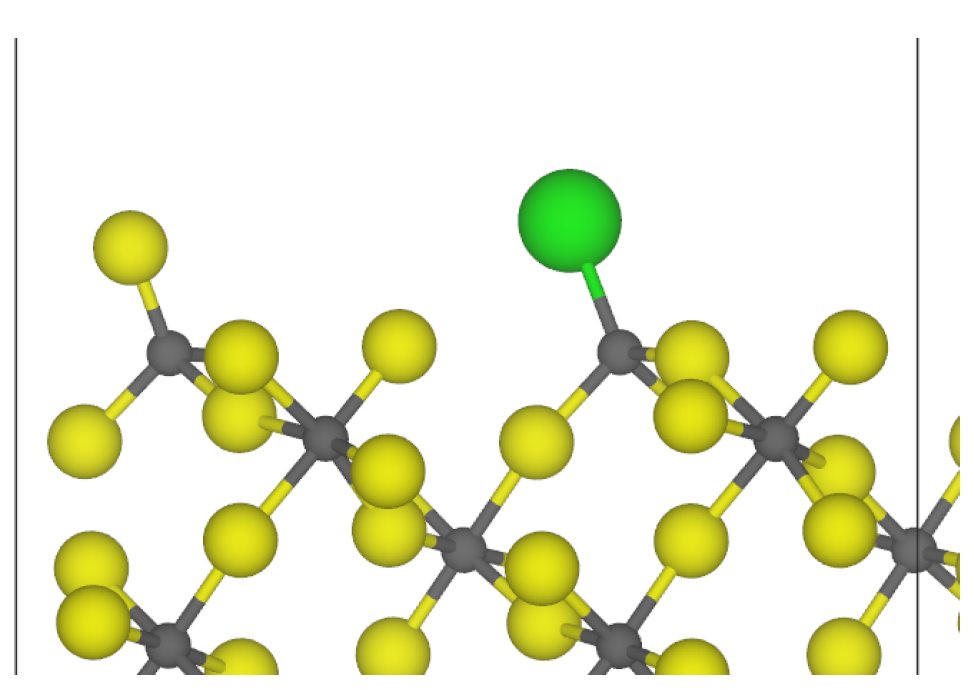
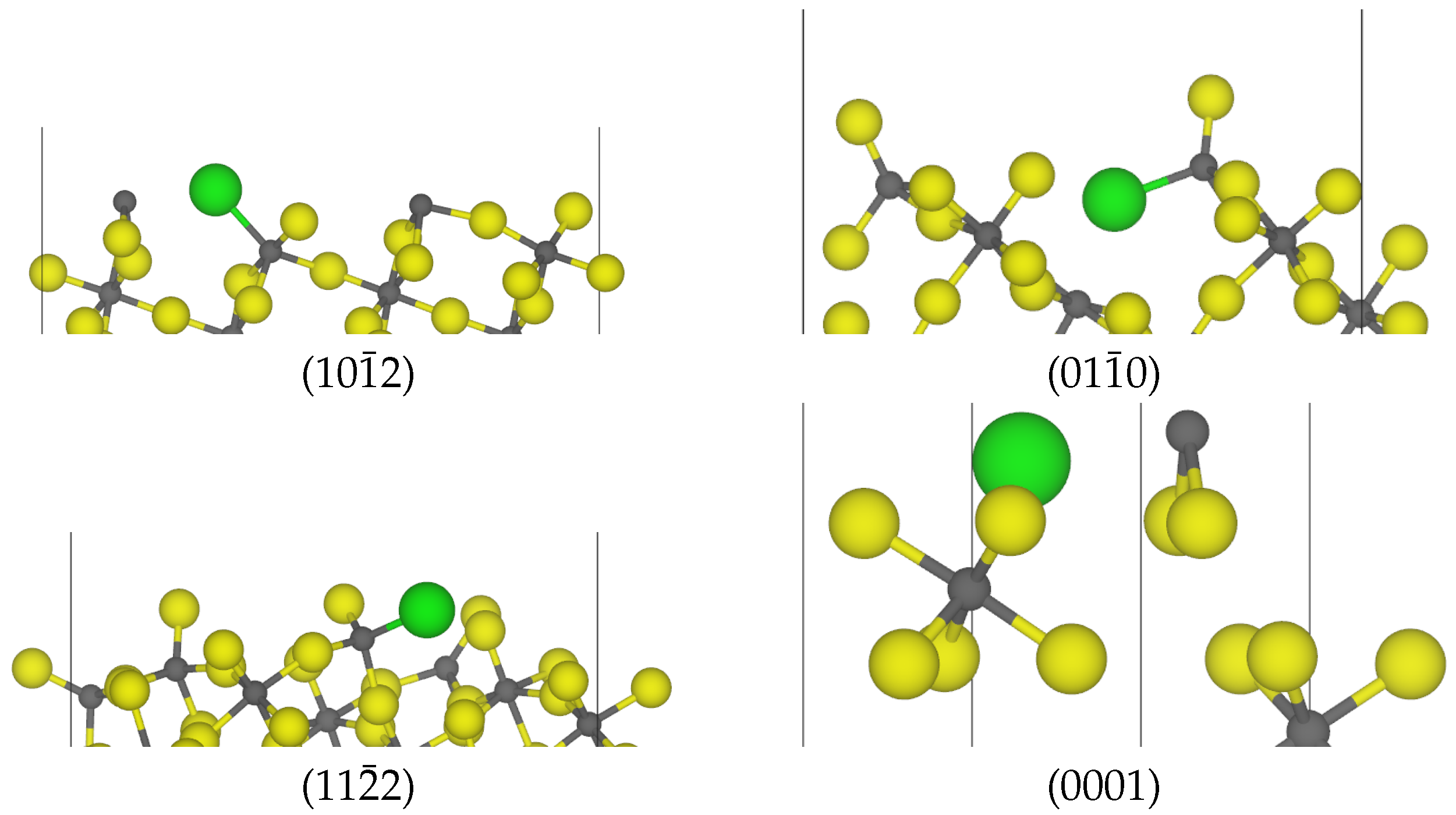
3.2. Chlorine Substitution of -AlF3
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Védrine, J.C. Heterogeneous Catalysis on Metal Oxides. Catalysts 2017, 7, 341. [Google Scholar] [CrossRef]
- Feynman, R.P. There’s plenty of room at the bottom. J. Microelectromech. Syst. 1992, 1, 60–66. [Google Scholar] [CrossRef]
- Birringer, R.; Gleiter, H.; Klein, H.P.; Marquardt, P. Nanocrystalline materials an approach to a novel solid structure with gas-like disorder? Phys. Lett. A 1984, 102, 365–369. [Google Scholar] [CrossRef]
- Murray, C.B.; Kagan, C.R.; Bawendi, M.G. Synthesis and Characterization of Monodisperse Nanocrystals and Close-Packed Nanocrystal Assemblies. Annl. Rev. Mat. Sci. 2000, 30, 545–610. [Google Scholar] [CrossRef]
- Kosmala, A.; Wright, R.; Zhang, Q.; Kirby, P. Synthesis of silver nano particles and fabrication of aqueous Ag inks for inkjet printing. Mater. Chem. Phys. 2011, 129, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Holzinger, M.; Le Goff, A.; Cosnier, S. Nanomaterials for biosensing applications: A review. Front. Chem. 2014, 2, 63. [Google Scholar] [CrossRef] [PubMed]
- Cushing, B.L.; Kolesnichenko, V.L.; O’Connor, C.J. Recent Advances in the Liquid-Phase Syntheses of Inorganic Nanoparticles. Chem. Rev. 2004, 104, 3893–3946. [Google Scholar] [CrossRef] [PubMed]
- Millstone, J.E.; Kavulak, D.F.J.; Woo, C.H.; Holcombe, T.W.; Westling, E.J.; Briseno, A.L.; Toney, M.F.; Fréchet, J.M.J. Synthesis, Properties, and Electronic Applications of Size-Controlled Poly(3-hexylthiophene) Nanoparticles. Langmuir 2010, 26, 13056–13061. [Google Scholar] [CrossRef] [PubMed]
- Khlebtsov, N.G.; Dykman, L.A. Optical properties and biomedical applications of plasmonic nanoparticles. J. Quant. Spectrosc. Radiat. Transf. 2010, 111, 1–35. [Google Scholar] [CrossRef]
- Unser, S.; Bruzas, I.; He, J.; Sagle, L. Localized Surface Plasmon Resonance Biosensing: Current Challenges and Approaches. Sensors 2015, 15, 15684–15716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, A.G.; Azoia, N.C.G.A.; Cavaco-Paulo, A. Albumin-Based Nanodevices as Drug Carriers. Curr. Pharm. Des. 2016, 22, 1371–1390. [Google Scholar] [CrossRef] [PubMed]
- Martis, E.; Badve, R.; Degwekar, M. Nanotechnology based devices and applications in medicine: An overview. Chron. Young Sci. 2012, 3, 68–73. [Google Scholar] [CrossRef]
- Nikalje, A.P. Nanotechnology and its Applications in Medicine. Med. Chem. 2015, 5, 81–89. [Google Scholar] [CrossRef]
- Reddy, M.V.; Subba Rao, G.V.; Chowdari, B.V.R. Metal Oxides and Oxysalts as Anode Materials for Li Ion Batteries. Chem. Rev. 2013, 113, 5364–5457. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Marks, T.J.; Fachetti, A. Metal oxides for optoelectronic applications. Nat. Mat. 2016, 15, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Toh, R.J.; Sofer, Z.; Pumera, M. Transition Metal Oxides for the Oxygen Reduction Reaction: Influence of the Oxidation States of the Metal and its Position on the Periodic Table. Chemphyschem 2015, 16, 3527–3531. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Robert, W.; Molt, J.; Blackburn, G.M. Metal Fluorides: Tools for Structural and Computational Analysis of Phosphoryl Transfer Enzymes. Top. Curr. Chem. 2017, 375, 1–31. [Google Scholar] [CrossRef]
- Lee, Y.; Sun, H.; Young, M.J.; George, S.M. Atomic Layer Deposition of Metal Fluorides Using HF–Pyridine as the Fluorine Precursor. Chem. Mat. 2016, 28, 2022–2032. [Google Scholar] [CrossRef]
- Wang, F.; Kim, S.W.; Seo, D.H.; Kang, K.; Wang, L.; Su, D.; Vago, J.J.; Wang, J.; Graetz, J. Ternary metal fluorides as high-energy cathodes with low cycling hysteresis. Nat. Commun. 2015, 6, 6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüdiger, S.; Kemnitz, E. The fluorolytic sol–gel route to metal fluorides—A versatile process opening a variety of application fields. Dalton Trans. 2008, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Kemnitz, E.; Groß, U.; Rüdiger, S.; Shekar, C.S. Amorphous Metal Fluorides with Extraordinary High Surface Areas. Ang. Chem. Int. Ed. 2003, 42, 4251–4254. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, S.K.; Groß, U.; Feist, M.; Prescott, H.A.; Shekar, S.C.; Troyanov, S.I.; Kemnitz, E. Non-aqueous synthesis of high surface area aluminum fluoride—A mechanistic investigation. J. Mater. Chem. 2005, 15, 588–597. [Google Scholar] [CrossRef]
- Rüdiger, S.; Groß, U.; Kemnitz, E. Non-aqueous sol–gel synthesis of nano-structured metal fluorides. J. Fluor. Chem. 2007, 128, 353–368. [Google Scholar] [CrossRef]
- Nickkho-Amiry, M.; Eltanany, G.; Wuttke, S.; Rüdiger, S.; Kemnitz, E.; Winfield, J.M. A comparative study of surface acidity in the amorphous, high surface area solids, aluminum fluoride, magnesium fluoride and magnesium fluoride containing iron(III) or aluminum(III) fluorides. J. Fluor. Chem. 2008, 129, 366–375. [Google Scholar] [CrossRef]
- Kemnitz, E. Nanoscale metal fluorides: A new class of heterogeneous catalysts. Catal. Sci. Technol. 2015, 5, 786–806. [Google Scholar] [CrossRef]
- Schacht, J.; Budau, J.H.; Gaston, N.; Paulus, B. Aluminum oxo-fluoride clusters: A first principle investigation of stability, synthetic considerations, and the interaction with water. J. Comp. Chem. 2018, 39, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wuttke, S.; Vimont, A.; Daturi, M.; Lavalley, J.C.; Teinz, K.; Kemnitz, E. Novel sol–gel prepared zinc fluoride: Synthesis, characterisation and acid–base sites analysis. J. Mater. Chem. 2012, 22, 14587–14593. [Google Scholar] [CrossRef]
- Rehmer, A.; Scheurell, K.; Kemnitz, E. Formation of nanoscopic CaF2 via a fluorolytic sol–gel process for antireflective coatings. J. Mater. Chem. C 2015, 3, 1716–1723. [Google Scholar] [CrossRef]
- Schmidt, L.; Emmerling, F.; Kirmse, H.; Kemnitz, E. Sol–gel synthesis and characterisation of nanoscopic strontium fluoride. RSC Adv. 2014, 4, 32–38. [Google Scholar] [CrossRef]
- Candu, N.; Wuttke, S.; Kemnitz, E.; Coman, S.; Parvulescu, V. Friedel-Crafts alkylations on nanoscopic inorganic fluorides. Recent Developments in Model Catalysis—Closing the Gap to Technical Applications. Appl. Catal. A Gen. 2011, 391, 169–174. [Google Scholar] [CrossRef]
- Coman, S.; Wuttke, S.; Vimont, A.; Daturi, M.; Kemnitz, E. Catalytic Performance of Nanoscopic, Aluminium Trifluoride-Based Catalysts in the Synthesis of (all-rac)-α-Tocopherol. Adv. Synth. Catal. 2008, 350, 2517–2524. [Google Scholar] [CrossRef]
- Coman, S.; Parvulescu, V.; Wuttke, S.; Kemnitz, E. Synthesis of Vitamin K1 and K1-Chromanol by Friedel–Crafts Alkylation in Heterogeneous Catalysis. Chem. Cat Chem. 2010, 2, 92–97. [Google Scholar] [CrossRef]
- Negoi, A.; Wuttke, S.; Kemnitz, E.; Macovei, D.; Parvulescu, V.I.; Teodorescu, C.M.; Coman, S.M. One-Pot Synthesis of Menthol Catalyzed by a Highly Diastereoselective Au/MgF2 Catalyst. Ang. Chem. Int. Ed. 2010, 49, 8134–8138. [Google Scholar] [CrossRef] [PubMed]
- Coman, S.M.; Patil, P.; Wuttke, S.; Kemnitz, E. Cyclisation of citronellal over heterogeneous inorganic fluorides—Highly chemo- and diastereoselective catalysts for (±)-isopulegol. Chem. Commun. 2009, 460–462. [Google Scholar] [CrossRef] [PubMed]
- Troncea, S.B.; Wuttke, S.; Kemnitz, E.; Coman, S.M.; Parvulescu, V.I. Hydroxylated magnesium fluorides as environmentally friendly catalysts for glycerol acetylation. Appl. Catal. B Environ. 2011, 107, 260–267. [Google Scholar] [CrossRef]
- Kemnitz, E.; Wuttke, S.; Coman, S.M. Tailor-Made MgF2-Based Catalysts by Sol–Gel Synthesis. Eur. J. Inorg. Chem. 2011, 2011, 4773–4794. [Google Scholar] [CrossRef]
- Prescott, H.A.; Li, Z.J.; Kemnitz, E.; Deutsch, J.; Lieske, H. New magnesium oxide fluorides with hydroxy groups as catalysts for Michael additions. J. Mater. Chem. 2005, 15, 4616–4628. [Google Scholar] [CrossRef]
- Wuttke, S.; Coman, S.; Kröhnert, J.; Jentoft, F.; Kemnitz, E. Sol–gel prepared nanoscopic metal fluorides—A new class of tunable acid-base catalysts. Catal. Today 2010, 152, 2–10. [Google Scholar] [CrossRef]
- Wuttke, S.; Coman, S.; Kröhnert, J.; Jentoft, F.; Kemnitz, E. Catalysis by Acids and Bases: New materials and surface studies ABC-6. In Proceedings of the 6th World Congress on Catalysis by Acids and Bases, Genova, Italy, 10–14 May 2009. [Google Scholar]
- Astruc, A.; Cochon, C.; Dessources, S.; Célérier, S.; Brunet, S. High specific surface area metal fluorides as catalysts for the fluorination of 2-chloropyridine by HF. Appl. Catal. A Gen. 2013, 453, 20–27. [Google Scholar] [CrossRef]
- Krüger, H.; Hertwig, A.; Beck, U.; Kemnitz, E. Low temperature sol–gel metal oxide and fluoride layer stacks for optical applications. Thin Solid Films 2010, 518, 6080–6086. [Google Scholar] [CrossRef]
- Noack, J.; Schmidt, L.; Gläsel, H.J.; Bauer, M.; Kemnitz, E. Inorganic-organic nanocomposites based on sol–gel derived magnesium fluoride. Nanoscale 2011, 3, 4774–4779. [Google Scholar] [CrossRef] [PubMed]
- Teinz, K.; Wuttke, S.; Börno, F.; Eicher, J.; Kemnitz, E. Highly selective metal fluoride catalysts for the dehydrohalogenation of 3-chloro-1,1,1,3-tetrafluorobutane. J. Catal. 2011, 282, 175–182. [Google Scholar] [CrossRef]
- Kobayashi, S.; Hamada, T.; Manabe, K. The Catalytic Asymmetric Mannich-Type Reactions in Aqueous Media. J. Am. Chem. Soc. 2002, 124, 5640–5641. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.L.; Mukhopadhyay, S.; Wander, A.; Searle, B.G.; Harrison, N.M. Structure and Stability of α-AlF3 Surfaces. J. Phys. Chem. C 2009, 113, 4976–4983. [Google Scholar] [CrossRef]
- Bailey, C.L.; Wander, A.; Mukhopadhyay, S.; Searle, B.G.; Harrison, N.M. Characterization of Lewis acid sites on the (100) surface of β-AlF3: Ab initio calculations of NH3 adsorption. J. Phys. Chem. 2008, 128, 224703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huesges, Z.; Müller, C.; Paulus, B.; Hough, C.; Harrison, N.; Kemnitz, E. Characterising MgF2 surfaces with CO adsorption calculations. Surf. Sci. 2013, 609, 73–77. [Google Scholar] [CrossRef]
- Hammerschmidt, L.; Müller, C.; Paulus, B. Electron correlation contribution to the physisorption of CO on MgF2(110). J. Chem. Phys. 2012, 136, 124117. [Google Scholar] [CrossRef] [PubMed]
- Kaawar, Z.; Paulus, B. A computational study of the structure of zinc fluoride surfaces. AIP Conf. Proc. 2015, 1653, 020051. [Google Scholar] [CrossRef] [Green Version]
- Kaawar, Z.; Müller, C.; Paulus, B. Theoretical investigations of the CO adsorption on ZnF2 surfaces. Surf. Sci. 2017, 656, 48–53. [Google Scholar] [CrossRef]
- Barth, J.; Johnson, R.L.; Cardona, M.; Fuchs, D.; Bradshaw, A.M. Dielectric function of CaF2 between 10 and 35 eV. Phys. Rev. B 1990, 41, 3291–3294. [Google Scholar] [CrossRef]
- Camy, P.; Doualan, J.; Renard, S.; Braud, A.; Ménard, V.; Moncorgé, R. Tm3+:CaF2 for 1.9 µm laser operation. Optics Comm. 2004, 236, 395–402. [Google Scholar] [CrossRef]
- Kumar, G.A.; Riman, R.; Chae, S.C.; Jang, Y.N.; Bae, I.K.; Moon, H.S. Synthesis and spectroscopic characterization of CaF2:Er3+ single crystal for highly efficient 1.53 µm amplification. J. App. Phys. 2004, 95, 3243–3249. [Google Scholar] [CrossRef]
- Tsujibayashi, T.; Toyoda, K.; Sakuragi, S.; Kamada, M.; Itoh, M. Spectral profile of the two-photon absorption coefficients in CaF2 and BaF2. App. Phys. Lett. 2002, 80, 2883–2885. [Google Scholar] [CrossRef]
- Giessibl, F.J.; Reichling, M. Investigating atomic details of the CaF2(111) surface with a qPlus sensor. Nanotechnology 2005, 16, S118. [Google Scholar] [CrossRef]
- Kaawar, Z.; Mahn, S.; Kemnitz, E.; Paulus, B. On the Morphology of Group II Metal Fluoride Nanocrystals at Finite Temperature and Partial Pressure of HF. Molecules 2017, 22, 663. [Google Scholar] [CrossRef] [PubMed]
- Kawaar, Z. Theoretical Investigations of Bulk and Surface Properties of Group II and Group XII Metal Fluorides. Ph.D. Thesis, Department of Biology, Chemistry and Pharmacy of Freie Universität Berlin, Berlin, Germany, 2018. [Google Scholar]
- Krahl, T.; Vimont, A.; Eltanany, G.; Daturi, M.; Kemnitz, E. Determination of the Acidity of High Surface AlF3 by IR Spectroscopy of Adsorbed CO Probe Molecules. J. Phys. Chem. C 2007, 111, 18317–18325. [Google Scholar] [CrossRef]
- Kemnitz, E.; Menz, D.H. Fluorinated metal oxides and metal fluorides as heterogeneous catalysts. Prog. Solid State Chem. 1998, 26, 97–153. [Google Scholar] [CrossRef]
- Daniel, P.; Bulou, A.; Rousseau, M.; Nouet, J.; Fourquet, J.L.; Leblanc, M.; Burriel, R. A study of the structural phase transitions in AlF 3: X-ray powder diffraction, differential scanning calorimetry (DSC) and Raman scattering investigations of the lattice dynamics and phonon spectrum. J. Phys. Condens. Matter 1990, 2, 5663. [Google Scholar] [CrossRef]
- Reuter, K.; Scheffler, M. Composition, structure, and stability of RuO2(110) as a function of oxygen pressure. Phys. Rev. B 2001, 65, 035406. [Google Scholar] [CrossRef]
- Geysermans, P.; Finocchi, F.; Goniakowski, J.; Hacquart, R.; Jupille, J. Combination of (100), (110) and (111) facets in MgO crystals shapes from dry to wet environment. Phys. Chem. Chem. Phys. 2009, 11, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Stampfl, C.; Kreuzer, H.J.; Payne, S.H.; Pfnür, H.; Scheffler, M. First-Principles Theory of Surface Thermodynamics and Kinetics. Phys. Rev. Lett. 1999, 83, 2993–2996. [Google Scholar] [CrossRef] [Green Version]
- Kanaki, E.; Gohr, S.; Müller, C.; Paulus, B. Theoretical study on the morphology of MgF2 nanocrystals at finite temperature and pressure. Surf. Sci. 2015, 632, 158–163. [Google Scholar] [CrossRef]
- Fortes, A.D.; Wood, I.G.; Vočadlo, L.; Knight, K.S.; Marshall, W.G.; Tucker, M.G.; Fernandez-Alonso, F. Phase behavior and thermoelastic properties of perdeuterated ammonia hydrate and ice polymorphs from 0 to 2GPa. J. App. Crystall. 2009, 42, 846–866. [Google Scholar] [CrossRef]
- Kokalj, A. Computer graphics and graphical user interfaces as tools in simulations of matter at the atomic scale. Comp. Mat. Sci. 2003, 28, 155–168. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Wirth, J.; Schacht, J.; Saalfrank, P.; Paulus, B. Fluorination of the Hydroxylated α-Al2O3 (0001) and Its Implications for Water Adsorption: A Theoretical Study. J. Phys. Chem. C 2016, 120, 9713–9718. [Google Scholar] [CrossRef]
- Budau, J.H.; Paulus, B.; Steenbergen, K.G. Theoretical investigation of the crystal structure of AlOF. Chem. Phys. 2017, 491, 112–117. [Google Scholar] [CrossRef]
- Li, X.; Paier, J.; Sauer, J.; Mirabella, F.; Zaki, E.; Ivars-Barcelo, F.; Shaikhutdinov, S.; Freund, H.J. Surface Termination of Fe3O4(111) Films Studied by CO Adsorption Revisited. J. Phys. Chem. B 2018, 122, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Vandichel, M.; Biswas, S.; Leus, K.; Paier, J.; Sauer, J.; Verstraelen, T.; Van Der Voort, P.; Waroquier, M.; Van Speybroeck, V. Catalytic Performance of Vanadium MIL-47 and Linker-Substituted Variants in the Oxidation of Cyclohexene: A Combined Theoretical and Experimental Approach. Chem. Plus Chem. 2014, 79, 1183–1197. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. It VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Wulff, G. Zur Frage der Geschwindigkeit des Wachsthums und der Auflösung der Krystallflächen. Z. Kristall. Crystal. Mat. 1901, 34. [Google Scholar] [CrossRef]
- Gibbs, J.W. The Collected Works of J. Willard Gibbs; Longmans, Green: New York, NY, USA, 1928. [Google Scholar]
- Von Laue, M. Der Wulffsche Satz für die Gleidigewichtsform von Kristallen. Z. Kristall. Crystal. Mat. 1943, 105. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 84204–84210. [Google Scholar] [CrossRef] [PubMed]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comp. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mat. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
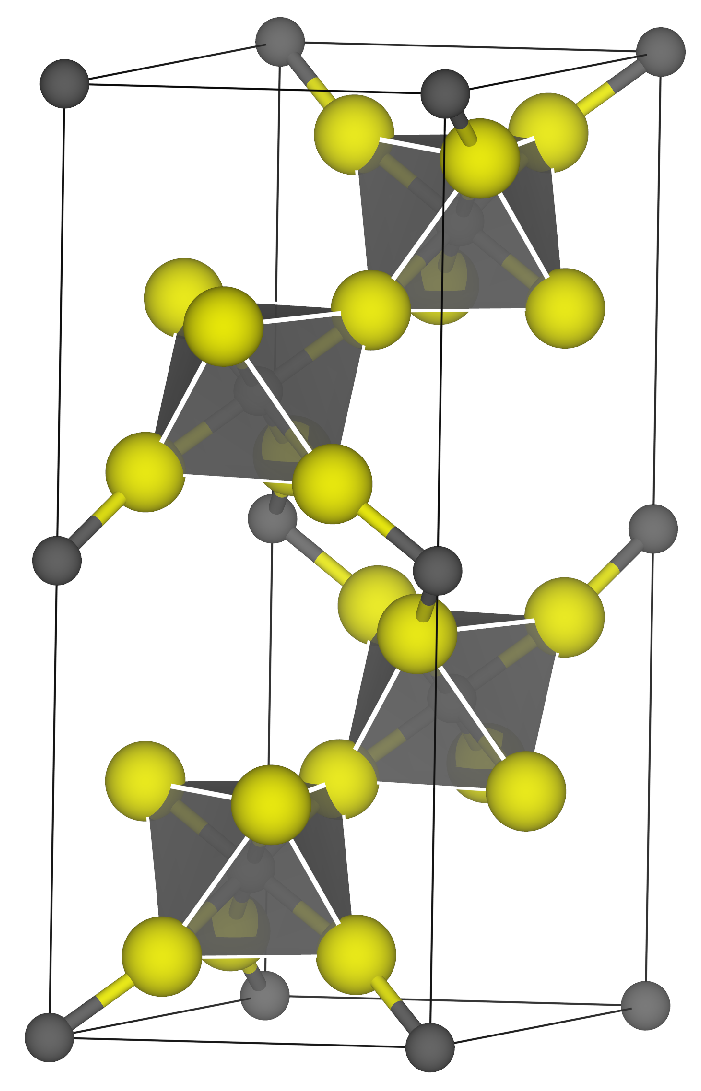
| Surface | Termination | Energy (J m−2) | Charge/e |
|---|---|---|---|
| (0001) | 1 | 6.84 | −3.00 |
| 2 | 1.76 | +3.00 | |
| () | 1 | 3.62 | −4.00 |
| 2 | 1.01 | 0.00 | |
| 3 | 1.75 | +4.00 | |
| () | 1 | 7.72 | −6.00 |
| 2 | 5.48 | −4.00 | |
| 3 | 1.05 | 0.00 | |
| 4 | 1.94 | +4.00 | |
| 5 | 1.66 | +6.00 | |
| () | 1 | 6.69 | −3.00 |
| 2 | 2.87 | −1.00 | |
| 3 | 1.73 | +1.00 | |
| 4 | 1.73 | +3.00 | |
| () | 1 | — | −3.00 |
| 2 | — | −1.00 | |
| 3 | 1.37 | +1.00 | |
| 4 | 1.39 | +3.00 | |
| () | 1 | — | −3.00 |
| 2 | 1.22 | +1.00 | |
| 3 | 1.31 | +3.00 | |
| () | 1 | 3.87 | −3.00 |
| 2 | 1.91 | −1.00 | |
| 3 | 1.41 | +1.00 | |
| 4 | 1.43 | +3.00 | |
| () | 1 | 4.22 | −6.00 |
| 2 | 2.09 | −2.00 | |
| 3 | 2.18 | +2.00 | |
| 4 | 1.49 | +6.00 | |
| () | 1 | 10.95 | −6.00 |
| 2 | 4.11 | −2.00 | |
| 3 | 1.92 | +2.00 | |
| 4 | 1.95 | +6.00 | |
| () | 1 | — | +6.00 |
| 2 | — | +4.00 | |
| 3 | 1.29 | 0.00 | |
| 4 | 1.29 | +4.00 | |
| 5 | 1.43 | +6.00 |
| -AlF3 | ACF | Reaction Energy | |||||
|---|---|---|---|---|---|---|---|
| Surface | Esurf | % Area | CN | Substitution | Esurf | % Area | kcal mol−1 |
| () | 1.01 | 35.33 | 4 | Terminal | 1.04 | 40.72 | 44.44 |
| Bridging | 1.30 | — | 67.39 | ||||
| () | 1.05 | 16.23 | 5 | Terminal | 1.17 | 2.70 | 45.52 |
| Bridging | 1.96 | — | 86.05 | ||||
| () | 1.22 | 0.00 | 4;5 | Terminal | 1.25 | 1.64 | 45.69 |
| () | 1.29 | 28.82 | 4;5 | Terminal | 1.33 | 36.36 | 45.90 |
| Bridging | 1.41 | — | 59.16 | ||||
| () | 1.41 | 0.00 | 4;5 | Terminal | 1.50 | 0.00 | 44.71 |
| () | 1.48 | 8.25 | 3;5 | Terminal | 1.56 | 17.40 | 47.54 |
| Bridging | 1.63 | — | 55.41 | ||||
| () | 1.73 | 0.00 | 4;3 | Bridging | 1.88 | 0.00 | 41.69 |
| (0001) | 1.76 | 11.36 | 3 | Bridging | 2.49 | 1.19 | 60.55 |
| () | 1.92 | 0.00 | 5 | Bridging | 2.24 | 0.00 | 52.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandharkar, R.; Becker, C.; Budau, J.H.; Kaawar, Z.; Paulus, B. A Computational Study of AlF3 and ACF Surfaces. Inorganics 2018, 6, 124. https://doi.org/10.3390/inorganics6040124
Pandharkar R, Becker C, Budau JH, Kaawar Z, Paulus B. A Computational Study of AlF3 and ACF Surfaces. Inorganics. 2018; 6(4):124. https://doi.org/10.3390/inorganics6040124
Chicago/Turabian StylePandharkar, Riddhish, Christian Becker, Johannes Horst Budau, Zeinab Kaawar, and Beate Paulus. 2018. "A Computational Study of AlF3 and ACF Surfaces" Inorganics 6, no. 4: 124. https://doi.org/10.3390/inorganics6040124
APA StylePandharkar, R., Becker, C., Budau, J. H., Kaawar, Z., & Paulus, B. (2018). A Computational Study of AlF3 and ACF Surfaces. Inorganics, 6(4), 124. https://doi.org/10.3390/inorganics6040124