Abstract
Quasi-linear anionic 3d-metal(I) silylamides are a new and promising class of molecules. Due to their highly negative reduction potential we wanted to test their capability to reduce substrates under coordination of their monoanionic radicaloid form. In a proof of principle study, we present the results of the reaction of metal(I) silylamides of chromium to cobalt with 2,2′-bipyridine (bipy), the redox non-innocence and reducibility of which was already established. In the course of these studies complexes of the type K{18-crown-6}[M(hmds)2(bipy)] (hmds = –N(SiMe3)2) were obtained. These compounds were isolated and thoroughly characterized to confirm the electron transfer onto the bipyridine ligand, which now acts as a radical monoanion. For comparison of the structural changes of the bipyridine ligand, the analogous zinc complexes were also synthesized. Overall our results indicate that anionic metal(I) silylamides are capable of reducing and ligate substrates, even when the electrochemical reduction potential of the latter is by up to 1 V higher.
1. Introduction
Complexes with two-coordinate 3d-metal(I) ions are a rare class of molecules in coordination chemistry [1,2,3,4,5,6,7,8,9,10,11,12,13]. They combine an uncommon coordination motif with an, for these metals, unusual oxidation state (with the exception of Cu and to a certain extent nickel). Their scarcity can be explained by the difficulties of stabilizing the electronically and coordinatively unsaturated metal ion. Thus, their isolation usually relies on the use of sterically encumbering and/or electronically stabilizing ligands, such as bulky amides or N-heterocyclic carbenes. Given their only recent history, little is known about their physical properties as well as chemical behavior towards substrates and small molecules. However, first reports indicate a high potential thereof. For example, in case of iron complexes, they can exhibit extraordinary single molecule magnetic behavior, which can be directly connected to the linear ligand arrangement as well as the oxidation state +1 [6,8]. Further, such complexes can mediate the cleavage of H2 [12] and C–halide bonds [14] or the trimerization of alkynes [15].
We recently showed that it is possible to obtain quasi-linear 3d-metal(I) complexes K{18c6}[MI(hmds)2] (M = Cr, Fe, Co; 18c6 = 18-crown-6) using the comparably unencumbering hmds ligand set (hmds = –N(SiMe3)2) [6,13]. As a result of the sterically uncongested metal center, the labile manganese complex K{18c6}[MnI(hmds)2], which is supposedly monomeric in solution, forms a metal−metal bonded dimer in the solid state (Scheme 1). Despite the highly accessible metal sites of these metal(I) complexes, they seem surprisingly inert towards Lewis bases such as phosphines, phosphites, NHCs or THF (exemplarily shown for iron) [6], which is in contrast to the behavior of other two-coordinate metal(I) fragments [1,16,17,18,19,20,21]. Given this observed reluctance to obtain further electron density upon Lewis base coordination, we were interested in the formal transfer of an electron from the metal(I) ion onto substrates to form metal(II) complexes bearing an anionic radicaloid ligand. In addition, we were interested in elaborating the synthetic usability of the elusive manganese(I) silylamide [MnI(hmds)2]− (Scheme 1), which cannot be isolated due to its extreme sensitivity.
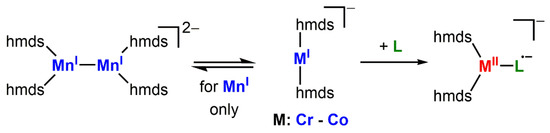
Scheme 1.
Metal(I) hexamethyldisilazanides (hmds = –N(SiMe3)2)2 and their expected use for reduction and ligation of a substrate L.
In a proof of principle study we thus chose 2,2′-biypyridine (bipy), which is widely used as a chelating donor ligand [22]. Further, it can act as a π-acceptor that can take up one or two electrons to form either the 2,2′-bipyridine radical monoanion [bipy]·− or the diamagnetic dianion [bipy]2− when subjected to highly reducing agents such as alkaline metals. This has been shown for the alkali metal salts of 2,2′- as well as 4,4′-bipyridine [23,24] and more importantly for 2,2′-bipyridine containing metal complexes [25,26,27,28,29,30,31]. Within these complexes the exact determination of the electronic structure of the [M(bipy)] unit is usually not trivial leading to the generally accepted classification of 2,2′-bipyridine as a redox non-innocent ligand [32,33]. Thereby, it has been shown that the structural features of the central C–C bond within the bipyridine ligand can be a strong indicator of its redox state [32,34,35]. This has been showcased for four-coordinate [M(mes)2(bipy)] (mes = 2,4,6-trimethylphenyl), where reduction with potassium graphite in the presence of either 18-crown-6 or crypt.222 led to a mainly bipyridine-based reduction and formation of K{L}[M(mes)2(bipy)] (L = 18-crown-6 or crypt.222) [34,35].
Given this background, we were curious if and how the above-mentioned two-coordinate metal(I) silylamides coordinate 2,2′-bipyridine. Thereby it is important to note that the reported reduction potential of the free bipy0/bipy·−-redox couple is fairly negative (E1/2 = −2.53 V vs Fc/Fc+ in MeCN) [36] in comparison to those of the two-coordinate metal(I) complexes (Epc of −1.5 V for Co to −2.5 V for Mn) [6,13]. We herein report on the reaction of K{18c6}[MI(hmds)2] (M = Cr–Co) with 2,2′-bipyridine, which leads to the formation of the respective bipyridine complexes K{18c6}[M(hmds)2(bipy)]. These complexes were examined by single-crystal X-ray diffraction analysis and 1H-NMR/UV–Vis spectroscopy and for their magnetic as well as electrochemical properties. In comparison with their neutral metal(II) counterparts and the additional synthesis of the zinc derivatives, the anionic complexes are overall best described as [MII(hmds)2(bipy·−)]−.
2. Results
2.1. Synthesis and Structural Characterisation
The respective metal(I) hmds complex (Cr–Co), either isolated (Fe and Co) or synthesized in situ (Cr and Mn), was treated with one equivalent of 2,2′-bipyridine in Et2O. In all cases an immediate reaction was visible by a change of color to intense dark green (Cr, Fe and Co) or red (Mn). These mixtures were filtered, layered with pentane and stored at −35 °C. After a few days crystalline samples of the complexes (1 (Cr−), 2 (Mn−), 3 (Fe−) and 4 (Co−)) were obtained in moderate to good yields (Scheme 2).
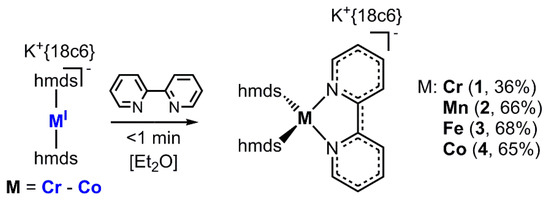
Scheme 2.
General synthetic approach for the complexes 1–4 (18c6 = 18-crown-6).
X-ray diffraction analysis confirmed their structural identity (Figure 1). Important structural parameters of the complexes 1–4 in the solid state are summarized in Table 1.
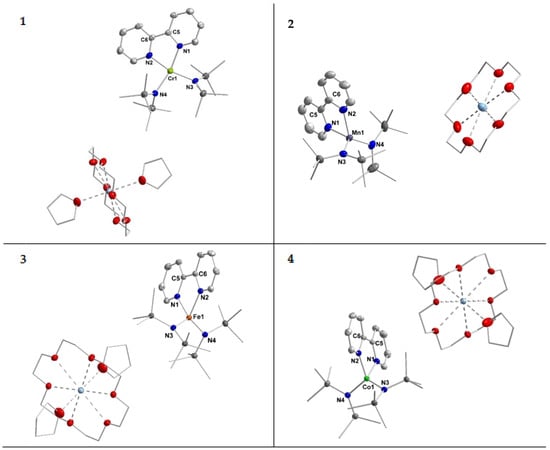
Figure 1.
Molecular structures of compounds 1–4. H atoms are omitted for clarity and ellipsoids are shown at 50% probability.

Table 1.
Important structural metrics of the complex anions of the compounds 1–4.
In the solid state the complex anions of compounds 1–4 exhibit a four-coordinate metal ion which is ligated by two hmds units and the bipyridine ligand. For the chromium compound 1 the torsion angle between the planes defined by N3–M1–N4 and N1–M1–N2 amounts to 23°. Given the respective τ’4-parameter of 0.26, this complex is best described as distorted square planar. From a ligand field perspective this indicates a chromium(II) ion (d4 configuration), which usually prefers a square planar ligand environment such as in [Cr(hmds)2(thf)2] or [CrCl4]2− [37,38,39]. For the manganese, iron and cobalt complexes the torsion angle amounts to 67.06° (2 (Mn−)), 73.98° (3 (Fe−)) and 75.3° (4 (Co−)). Together with the respective τ’4-parameters of 0.78, 0.77 and 0.81 these complexes are overall best described as distorted tetrahedral.
The M–Nhmds bond lengths get slightly shorter along the series from chromium (2.09 Å) to cobalt (2.00 Å). For the M–Nbipy distances a contraction is also observed from manganese (2.15 Å) to cobalt (2.00 Å), whereas for chromium a distance of 2.08 Å is observed. This behavior is in general agreement with the ion radii of the respective metal(II) ion. The lengths of the central C–C bond of the bipyridine ligand increases slightly from 1.417(6) Å for chromium to 1.432(3) Å for cobalt. The C–C bond lengths are thereby in the range of ligated 2,2′-bipyridine radical anions within isolable complexes [32,34,35,40,41].
To further substantiate the presence of a bipyridine radical anion in the complexes 1–4 on a structural level, we compared their structural features to those of their neutral counterparts, [MII(hmds)2(bipy)]. For the chromium (5) [42], manganese (6) [43] and iron (7) [43] complexes the synthesis and solid state structures were already described in the literature. As further analysis of their spectroscopic properties was lacking, their synthesis was reproduced by the reaction of the respective hmds salt with 2,2′-bipyridine in Et2O. Analogously, the missing cobalt derivative 8 was obtained as a dark red crystalline solid in moderate yield (Scheme 3; Figure 2, left). Further, we sought the isolation of the respective zinc complexes, as for zinc the reduction of the metal ion is highly unlikely due to the high stability of the +2 oxidation state. Zn(hmds)2 was thus reacted with 2,2′-bipyridine, giving [Zn(hmds)2(bipy)] (9) as pale-yellow crystals (63% yield; Figure 2, middle).
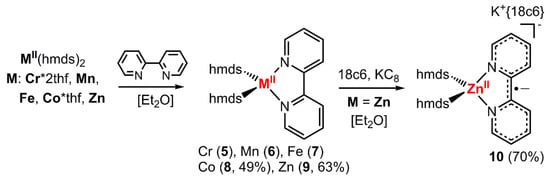
Scheme 3.
General synthetic approach for the neutral complexes 5–9 (known for Cr, Mn and Fe) as well as the reduced zinc complex 10 (Zn−).
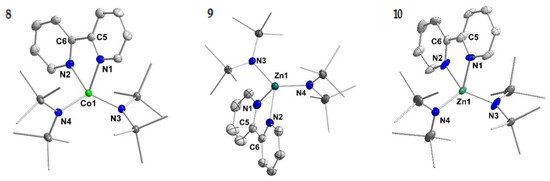
Figure 2.
Molecular structure of compounds 8–10. H atoms are omitted for clarity and ellipsoids are shown at 50% probability. The K{18c6}-cation is not shown for 10.
The reduction of compound 9 (Zn) with one equivalent KC8 in the presence of 18-crown-6 lead to the isolation of rose crystals of K{18c6}[Zn(hmds)2(bipy)] (10; Figure 2, right). Similarly, compounds 1–4 could also be obtained by reduction of the respective, preformed [M(hmds)2(bipy)] complex.
Important structural features of 5–10 are given in Table 2. The structure of 10 suffers from disorder of the complex over two positions. Structural metrics are given for one of the molecules and should be treated with care. For the neutral metal(II) compounds 5–9 the following trends are observed: The respective metal nitrogen distances of bipyridine and the hmds ligands shorten along the series, similar to observations made for the reduced complexes. The central C–C bond of the bipyridine ligand also shortens from 1.502(8) Å for the manganese complex 6 (Mn) to 1.474(7) Å for the zinc complex 9 (Zn). The chromium complex 5 (Cr) shows again deviations from this trend with a C–C bond length of 1.481(3) Å. Further, for all neutral complexes but chromium a distorted tetrahedral coordination environment is observed with a torsion angle that increases from 65° to 73° for the manganese to zinc complex. In contrast, the chromium complex 5 (Cr) is better described as distorted square planar with a torsion angle of 20°.
Table 2.
Important structural metrics of compounds 5–9 as well as of the complex anion of K{18c6}[Zn(hmds)2bipy], 10.
In comparison with the neutral compounds, the central C–C bond lengths of the bipyridine ligand are shorter by between 0.05 Å (Zn) and 0.08 Å (Mn) in the reduced complexes. In addition, the reduced complexes exhibit shorter M–Nbipy bond lengths, whereas those of the M–Nhmds bonds are slightly elongated. Otherwise, the general coordination geometry around the metal are largely the same for the neutral and the reduced complexes. Given these geometric features, the anionic complexes 1–4 and 10 (Zn−) are likely to contain a monoanionic 2,2′-bipyridine ligand.
2.2. 1H-NMR Spectroscopic Features
To gain insights into changes of the electronic situation of the anionic complexes, we turned to the 1H NMR spectroscopic features of the reduced as well as the neutral complexes (Table 3). Providing their mostly paramagnetic nature, the proton signals of the hmds as well as the bipyridine ligands might be indicative of changes in the electronic properties of the metal center. Thereby it has to be noted that the respective proton NMR features of the literature known compounds 5 (Cr), 6 (Mn) and 7 (Fe) were not reported.
Table 3.
1H-NMR spectroscopic features of the compounds 1–10 in THF-d8 (500 MHz).
For the manganese (2 (Mn−) and 6 (Mn)) and chromium complexes (1 (Cr−) and 5 (Cr)) the proton NMR spectra showed, if at all, very broad and overlapping signals, which rendered their spectra uninformative (for spectra see Supplementary Materials). The iron complex 3 (Fe−) is represented by four signals at −268.1, 178.2, 77.9 and 64.5 ppm for the bipyridine ligand (Figure 3, top). The position of the SiMe3 signal is very broad and positioned at approximately 3.58 ppm and is overlapped by the signals belonging to the used solvent (THF-d8) and 18-crown-6. In comparison, the 1H-NMR spectrum of the neutral iron(II) complex 7 (Fe) shows rather sharp signals at 10.95 ppm for the SiMe3 unit and at 64.09, 52.13 and −19.13 ppm for the bipyridine ligand (Figure 3, bottom).
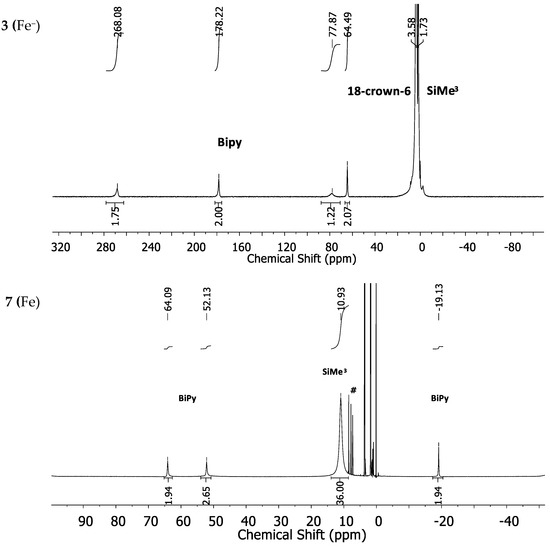
Figure 3.
1H-NMR spectra of the iron complexes 3 (Fe−) (top) and 7 (Fe) (bottom) in THF-d8. # denotes free 2,2′-bipyridine as an impurity.
The expected fourth signal of the bipyridine ligand could not be located in the region between ±400 ppm. It is probably subject to large line broadening and/or may be located beneath other signals. The cobalt complex 4 (Co−) shows four strongly low-field shifted 1H-NMR signals for the bipyridine ligand at 155.8, 143.95, 104.3 and 50.26 ppm (Figure 4, top). The signal for the SiMe3 groups is situated at 2.47 ppm whereas the one belonging to the crown-ether is approximately at its diamagnetic position. The neutral cobalt complex 8 (Co) exhibits sharper and less paramagnetically shifted signals at 59.7, 50.94, 42.65 and −21.01 ppm for the bipyridine ligand. The signal belonging to the SiMe3 protons is shifted further away (8.11 ppm) from its diamagnetic position (Figure 4, bottom) in comparison with complex 4 (Co−).
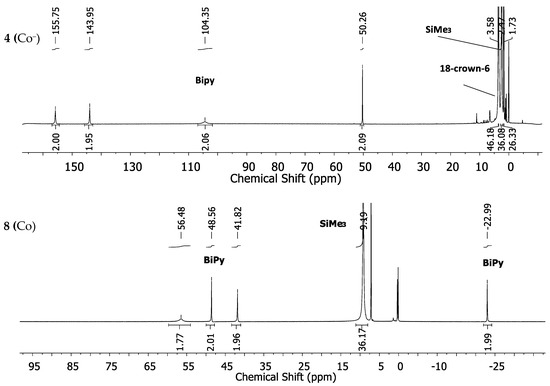
Figure 4.
1H-NMR spectra of the cobalt complexes 4 (Co−) (top) and 8 (Co) (bottom) in THF-d8.
The proton signals of the bipyridine ligand of the reduced Zn complex 10 (Zn−) are shifted to a higher field in comparison to their position found for the diamagnetic, neutral derivative 9 (Zn). The signals are moderately broadened and show reduced intensity.
The signal belonging to the SiMe3 groups is also considerably shifted by approximately 0.6 ppm to a lower field and exhibits a surprisingly large line width of 234 Hz, surpassing the ones found for the more paramagnetic cobalt complexes. Overall, for the anionic complexes, larger paramagnetic shifts and line-broadening are observed for the bipyridine protons in comparison with the respective neutral complexes. In contrast, for the hmds protons for each metal, diverging trends were observed.
2.3. UV/Vis-Spectroscopic Features
To get further insights into the electronic structure of the reduced complexes 1–4 and 10, their UV/Vis spectra were recorded together with those of their non-reduced counterparts. The presence of reduced 2,2-bipyridine can be deducted from two strong bands at 532 and 562 nm and three bands between 700 and 1000 nm, as shown for in situ generated Na(bipy) in THF [44]. For 2,2-bipyridyl radical anion containing metal complexes the situation is similar although the two high-energy bands are often not resolved [30,31,41,45]. The high energy transitions are tentatively assigned to a π→π*-based transition whereas the ones in the low energy region are due to a π∗→π* centered transition [44]. To corroborate the UV/Vis-spectroscopic signature of the 2,2-bipyridyl radical anion under our reaction conditions, we attempted the reduction of 2,2′-bipyridine with KC8 in Et2O in the absence of any 3d-metal complex. However, no change of color was observed. This is in contrast to the reports of isolation of the monopotassium salt of 2,2-bipyridine by the group of Goicoechea and is presumably due to stabilizing effects of the used ethylene diamine solvent in their case. When the reduction of 2,2-bipyridine is performed in the presence of 18-crown-6 the instant formation of a violet solution was observed. The isolation of the presumed K{18-crown-6} stabilized 2,2′-bipyridyl anion failed in our hands, as rapid discoloring of the solution was observed within 60 s. Nonetheless, an immediately recorded UV/Vis spectrum showed two sharp bands at 542 and 577 nm and some very weak bands in the low energy region between 800 and 1000 nm. The position of the signals are similar to those of Na(bipy) in THF, although in our case the low-energy bands were substantially weaker in relative intensity.
Given this background, we started with the zinc derivatives, as in this case the bipyridine centered reduction is highly plausible. The neutral zinc(II) complex 9 (Zn) shows only a weak absorption band around 350 nm, which reflects its pale yellow color (Figure 5). In the case of the reduced zinc complex 10 (Zn−) this band is more pronounced. Furthermore, two distinct sharp bands at 505 and 535 nm and three broad bands in the range of 700−1000 nm were observed, which can be attributed to the bipyridyl radical anion (Table 4). At 914 nm an additional small band was detected which was already observed in the case of the neutral complex 9 (Zn).
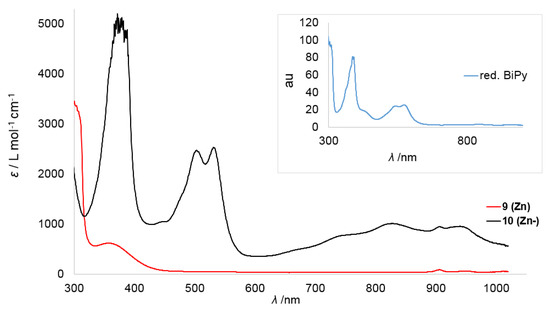
Figure 5.
Overlay of the UV/Vis spectra of the Zn complexes 9 (Zn) and 10 (Zn−) in Et2O. The inset belongs to the in situ spectrum of 2,2′-bipyridine reduced by KC8 in the presence of 18-crown-6 in Et2O after approximately 3 min.
Table 4.
UV/Vis spectroscopic features of complexes 1–10 in Et2O. Bold numbers highlight the two optical bands attributed to the π→π*-based transition of the reduced bipyridine ligand.
Overall, the coordination of the supposed bipyridine radical anion to the zinc ion lead to a slight blue-shift of the two high-energy bands with only marginal change in the separation in between them (Δλ ≈ 30 nm). The bands in the region of 700–1000 nm are more pronounced than for “K{18c6}[bipy]” and their intensity is in line with the reports on Na(bipy).
Next, we examined the compounds 1–8 whose main optical transitions are shown in Table 4.
For the reduced manganese and chromium complexes 2 (Mn−) (Figure 6) and 1 (Cr−) (see Supplementary Materials for spectrum) the two high energy bands were somewhat blue-shifted (Mn: 487 and 523 nm, Δλ ≈ 35 nm; Cr: 488 and 525 nm, Δλ ≈ 35 nm). For the iron complex 3 (Fe−) the intensity of these bands is reduced, which impeded their identification (460 and 497 nm, Δλ ≈ 30 nm), whereas for compound 4 (Co−) (see Supplementary Materials for spectra) no clear assignment was possible. The low-energy region between 700 and 1000 nm shows pronounced bands for complexes 2 (Mn−) and 4 (Co−), whereas the spectra of compounds 1 (Cr−) and 3 (Fe−) reveal only a broad absorption.
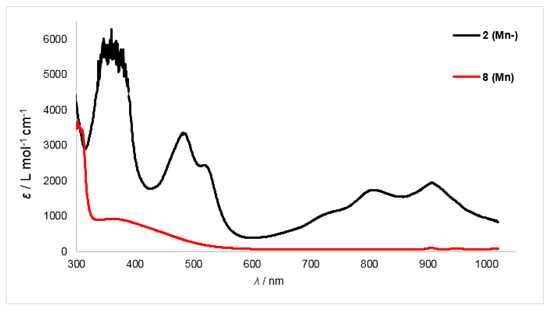
Figure 6.
Overlay of UV/Vis spectra of the manganese complexes 2 (Mn−) and 6 (Mn).
2.4. Magnetic Properties
Given the paramagnetic nature of the anionic complexes, their magnetic properties in solid state were evaluated using a physical properties measurement system and in solution via the Evans method [46,47]. The structural and UV/Vis spectroscopic features of the anionic complexes already hinted to their likely formulation as a [MII/bipy·−] system. As such, two limiting cases can be assumed: First, a metal(II) ion that is strongly ferromagnetically coupled to the unpaired electron of the bipyridyl radical anion, which gives Stot = SM + S1/2 as the upper limit. On the other hand, antiferromagnetic coupling leads to Stot = SM − S1/2. Further, the absence of significant coupling between the spin centers would give Stot = SM × S1/2, with a value for the magnetic susceptibility that lies between those limiting cases. For the metal(II) ions a high-spin configuration is assumed, given the low-coordinate and rather weak ligand field environment of these complexes. The magnetic susceptibilities of compounds 1–4 and 10 (Zn−) were measured in the solid state in a field of H = 1 T in a temperature range of 3–300 K (Figure 7). The magnetic data were corrected for diamagnetic contributions from the sample holder and the diamagnetic susceptibility of the respective compound using Pascal constants. The paramagnetic susceptibility χpara was fitted using the Curie-Weiss Law (neff = effective magnetic moment in Bohr’s magnetons per formula unit, Θ = Weiss temperature) with contributions from a temperature independent paramagnetism χTIP using the overall equation χparaT = (χTIP + χCW)T. Obtained results (Table 5) are thus discussed within the simplified framework of the presented limiting cases with a focus on the room-temperature χT values and the general curvature of the χT vs T slopes. A more detailed description of this spin-system would necessitate more experimental magnetic data which lay beyond the scope of this study.
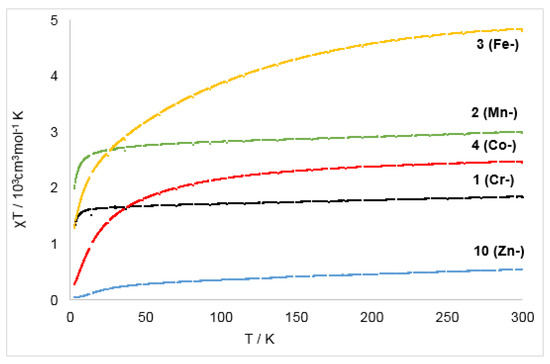
Figure 7.
Temperature magnetic susceptibility (χT vs T) of the complexes 1–4 and 10 from 3 K to 300 K at 1 T.
Table 5.
Calculated Θ and χTIM values for the anionic complexes 1–4 and 10 obtained from fitting of their variable temperature magnetic data to the Curie–Weiss Law.
For the zinc complex 10 (Zn−) the χT vs T slope showed a gradual increase when going from 3 K to 50 K, after which the curve flattens, reaching a value of 0.54 cm3·mol−1·K at 300 K (1.72 μB). The value is close to the measured magnetic moment of 1.81 μB in solution using the Evans method and is in line with a S = ½ system. The slight, steady increase from 50 K to 300 K can be explained by contributions from a temperature independent paramagnetism, whereas the decrease below 50 K is unexpected for an isolated S = ½ system. This can be reasoned by significant intermolecular magnetic coupling between the ligating bipyridyl radical anions whose phenyl rings are in proximity in the solid state (minimum C–C distance of 3.99 Å). Such an intermolecular interaction is known for purely organic radicals, which can also exhibit intermolecular ferro- as well as antiferromagnetic coupling [48,49,50].
For the chromium complex 1 (Cr−) the slope increased rapidly from 2 K to 20 K, where it reaches a plateau at a χT value of 1.84 cm3·mol−1·K (300 K, μeff = 3.66 μB). The value at room temperature fits well the magnetic moment in solution (μeff = 3.60 μB, Evans method). It is far off the one for a possible formulation as a chromium(I) complex for which a spin-only value of 5.97 (S = 5/2) is likely, as observed for the chromium(I) precursor [Cr(hmds)2]− [13]. Thus, the observed value is best explained by a strongly antiferromagnetically coupled [CrII/bipy·−]. The slope of the manganese complex 2 (Mn−) shows similar behavior. Its effective magnetic moment of 4.71 μB in solid state is slightly higher than the one in solution (4.46 μB) which might be indicative of a paramagnetic impurity. Nonetheless, the value fits best for the description for complex 2 (Mn−) as a strongly antiferromagnetically coupled ([MnII/bipy·−]) system.
The χT(T) slope of the reduced iron and cobalt complexes 3 (Fe−) and 4 (Co−) increased steadily and merely plateaued at 300 K, reaching 4.80 cm3·mol−1·K (μeff = 6.67 μB) for complex 3 (Fe−) and 2.45 cm3·mol−1·K (μeff = 4.61 μB) for complex 4 (Co−). These very high values are suspicious as they exceed those of their respective quasi-linear metal(I) precursor complexes, which already show free ion behavior with significant spin-orbit coupling [6,13]. Together with the significantly lower magnetic susceptibilities in solution at ambient temperatures (3 (Fe−): μeff = 4.12 μB; 4 (Co−): μeff = 3.52 μB), this points to the presence of a superparamagnetic impurity. A similar observation of unrealistically high χT(T) values was already made for the used two-coordinate iron(I) precursor, which could be attributed to an otherwise undetectable paramagnetic impurity [6]. Given the magnetic moments in solution for complexes 3 (Fe−) and 4 (Co−), they also contain most likely high-spin metal(II) ions antiferromagnetically coupled to the 2,2′-bipyridine radical anion [23].
2.5. Electrochemistry
Additional information on the electron transfer onto the bipyridine ligand was sought using electrochemical means. When the reduced complexes 1–4 and 10 are subjected to cyclovoltammetric measurements (1 mM, THF, 500 mV/s, 0.1 M NBu4PF6, vs Fc/Fc+), no clear redox event is observed in the expected region between −1 V to −3 V (exemplarily shown for 2 (Mn−) in Figure 8, see Supplementary material for further details). This is surprising, as bipyridine complexes can exhibit a rich electrochemistry with reversible redox events in this region (see the introduction). Upon examination of the neutral complexes 6–9, each showed a reversible redox event between E1/2 = −2.48 V (6 (Mn)) and −2.81 V (9 (Zn), Table 6). In contrast, the chromium complex 5 (Cr) showed no comparable redox event. As the reported E1/2 = −2.3 V of the bipy/bipy− redox couple of free 2,2′-bipyridine is low in comparison with that of compounds 6–9, we analyzed 2,2′-bipyridine under our conditions, which gave E1/2 = −2.67 V. Thus, the measured values for E1/2 of complexes 6–9 vary only little with respect to the identity of the metal of the employed [M(hmds)2(bipy)] complex and are close to the value of free 2,2′-bipyridine. This indicates a bipyridine centered reduction for these compounds. Compounds 6–8 show a second irreversible reduction around −3.0 V. This reduction wave is also rather unaffected by the identity of the metal in those complexes and is presumably due to a second bipyridine centered reduction, which would lead to the diamagnetic bipyridyl dianion [23].
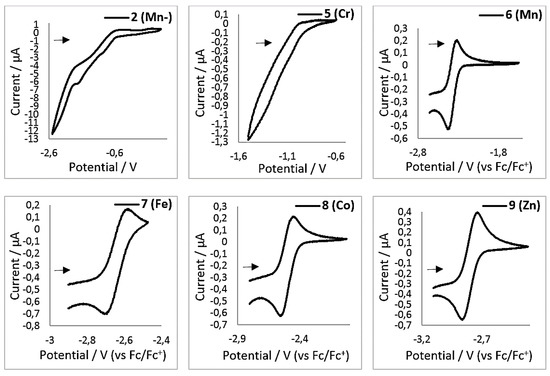
Figure 8.
Cyclic voltammograms for complexes 2, 5–9 in THF (1 mM, 500 mV/s, 0.1 M [NBu4][PF6]).
Table 6.
Cyclovoltammetric data for compounds 6–9 and 2,2′-bipyridine.
3. Discussion
The quasi-linear metal(I) complexes K{18c6}[M(hmds)2] reacted instantaneously with 2,2-bipyridine, giving four-coordinate complexes of the type K{18c6}[M(hmds)2(bipy)]. This is remarkable given the high reduction potential of free 2,2′-bipyridine (E1/2 = −2.3 V vs Fc/Fc+). Within complexes the reduction of bipyridine is usually found to occur in a range between −0.8 V and −2.5 V depending on the metal and the charge of the complex [30,40,51,52,53,54,55,56]. Thereby, lower potentials are observed mainly for neutral or anionic complexes, such as [Mn(bipy)(CO)3Br] (approximately −2.0 V vs Fc/Fc+). Our results implicate that, despite the lack of an observable interaction of [MI(hmds)2]− with Lewis bases, as examined exemplarily for iron, an inner-sphere electron transfer mechanism is likely to take place upon temporary bipyridine coordination. The structural features of K{18c6}[M(hmds)2(bipy)], especially those of the central C–C bond of the bipyridine ligand, are indicative of the reduction of the bipyridine to its monoanionic state. This notion is supported by the lack of significant geometric differences around the metal ion in comparison with their neutral, non-reduced counterparts. This holds especially true for the chromium complex, which retains its distorted square planar geometry in the neutral as well as the reduced state, which is typical for four-coordinate chromium(II) ions.
Electrochemical examination of the reduced complexes [M(hmds)2(bipy)]− by cyclovoltammetry surprisingly showed no redox event between −1 V and −3 V (vs. Fc/Fc+), which is the expected range of bipyridyl radical anion containing complexes (see the introduction). This may be the result of the highly sensitive nature of these compounds. In contrast, their neutral counterparts 6–9, (with the exception of 5 (Cr)) exhibit a reversible redox event in the range of E1/2 = −2.48 V (2 (Mn)) and E1/2 = −2.81 V (9 (Zn)), which can be attributed to the [M(hmds)2(bipy)]0/[M(hmds)2(bipy)]−1 redox pair. As the redox events are largely independent from the identity of the metal and similar to the one of free 2,2′-bipyridine (E1/2 = −2.67 V), it is highly indicative of a largely bipyridine centered reduction in these complexes. It is important to note that these E1/2 values are partially more negative than the ones of the respective quasi-linear metal(I) silylamides (Epc = −2.45 V (MnI), −1.98 V (FeI) and −1.47 V (CoI)) [6,13]. For chromium no clear redox event is visible in the examined region.
1H-NMR spectroscopic examination of compounds 1–8 revealed highly paramagnetically influenced signals for all those complexes, whereas those of chromium (1 (Cr−) and 5 (Cr)) and manganese (2 (Mn−) and 6 (Mn)) are mostly uninformative due to the extent of the paramagnetism. For the iron and cobalt complexes 3 (Fe−) and 4 (Co−) generally larger isotropic shifts and line broadening for the bipyridine protons are observed in comparison to their respective neutral counterparts 7 (Fe) and 8 (Co). Whereas the zinc complex 9 (Zn) is diamagnetic with well resolved proton signals; in the reduced zinc complex 10 (Zn−) these signals are subject to an isotropic shift and significant line broadening. For the signals of the SiMe3 protons of the complexes, no comparable trend is observed for each complex pair. Generally a higher unpaired electron count is connected to a more paramagnetically distorted 1H-NMR spectrum for otherwise structurally identical compounds [57], as for example seen for the metal(I) precursors [6,13] or the [M(hmds)2(bipy)] complexes themselves (vide supra). In the hypothetical case of a largely metal-based reduction, this would lead to an overall reduction of the respective spin state, which generally gives sharper and lesser isotropic shifted proton signals. Therefore, in the reduced complexes the bipyridine protons likely experience not only the presence of the paramagnetic metal center but also the proximity of the electron of the bipyridyl radical anion. However, one has to bear in mind that this is only a simplified approach as the exact extent of magnetic relaxation as well as that the isotropic shift is dependent on the orientation of the d-orbital towards the respective proton to which the electron is added (pseudocontact shift) and the change in the hyperfine coupling between the protons and the unpaired electrons of metal (contact shift) [58].
UV/Vis spectroscopy showed for most reduced complexes (1 (Cr−), 2 (Mn−), 10 (Zn−) and to a lesser extent for 3 (Fe−)) two distinct bands between 450 and 550 nm. These can also be found for the free 2,2′-bipyridine radical anion and are attributed to a pi–pi* transition [44]. Further, complexes 2 (Mn−), 4 (Co−) and 10 (Zn−) show strong additional absorption bands in the range of 700 to 1000 nm, which originate from a pi–pi* transition within the bipyridyl radical anion. This means that for all reduced complexes UV/Vis spectroscopy reveals the presence of a bipyridyl radical anion. Thereby, its signature is most pronounced for the manganese (2 (Mn−)) and zinc (2 (Zn−)) derivatives. It indicates in these cases a mainly electrostatic interaction between the metal ion and the radical anion. For cobalt, iron and chromium, the respective transitions belonging to the bipyridine radical anion are broader and less pronounced, which might be attributed to additional orbital interactions with the d-metal ion.
The examination of the magnetic features of the anionic complexes in the solid state show for the chromium (1 (Cr−)) and manganese (2 (Mn−)) complexes that these systems are best described as strongly antiferromagnetically coupled metal(II) bipyridine radical anions. This is consistent with the situation found for the respective [M(mes)2(bipy)]− complexes (M = Cr and Mn) [35]. For the cobalt and iron complexes 3 (Fe−) and 4 (Co−) the situation is more complicated, as solid state magnetic measurements of otherwise analytically pure samples gave rather high χT(T) values at room temperature, which are materially higher than those found in solution by Evan’s method. This indicates an inseparable superparamagnetical impurity, which makes the solid state measurements ambiguous. The magnetic susceptibility in solution for complexes 3 (Fe−) and 4 (Co−) amount to 4.12 μB and 3.57 μB. These values are indicative of a strongly antiferromagnetically coupled [MII(hmds)2(bipy·−)]− system (μtheor.(S = |2 − 1/2|) = 3.87 μB for 3 (Fe−); μtheor. (S = |3/2 − 1/2|) = 2.83 μB for 4 (Co−)) with additional orbital contributions [34]. The room temperature magnetic susceptibility of 3 (Fe−) is in line with that found for [Fe(mes)2(bipy)]− [34], whereas the value found for complex 4 (Co−) is significantly higher than the one obtained for the pseudo square-planar [Co(mes)2(bipy)]− (μeff = 1.88 μB). For the latter, a mixture of a singlet and a triplet state was assumed [35]. The comparable high value found for complex 4 (Co−) is probably due to a different complex geometry (distorted tetrahedral) giving the presence of a high spin cobalt(II) ion. Thereby, the difference in geometry between [Co(mes)2(bipy)]− and 4 (Co−) can be attributed to the higher ligand field imposed by the mesityl groups.
Taken together, the reduced complexes [M(hmds)2(bipy)]− are best described as metal(II) complexes bearing a monoanionic bipyridine radical, which is deducted from the combined use of a variety of analytical techniques. The outcome is in line with a study on related [M(mes)2(bipy)]− complexes (M = Cr–Ni; mes = 2,4,6-trimethylbenzyl), which, however, were obtained from a reduction of the preformed [M(mes)2(bipy)] parent complexes with KC8 in the presence of crypt.222 [34,35].
4. Materials and Methods
All manipulations were carried out in a glovebox under a dry argon atmosphere, unless indicated otherwise. Used solvents were dried by continuous distillation over sodium metal for several days, degassed via three freeze-pump cycles and stored over molecular sieves 4 Å. Deuterated solvents were used as received, degassed via three freeze-pump cycles and stored over molecular sieves 4 Å. The 1H-NMR spectra were recorded on a Bruker AV 500 or a Bruker HD 500 NMR spectrometer (Bruker Corporation, Billerica, MA, USA). Chemical shifts are reported in ppm relative to the residual proton signals of the solvent (for 1H) or relative to the signal of the solvent itself (13C). w1/2 is the line width of a signal at half its maximum intensity. Integrals of the broad signals of the hmds units were obtained directly or by peak fitting (in case of overlapping signals) using the MestreNova software package (Mestrelab, Santiago de Compostela, Spain). IR measurements were conducted on a Bruker Alpha ATR-IR spectrometer (Bruker Corporation, Billerica, MA, USA). Elemental analyses were performed by the “in-house” service of the Chemistry Department of the Philipps University Marburg, Germany using a CHN(S) analyzer vario MICRO Cube (Elementar Analysensysteme GmbH, Langenselbold, Germany). UV/Vis-spectra were recorded on an AnalytikJena Specord S600 diode array spectrometer (AnalytikJena, Jena, Germany). The dc susceptibility data was obtained in a range of 3 K to 300 K at 1 T on a physical properties measurement system from LQT/QD (Quantum Design, San Diego, CA, USA). The samples were mounted in a plastic sample holder. The Curie-Weiss Law was used for fitting the magnetic data, whereas the diamagnetic correction was obtained using tabulated pascal constants as well as experimental data for the sample holder. Cyclic voltammograms were recorded on a RHD Instruments measuring cell of the type Microcell HC and a Pt electrode (RHD Instruments GmbH, Darmstadt, Germany). The potentiostat is a Methrom autolab type PGStat 204 (Methrom AG, Herisau, Switzerland). Cyclic voltammetry measurements were carried out under Argon at 500 mV/s scan rate in THF/0.1 M [NBu4][PF6]. Potentials are referenced to Fc/Fc+, which was added after the respective measurement. 2,2′-bipyridine and Nbu4PF6 were obtained commercially and used as received, unless noted otherwise. Nbu4PF6 was dried in vacuo at 100 °C for several hours prior to use. [M(N(SiMe3)2)2] (Mn and Fe) [59,60], [Cr(N(SiMe3)2)2(thf)2] [61], [Co(N(SiMe3)2)2(thf)] [59], K{18c6}[M(N(SiMe3)2)2] (Fe and Co) [6,13] and [M(N(SiMe3)2)2(bipy)] (Cr–Fe) [43,62], were prepared according to literature procedures. K{18c6}[M(N(SiMe3)2)2] (Cr and Mn) were synthesized in situ according the published procedure for K{18c6}[Mn(N(SiMe3)2)2] and used as such [13].
4.1. Synthesis of Complexes
[K{18c6}][Cr(N(SiMe3)2)2(bipy)] (1): [Cr(N(SiMe3)2)2*(THF)2] (103.4 mg, 0.20 mmol, 1.0 equivalent) and 18-crown-6 (60.6 mg, 0.2 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O. The addition of KC8 (27.0 mg. 0.2 mmol, 1.0 equivalent) led to an immediate color change to dark orange. After stirring for 5 min, the mixture was filtered, and the filtrate was given to 2,2’-bipyridine (31.2 mg, 0.20 mmol, 1.0 equivalent), this led to an immediate color change to dark green. The solution was stirred for 5 min and layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark green crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 1 as a dark green crystalline solid (60.2 mg, 0.072 mmol, 36%). 1H-NMR ([D8]THF, 500.1 MHz, 298 K, ppm): No useful signal attribution possible; IR (ATR, cm−1): ν = 2939 (w), 2892 (m), 1492 (m), 1350 (w), 1231 (m), 1104 (vs), 1004 (vs), 961 (s), 890 (m), 869 (m), 819 (vs), 716 (m), 654 (m), 604 (w), 465 (w), 425 (w); elemental analysis calculated (C34H68CrKN4O6Si4 832.37 g/mol) C 49.06 H 8.23 N 6.26; experimental C 49.01 H 8.15 N 6.36. μeff = 3.60 μB ([D8]THF). Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 1 in Et2O at −35 °C.
[K{18c6}][Mn(N(SiMe3)2)2(bipy)] (2): Mn(N(SiMe3)2)2 (75.1 mg, 0.20 mmol, 1.0 equivalent) and 18-crown-6 (63.7 mg, 0.2 mmol, 1.05 equivalent) were dissolved in 3 mL of Et2O. The addition of KC8 (32.4 mg. 0.2 mmol, 1.5 equivalent) led to an immediate color change to dark purple. After stirring for 5 min, the mixture was filtered, and the filtrate was given to 2,2´-bipyridine (31.2 mg, 0.20 mmol, 1.0 equivalent), leading to an immediate color change to dark red. The solution was stirred for 5 min and layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark red crystalline solid. The solution was removed via a Pasteur pipette. The residue was washed with 2 × 5 mL of pentane and dried under reduced pressure to afford compound 2 as a dark red crystalline solid (110.9 mg, 0.13 mmol, 66%). 1H-NMR ([D8]THF, 500.1 MHz, 298 K, ppm): δ = 3.62 (s, 24H, w½ = 29 Hz, OCH2), 19.90 (s, 36H, w½ = 2312 Hz, Si(CH3)3); IR (ATR, cm−1): ν = 2940 (w), 2889(w), 1453 (m), 1351 (m), 1232 (s), 1001 (s), 961 (s), 890 (m), 869 (m), 819 (vs), 771 (vs), 724 (m), 703 (m), 659 (m), 604 (w), 528 (w), 449 (w), 415 (w); elemental analysis calculated (C34H68MnKN4O6Si4 832.37 g/mol) C 48.89 H 8.21 N 6.71; experimental C 47.61 H 8.09 N 6.35. A lower carbon value of 1% was consistently observed upon several attempts (>5), despite using different, freshly prepared crystalline batches of 8. We attribute this to possible carbide formation. Μeff = 3.46 μB ([D8]THF). Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 2 in Et2O at −35 °C.
[K{18c6}][Fe(N(SiMe3)2)2(bipy)] (3): [K{18c6}][Fe(N(SiMe3)2)2] (135.9 mg, 0.20 mmol, 1.0 equivalent) and 2,2´-bipyridine (31.2 mg, 0.20 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from brownish green to dark green. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark green crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 3 as a dark green crystalline solid (113.8 mg, 0.14 mmol, 68%). 1H-NMR ([D8]THF, 500.3 MHz, 298 K, ppm): δ = 1.73 (s, 36H, Si(CH3)3), 3.58 (s, 24H, OCH2) 64.49 (s, 2H, w½ = 154 Hz, CHbipy), 77.87 (s, 2H, w½ = 1534 Hz, CHbipy), 178.22 (s, 2H, w½ = 478 Hz, CHbipy), 268.08 (s, 2H, w½ = 711 Hz, CHbipy); IR (ATR, cm−1): ν = 2941 (w), 2890 (w), 1536 (w), 1493 (m), 1350 (m), 1229 (m), 1001 (s), 961 (s), 889 (m), 869 (m), 820 (vs), 774 (m), 728 (m), 703 (m), 640 (m), 601 (w), 528 (w), 455(w), 417 (w); elemental analysis calculated (C34H68FeKN4O6Si4 836.22 g/mol) C 48.84 H 8.20 N 6.70; experimental C 48.63 H 8.06 N 6.96. μeff = 4.12 μB ([D8]THF). Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 3 in Et2O at −35 °C.
[K{18c6}][Co(N(SiMe3)2)2(bipy)] (4): [K{18c6}][Co(N(SiMe3)2)2] (68.3 mg, 0.10 mmol, 1.0 equivalent) and 2,2´-bipyridine (15.6 mg, 0.10 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from pale yellow-green to dark green. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark green crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 4 as a dark green crystalline solid (54.8 mg, 0.065 mmol, 65%). 1H-NMR ([D8]THF, 500.1 MHz, 298 K, ppm): δ = 2.47 (bs, 36H, w½ = 83 Hz, Si(CH3)3), 3.58 (s, 24H, w½ = 29 Hz, OCH2), 50.26 (s, 2H, w½ = 25 Hz, CHbipy), 104.35 (bs, 2H, w½ = 749 Hz, CHbipy), 143.95 (s, 2H, w½ = 85 Hz, CHbipy), 155.75 (s, 2H, w½ = 100 Hz, CHbipy); IR (ATR, cm−1): ν = 2940 (m), 2887 (m), 1565 (w), 1552 (w), 1503 (m), 1470 (m), 1451 (m), 137 (w), 1350 (m), 1293 (m), 1227 (m), 1103 (vs), 1009 (s), 958 (s), 866 (m), 817 (vs), 687 (m), 653 (w), 605 (m), 528 (m), 462 (w), 419 (w); elemental analysis calculated (C34H68CoKN4O6Si4 839.31 g/mol) C 48.66 H 8.17 N 6.68; experimental C 48.98 H 7.79 N 6.83. μeff = 3.52 μB ([D8]THF). Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 4 in Et2O at −35 °C.
[Cr(N(SiMe3)2)2(bipy)] (5) [42]: [Cr(N(SiMe3)2)2*(THF)2] (206.8 mg, 0.4 mmol, 1.0 equivalent) and 2,2´-bipyridine (62.5 mg, 0.4 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from orange to dark violet. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark violet crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 5 as a dark violet crystalline solid (184.0 mg, 0.35 mmol, 87%). 1H-NMR ([D8]THF, 500.1 MHz, 300 K, ppm): δ = −71.19 (s, 2H, w½ = 931 Hz, CHbipy), 16.47 (s, 2H, w½ = 258 Hz, CHbipy), 40.78 (s, 36H, w½ = 2539 Hz, Si(CH3)3), 47.05 (s, 2H, w½ = 979 Hz, CHbipy), 53.82 (s, 2H, w½ = 1415 Hz, CHbipy); IR (ATR, cm−1): ν = 2939 (m), 2892 (w), 1601 (m), 1572 (w), 1466 (w), 1441 (m), 1305 (w), 1249 (s), 1234 (s), 1152 (w), 986 (vs), 886 (m), 864 (m), 813 (vs), 761 (vs), 733 (vs), 657 (s), 606 (m), 419 (m).
[Mn(N(SiMe3)2)2(bipy)] (6) [43]: Mn(N(SiMe3)2)2 (150.3 mg, 0.4 mmol, 1.0 equivalent) and 2,2´-bipyridine (62.5 mg, 0.4 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from colorless to dark orange. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark orange crystalline solid. The solution was removed via pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 6 as a light orange crystalline solid (194.0 mg, 0.36 mmol, 92%). 1H-NMR ([D8]THF, 500.1 MHz, 300 K, ppm): δ = 29.54 (s, 36H, w½ = 5723 Hz, Si(CH3)3), no further useful signal attribution is possible; IR (ATR, cm−1): ν = 2942 (m), 2890 (w), 1596 (m), 1441 (m), 1235 (s), 1155 (w), 993 (vs), 888 (w), 866 (s), 814 (vs), 775 (m), 756 (s), 736 (m), 703 (w), 662 (s), 625 (w) 610 (m).
[Fe(N(SiMe3)2)2(bipy)] (7) [43]: Fe(N(SiMe3)2)2 (149.9 mg, 0.4 mmol, 1.0 equivalent) and 2,2´-bipyridine (62.5 mg, 0.4 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from pale to dark green. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark green crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 7 as a dark green crystalline solid (198.0 mg, 0.37 mmol, 93%). 1H-NMR ([D8]THF, 500.1 MHz, 300 K, ppm): δ = −19.13 (s, 2H, w½ = 61 Hz, CHbipy), 10.93 (s, 36H, w½ = 425 Hz, Si(CH3)3), 52.13 (s, 2H, w½ = 128 Hz, CHbipy), 64.09 (s, 2H, w½ = 113 Hz, CHbipy), the fourth signal belonging to the bipyridine ligand was not observed, which is attributed to its paramagnetism; IR (ATR, cm−1): ν = 2942 (m), 2890 (w), 1597 (w), 1441 (m), 1236 (s), 974 (vs), 888 (m), 862 (m), 820 (vs), 779 (s), 755 (s), 708 (s), 662 (s), 621 (w), 610 (w).
[Co(N(SiMe3)2)2(bipy)] (8): [Co(N(SiMe3)2)2*thf] (460.0 mg, 1.0 mmol, 1.0 equivalent) and 2,2´-bipyridine (156.12 mg, 1.0 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from pale green to dark red. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark red crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 8 as a dark red crystalline solid (263.0 mg, 0.49 mmol, 49%). 1H-NMR ([D8]THF, 500.1 MHz, 300 K, ppm): δ = −21.01 (s, 2H, w½ = 27 Hz, CHbipy), 8.11 (s, 36H, w½ = 68 Hz, Si(CH3)3), 42.65 (s, 2H, w½ = 31 Hz, CHbipy), 50.94 (s, 2H, w½ = 25 Hz, CHbipy), 59.68 (s, 2H, w½ = 253 Hz, CHbipy); IR (ATR, cm−1): ν = 2941 (w), 2890 (m), 1598 (m), 1441 (m), 1235 (s), 1154 (m), 976 (vs), 888 (w), 860 (vs), 660 (m), 609 (w); elemental analysis calculated (C22H44CoN2Si4 535.19 g/mol) C 49.31 H 8.28 N 10.46; experimental C 48.65 H 8.20 N 10.50. A lower carbon value of 1% was consistently observed upon several attempts (>5), despite using different, freshly prepared crystalline batches of 8. We attribute this to possible carbide formation. LIFDI mass calc. (C22H44CoN2Si4 535.1975 m/z); exp. (C22H44CoN2Si4 535.1958 m/z). Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 8 in Et2O at −35 °C.
[Zn(N(SiMe3)2)2(bipy)] (9): Zn(N(SiMe3)2)2 (154.2 mg, 0.4 mmol, 1.0 equivalent) and 2,2´-bipyridine (62.5 mg, 1.0 mmol, 1.0 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from colorless to pale yellow. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a light yellow crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 9 as a light yellow crystalline solid (136.3 mg, 0.25 mmol, 63%). 1H-NMR ([D8]THF, 500.1 MHz, 300 K, ppm): δ = 0.11 (s, 36H, w½ = 2 Hz, Si(CH3)3), 7.64–7.85 (m, 2H, CHbipy), 8.17 (t, 3J =7.5 Hz, 2H, CHbipy), 8.44 (d, 3J =8.1 Hz, 2H, CHbipy), 9.92 (d, 3J = 4.7 Hz, 2H, CHbipy); IR (ATR, cm−1): ν = 2942 (m), 2892 (w), 1597 (m), 1474 (m), 1236 (s), 1154 (m), 984 (vs), 894 (m), 868 (s), 821 (vs), 757 (s), 737 (s), 662 (s), 609 (w), 427 (w); elemental analysis calculated (C22H44ZnN2Si4 542.34 g/mol) C 48.72 H 8.18 N 10.33; experimental C 48.26 H 7.89 N 10.43. HR-LIFDI-MS (calculated for C22H44ZnN2Si4: 540.1935 m/z) = 540.1932 m/z. Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 9 in Et2O at −35 °C.
[K{18c6}][Zn(N(SiMe3)2)2(bipy)] (10): Compound 9 (54.2 mg, 0.10 mmol, 1.0 equivalent), 18-crown-6 (26.4 mg, 0.1 mmol, 1.0 equivalent) and KC8 (20.3 mg, 0.1 mmol, 1.5 equivalent) were dissolved in 3 mL of Et2O, which led to an immediate color change from pale yellow to dark purple. After stirring for 5 min the solution was layered with 3 mL of pentane. Storing the solution at −35 °C for days yielded a dark red crystalline solid. The solution was removed via a Pasteur pipette. Washing of the residue with 2 × 5 mL of pentane and drying it under reduced pressure afforded compound 10 as a dark red crystalline solid (59.5 mg, 0.07 mmol, 70%). 1H-NMR ([D8]THF, 500.1 MHz, 298 K, ppm): δ = 0.53 (bs, 36H, w½ = 139 Hz, Si(CH3)3), 3.63 (s, 24H, w½ = 6 Hz, OCH2), 4.18 (s, 2H, w½ = 20 Hz, CHbipy), 4.95 (bs, 2H, w½ = 20 Hz, CHbipy), 5.81 (s, 2H, w½ = 24 Hz, CHbipy), 6.72 (s, 2H, w½ = 18 Hz, CHbipy); IR (ATR, cm−1): ν = 2941 (m), 2893 (m), 1570 (w), 1486 (m), 1473 (w), 1454 (w), 1351 (m), 1283 (w), 1233 (s), 1104 (vs), 989 (vs), 961 (s), 870 (s), 823 (vs), 780 (w), 748(m), 716 (m), 658 (m), 608 (m), 529 (m), 452 (w), 415 (w); elemental analysis calculated (C34H68ZnKN4O6Si4 845.75 g/mol) C 48.28 H 8.10 N 6.62; experimental C 48.49 H 7.86 N 6.59. μeff = 1.82 μB ([D8]THF). Crystals, suitable for X-ray diffraction analysis, were obtained from a pentane layered solution of compound 10 in Et2O at −35 °C.
4.2. X-ray Diffraction Data
Data for compounds 3 (CCDC 1918204), 4 (CCDC 1918205) and 10 (CCDC 1918207) were collected at 100 K on a Bruker Quest D8 diffractometer (Bruker Corporation, Billerica, MA, USA) using a graphite-monochromated Mo Kα radiation and equipped with an Oxford Instrument Cooler Device (Oxford Instruments, Abingdon, UK). Data for compounds 8 (CCDC 1918202), 1 (CCDC 1918208) and 9 (CCDC 1918206) were collected at 100 K on a STOE IPDS2 diffractometer (STOE & Cie GmbH, Darmstadt, Germany) and data for compound 2 (CCDC 1918203) were collected at 100 K on a STOE IPDS2T diffractometer using a graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and equipped with an Oxford Cryosystems Cryostream Cooler Device (Oxford Instruments, Abingdon, UK). The structures have been solved using either OLEX SHELXT V2014/1 [63] and refined by means of least-squares procedures on an F2 with the aid of the program SHELXL-2016/6 [64] included in the software package WinGX version 1.63 [65] or using CRYSTALS [66]. The atomic scattering factors were taken from International Tables for X-ray Crystallography [67]. All non-hydrogen atoms were refined anisotropically. All hydrogens atoms were refined by using a riding model. Absorption corrections were introduced by using the MULTISCAN and X-Red programs [68,69]. Drawings of molecules are performed with the program DIAMOND (Crystal Impact, Bonn, Germany) with 50% probability displacement ellipsoids for non-H atoms. Depiction of H atoms is omitted for clarity.
Crystal Data for 1 (C42H84CrKN4O8Si4 x (thf)2, 976.59 g/mol): Triclinic, space group P−1, a = 11.6269(2) Å, b = 21.5546(4) Å, c = 21.8560(4) Å, α = 95.633(2)°, β = 89.8920(10)°, γ = 90.377(2)°, V = 5450.83(17) Å3, Z = 4, T = 100.0 K, μ(Mo Kα) = 0.710 mm−1, ρcalc = 1.190 g/cm3, 88,325 reflections measured (3.504° ≤ 2Θ ≤ 50.0°), 19,148 unique (Rint = 0.0630, Rsigma = 0.0368), which were used in all calculations. The final R1 was 0.0512 (I > 2σ(I)) and wR2 was 0.1323 (all data).
Crystal data for 2 (C34H68MnKN4O6Si4, 835.32 g/mol): Monoclinic, space group C2, a = 34.8403(17) Å, b = 18.4715(11) Å, c = 14.5302(8) Å, α = 90.0, β = 96.555(4)°, γ = 90.0°, V = 9289.8(9) Å3, Z = 8, T = 100.0 K, μ(Mo Kα) = 0.710 mm−1, ρcalc = 1.194 g/cm3, 52,126 reflections measured (3.668° ≤ 2Θ ≤ 54.998°), 21354 unique (Rint = 0.0285, Rsigma = 0.0488), which were used in all calculations. The final R1 was 0.0376 (I > 2σ(I)) and wR2 was 0.0815 (all data).
Crystal data for 3 (C42H84FeKN4O8Si4 x (thf)2, 978.61 g/mol): Triclinic, space group P−1, a = 10.8613(5) Å, b = 13.2627(6) Å, c = 19.8669(9) Å, α = 77.5970(10)°, β = 80.062(2)°, γ = 83.452(2)°, V = 2744.3(2) Å3, Z = 2, T = 100.0 K, μ(Mo-Kα) = 0.710 mm−1, ρcalc = 1.184 g/cm3, 103,782 reflections measured (4.646 ° ≤ 2Θ ≤ 55.124°), 12,642 unique (Rint = 0.0332, Rsigma = 0.0201), which were used in all calculations. The final R1 was 0.0279 (I > 2σ(I)) and wR2 was 0.0658 (all data).
Crystal data for 4 (C42H84CoKN4O8Si4 x (thf)2, 978.61 g/mol): Triclinic, space group P−1, a = 10.8097(5) Å, b = 13.2648(6) Å, c = 19.9051(10) Å, α = 77.712 (2)°, β = 80.199(2)°, γ = 83.606(2)°, V = 2739.7(2) Å3, Z = 2, T = 100.0 K, μ(Mo Kα) = 0.710 mm−1, ρcalc = 1.192 g/cm3, 89,930 reflections measured (4.238° ≤ 2Θ ≤ 55.058°), 12,549 unique (Rint = 0.0853, Rsigma = 0.0565) which were used in all calculations. The final R1 was 0.0420 (I > 2σ(I)) and wR2 was 0.0831 (all data).
Crystal data for 8 (C22H44CoKN4Si4, 535.90 g/mol): Orthorhombic, space group Pca21, a = 18.7674(9) Å, b = 18.1030(8) Å, c = 17.5930(7) Å, α = β = γ = 90.0°, V = 5977.2(5) Å3, Z = 8, T = 100.0 K, μ(Mo Kα) = 0.710 mm−1, ρcalc = 1.191 g/cm3, 31,013 reflections measured (3.88° ≤ 2Θ ≤ 52.0°), 10,939 unique (Rint = 0.0280, Rsigma = 0.0312), which were used in all calculations. The final R1 was 0.0280 (I > 2σ(I)) and wR2 was 0.0689 (all data).
Crystal data for 9 (C22H44ZnKN4Si4, 542.34 g/mol): Orthorhombic, space group Pca21, a = 18.8806(12) Å, b = 18.0560(13) Å, c = 17.6133(17) Å, α = β = γ = 90.0°, V = 6004.5(8) Å3, Z = 8, T = 100.0 K, μ(Mo Kα) = 0.710 mm−1, ρcalc = 1.200 g/cm3, 52,603 reflections measured (4.314° ≤ 2Θ ≤ 51.996°), 11,789 unique (Rint = 0.0737, Rsigma = 0.0589), which were used in all calculations. The final R1 was 0.0340 (I > 2σ(I)) and wR2 was 0.0718 (all data).
Crystal data for 10 (C38H78MZnKN4O7Si4 x (Et2O), 919.87 g/mol): Monoclinic, space group P21/c, a = 12.1899(7) Å, b = 18.4365(10) Å, c = 22.8273(13) Å, α = 90.0, β = 95.432(2)°, γ = 90.0°, V = 5107.2(5) Å3, Z = 4, T = 100.0 K, μ(Mo Kα) = 0.710 mm−1, ρcalc = 1.196 g/cm3, 96,672 reflections measured (4.268° ≤ 2Θ ≤ 49.998°), 8987 unique (Rint = 0.0946, Rsigma = 0.0427) which were used in all calculations. The final R1 was 0.1288 (I > 2σ(I)) and wR2 was 0.2616 (all data).
5. Conclusions
We presented the facile reaction of two-coordinate 3d-metal(I) silylamides (Cr−Co) with 2,2′-bipyridine. Obtained complexes of the form K{18-crown-6}[M(hmds)2(bipy)] were structurally and spectroscopically characterized. Thereby, the isolation of the manganese complex [Mn(hmds)2(bipy)]− shows that the so-far elusive monomeric manganese(I) hexamethyldisilazide [Mn(hmds)2]− can be trapped in synthetically acceptable yields. The anionic complexes were examined with respect to their structural features in solid state as well as their 1H-NMR and UV/Vis spectroscopic and electrochemical signatures in conjunction with their magnetic properties. Comparisons with their neutral, not-reduced counterparts are drawn. Overall, the obtained anionic complexes are described best as [MII(hmds)2(bipy·−)]−; 2,2′-bipyridine is thus reduced to a certain extent upon coordination, even if the reduction potentials of the employed metal(I) complexes can be lower by up to 1 V than the one of free 2,2′-bipyridine (as in case of cobalt). This indicates that, although the used metal(I) complexes are reluctant to coordinate donor ligands, electron transfer to a substrate and its persistent coordination in a reduced state is possible. Ongoing studies now focus on the use of these metal(I) complexes as a valuable tool to obtain and stabilize more unusual organic radical anions.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/7/10/117/s1: The CIF and the checkCIF output files. 1H-NMR spectra, UV/Vis spectra and cyclovoltammograms of all compounds. Magnetic susceptibility data of compounds 1–4 and 10.
Author Contributions
Conceptualization, C.G.W.; Data curation, I.M., C.S., C.P., C.G.W.; Formal analysis, I.M., C.P., C.G.W.; Funding acquisition, C.G.W.; Investigation, I.M., C.S., C.G.W.; Project administration, C.G.W.; Resources, F.K., C.G.W.; Validation, C.G.W.; Visualization, I.M., C.S., C.G.W.; Writing—original draft, I.M., C.G.W.; Writing—review & editing, I.M., C.S., C.P., F.K., C.G.W.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG), grant number WE 5627/4-1.
Acknowledgments
The authors thank the Department of Chemistry of the Philipps University for support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Power, P.P. Stable two-coordinate, open-shell (d1-d9) transition metal complexes. Chem. Rev. 2012, 112, 3482–3507. [Google Scholar] [PubMed]
- Ung, G.; Rittle, J.; Soleilhavoup, M.; Bertrand, G.; Peters, J.C. Two-Coordinate Fe0 and Co0 Complexes Supported by Cyclic (alkyl) (amino) carbenes. Angew. Chem. Int. Ed. 2014, 53, 8427–8431. [Google Scholar]
- Roy, S.; Mondal, K.C.; Roesky, H.W. Cyclic Alkyl (amino) Carbene Stabilized Complexes with Low Coordinate Metals of Enduring Nature. Acc. Chem. Res. 2016, 49, 357–369. [Google Scholar] [PubMed]
- Mo, Z.; Ouyang, Z.; Wang, L.; Fillman, K.L.; Neidig, M.L.; Deng, L. Two- and three-coordinate formal iron(I) compounds featuring monodentate aminocarbene ligands. Org. Chem. Front. 2014, 1, 1040–1044. [Google Scholar]
- Samuel, P.P.; Mondal, K.C.; Amin Sk, N.; Roesky, H.W.; Carl, E.; Neufeld, R.; Stalke, D.; Demeshko, S.; Meyer, F.; Ungur, L.; et al. Electronic Structure and Slow Magnetic Relaxation of Low-Coordinate Cyclic Alkyl(amino) Carbene Stabilized Iron(I) Complexes. J. Am. Chem. Soc. 2014, 136, 11964–11971. [Google Scholar] [PubMed]
- Werncke, C.G.; Bunting, P.C.; Duhayon, C.; Long, J.R.; Bontemps, S.; Sabo-Etienne, S. Two-coordinate iron(I) complex [Fe{N(SiMe3)2}2]−: Synthesis, properties, and redox activity. Angew. Chem. Int. Ed. 2015, 54, 245–248. [Google Scholar]
- Lin, C.-Y.; Fettinger, J.C.; Grandjean, F.; Long, G.J.; Power, P.P. Synthesis, Structure, and Magnetic and Electrochemical Properties of Quasi-Linear and Linear Iron(I), Cobalt(I), and Nickel(I) Amido Complexes. Inorg. Chem. 2014, 53, 9400–9406. [Google Scholar] [PubMed]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron(I) complex. Nat. Chem. 2013, 5, 577–581. [Google Scholar]
- Lipschutz, M.I.; Yang, X.; Chatterjee, R.; Tilley, T.D. A Structurally Rigid Bis (amido) Ligand Framework in Low-Coordinate Ni(I), Ni(II), and Ni(III) Analogues Provides Access to a Ni(III) Methyl Complex via Oxidative Addition. J. Am. Chem. Soc. 2013, 135, 15298–15301. [Google Scholar]
- Cai, I.C.; Lipschutz, M.I.; Tilley, T.D. A bis (amido) ligand set that supports two-coordinate chromium in the +1, +2, and +3 oxidation states. Chem. Commun. 2014, 50, 13062–13065. [Google Scholar]
- Samuel, P.P.; Neufeld, R.; Chandra Mondal, K.; Roesky, H.W.; Herbst-Irmer, R.; Stalke, D.; Demeshko, S.; Meyer, F.; Rojisha, V.C.; De, S.; et al. Cr(I)Cl as well as Cr+ are stabilised between two cyclic alkyl amino carbenes. Chem. Sci. 2015, 6, 3148–3153. [Google Scholar] [PubMed]
- Samuel, P.P.; Mondal, K.C.; Roesky, H.W.; Hermann, M.; Frenking, G.; Demeshko, S.; Meyer, F.; Stückl, A.C.; Christian, J.H.; Dalal, N.S.; et al. Synthesis and Characterization of a Two-Coordinate Manganese Complex and its Reaction with Molecular Hydrogen at Room Temperature. Angew. Chem. Int. Ed. 2013, 52, 11817–11821. [Google Scholar]
- Werncke, C.G.; Suturina, E.; Bunting, P.C.; Vendier, L.; Long, J.R.; Atanasov, M.; Neese, F.; Sabo-Etienne, S.; Bontemps, S. Homoleptic Two-Coordinate Silylamido Complexes of Chromium(I), Manganese(I), and Cobalt(I). Chem. Eur. J. 2016, 22, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Werncke, C.G.; Pfeiffer, J.; Müller, I.; Vendier, L.; Sabo-Etienne, S.; Bontemps, S. C–Halide bond cleavage by a two-coordinate iron(I) complex. Dalton Trans. 2019, 48, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Lipschutz, M.I.; Tilley, T.D. Carbon–Carbon Cross-Coupling Reactions Catalyzed by a Two-Coordinate Nickel(II)-Bis (amido) Complex via Observable NiI, NiII, and NiIII Intermediates. Angew. Chem. Int. Ed. 2014, 53, 7290–7294. [Google Scholar]
- Lipschutz, M.I.; Chantarojsiri, T.; Dong, Y.; Tilley, T.D. Synthesis, characterization, and alkyne trimerization catalysis of a heteroleptic two-coordinate Fe(I) complex. J. Am. Chem. Soc. 2015, 137, 6366–6372. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Z.; Du, J.; Wang, L.; Kneebone, J.L.; Neidig, M.L.; Deng, L. Linear and T-Shaped Iron(I) Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis and Structure Characterization. Inorg. Chem. 2015, 54, 8808–8816. [Google Scholar] [CrossRef]
- Hicks, J.; Jones, C. Low-Coordinate Cobalt(I) Complexes Stabilized by an Extremely Bulky Amide Ligand. Organometallics 2015, 34, 2118–2121. [Google Scholar] [CrossRef]
- Pfirrmann, S.; Limberg, C.; Herwig, C.; Stösser, R.; Ziemer, B. A dinuclear nickel(I) dinitrogen complex and its reduction in single-electron steps. Angew. Chem. Int. Ed. 2009, 48, 3357–3361. [Google Scholar] [CrossRef]
- MacLeod, K.C.; Lewis, R.A.; DeRosha, D.E.; Mercado, B.Q.; Holland, P.L. C-H and C-N Activation at Redox-Active Pyridine Complexes of Iron. Angew. Chem. Int. Ed. 2017, 56, 1069–1072. [Google Scholar] [CrossRef]
- Dugan, T.R.; Sun, X.; Rybak-Akimova, E.V.; Olatunji-Ojo, O.; Cundari, T.R.; Holland, P.L. A Masked Two-Coordinate Cobalt(I) Complex That Activates C–F Bonds. J. Am. Chem. Soc. 2011, 133, 12418–12421. [Google Scholar] [CrossRef] [PubMed]
- Constable, E.C. Homoleptic Complexes of 2,2′-Bipyridine. In Advances in Inorganic Chemistry, 1st ed.; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1989; Volume 34, pp. 1–63. [Google Scholar]
- Gore-Randall, E.; Irwin, M.; Denning, M.S.; Goicoechea, J.M. Synthesis and characterization of alkali-metal salts of 2,2′- and 2,4′-bipyridyl radicals and dianions. Inorg. Chem. 2009, 48, 8304–8316. [Google Scholar] [CrossRef] [PubMed]
- Denning, M.S.; Irwin, M.; Goicoechea, J.M. Synthesis and characterization of the 4,4’-bipyridyl dianion and radical monoanion. A structural study. Inorg. Chem. 2008, 47, 6118–6120. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.; Lehn, J.-M.; Pauls, J.; Holl, S.; Krenzel, V. Sodium Salts of the Bipyridine Dianion: Polymer [(bpy)2−{Na+(dme)}2]∞, Cluster [(Na8O)6+Na+6(bpy)62−(tmeda)6], and Monomer [(bpy)2−{Na+(pmdta)}2]. Angew. Chem. Int. Ed. 1999, 38, 952–955. [Google Scholar] [CrossRef]
- Roitershtein, D.; Domingos, A.; Pereira, L.C.J.; Ascenso, J.R.; Marques, N. Coordination of 2,2′-bipyridyl and 1,10-phenanthroline to yttrium and lanthanum complexes based on a scorpionate ligand. Inorg. Chem. 2003, 42, 7666–7673. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, P.L.; Corey, E.R.; Corey, J.Y.; Hey, G.W. Synthesis and characterization of complexes of aluminum halide with 2,2′-bipyridine, 1,10-phenanthroline and 2,2′,2′′-terpyridine in acetonitrile. Inorg. Chem. 1977, 16, 462–467. [Google Scholar] [CrossRef]
- Nikiforov, G.B.; Roesky, H.W.; Noltemeyer, M.; Schmidt, H.-G. Reactivity of Ti(bipy)3 and preparation of the Li(THF)4[Al(bipy)2] complex with the dinegative bipy ligand. Polyhedron 2004, 23, 561–566. [Google Scholar] [CrossRef]
- Jacquot, L.; Xémard, M.; Clavaguéra, C.; Nocton, G. Multiple One-Electron Transfers in Bipyridine Complexes of Bis (phospholyl) Thulium. Organometallics 2014, 33, 4100–4106. [Google Scholar] [CrossRef]
- Ortu, F.; Liu, J.; Burton, M.; Fowler, J.M.; Formanuik, A.; Boulon, M.-E.; Chilton, N.F.; Mills, D.P. Analysis of Lanthanide-Radical Magnetic Interactions in Ce(III) 2,2′-Bipyridyl Complexes. Inorg. Chem. 2017, 56, 2496–2505. [Google Scholar] [CrossRef]
- Ortu, F.; Zhu, H.; Boulon, M.-E.; Mills, D. Synthesis and Reactivity of a Cerium(III) Scorpionate Complex Containing a Redox Non-Innocent 2,2′-Bipyridine Ligand. Inorganics 2015, 3, 534–553. [Google Scholar] [CrossRef]
- Scarborough, C.C.; Wieghardt, K. Electronic structure of 2,2′-bipyridine organotransition-metal complexes. Establishing the ligand oxidation level by density functional theoretical calculations. Inorg. Chem. 2011, 50, 9773–9793. [Google Scholar] [CrossRef] [PubMed]
- Wolff, C.; Gottschlich, A.; England, J.; Wieghardt, K.; Saak, W.; Haase, D.; Beckhaus, R. Molecular and Electronic Structures of Mononuclear and Dinuclear Titanium Complexes Containing π-Radical Anions of 2,2′-Bipyridine and 1,10-Phenanthroline: An Experimental and DFT Computational Study. Inorg. Chem. 2015, 54, 4811–4820. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.; Jenkins, R.K.; Denning, M.S.; Kramer, T.; Grandjean, F.; Long, G.J.; Herchel, R.; McGrady, J.E.; Goicoechea, J.M. Experimental and computational study of the structural and electronic properties of FeII(2,2′-bipyridine)(mes)2 and [FeII(2,2′-bipyridine)(mes)2]−, a complex containing a 2,2′-bipyridyl radical anion. Inorg. Chem. 2010, 49, 6160–6171. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.; Doyle, L.R.; Krämer, T.; Herchel, R.; McGrady, J.E.; Goicoechea, J.M. A homologous series of first-row transition-metal complexes of 2,2′-bipyridine and their ligand radical derivatives: Trends in structure, magnetism, and bonding. Inorg. Chem. 2012, 51, 12301–12312. [Google Scholar] [CrossRef] [PubMed]
- Tokel-Takvoryan, N.E.; Hemingway, R.E.; Bard, A.J. Electrogenerated chemiluminescence. XIII. Electrochemical and electrogenerated chemiluminescence studies of ruthenium chelates. J. Am. Chem. Soc. 1973, 95, 6582–6589. [Google Scholar] [CrossRef]
- Bradley, D.C.; Hursthouse, M.B.; Newing, C.W.; Welch, A.J. Square planar and tetrahedral chromium (II) complexes; crystal structure determinations. Chem. Commun. 1972, 567. [Google Scholar] [CrossRef]
- Cotton, F.A.; Rice, C.E.; Rice, G.W. The crystal and molecular structures of bis (2,4-pentanedionato) chromium. Inorg. Chim. Acta 1977, 24, 231–234. [Google Scholar] [CrossRef]
- Babar, M.A.; Ladd, M.F.C.; Larkworthy, L.F.; Povey, D.C.; Proctor, K.J.; Summers, L.J. The crystal structures of propane-1,3-diammonium tetrachlorochromate(II), a sheet ferromagnet, and bis (dimethylammonium) tetrachlorochromate(II) an antiferromagnetic compound containing isolated [Cr3Cl12]6− units. Chem. Commun. 1981, 1046. [Google Scholar] [CrossRef]
- Scarborough, C.C.; Sproules, S.; Weyhermüller, T.; DeBeer, S.; Wieghardt, K. Electronic and molecular structures of the members of the electron transfer series [Cr(tbpy)3]n (n = 3+, 2+, 1+, 0): An X-ray absorption spectroscopic and density functional theoretical study. Inorg. Chem. 2011, 50, 12446–12462. [Google Scholar] [CrossRef]
- Schultz, M.; Boncella, J.M.; Berg, D.J.; Tilley, T.D.; Andersen, R.A. Coordination of 2,2′-Bipyridyl and 1,10-Phenanthroline to Substituted Ytterbocenes: An Experimental Investigation of Spin Coupling in Lanthanide Complexes. Organometallics 2002, 21, 460–472. [Google Scholar] [CrossRef]
- Zhou, W.; Desnoyer, A.N.; Bailey, J.A.; Patrick, B.O.; Smith, K.M. Direct synthesis of ligand-based radicals by the addition of bipyridine to chromium(II) compounds. Inorg. Chem. 2013, 52, 2271–2273. [Google Scholar] [CrossRef] [PubMed]
- Margraf, G.; Schödel, F.; Sänger, I.; Bolte, M.; Wagner, M.; Lerner, H.-W. Eine elektrochemische und strukturelle Studie an den Eisensilylamiden Fe[N(SiMe3)2]2 und Fe[N(SiMe3)2]3 / An Electrochemical and Structural Study of the Iron Silylamides Fe[N(SiMe3)2]2 and Fe[N(SiMe3)2]3. Z. Naturforsch. B Chem. Sci. 2012, 67, 549–556. [Google Scholar] [CrossRef]
- König, E.; Kremer, S. The lower excited electronic states of singly and doubly reduced 2,2′-bipyridine. Chem. Phys. Lett. 1970, 5, 87–90. [Google Scholar] [CrossRef]
- Da Re, R.E.; Kuehl, C.J.; Brown, M.G.; Rocha, R.C.; Bauer, E.D.; John, K.D.; Morris, D.E.; Shreve, A.P.; Sarrao, J.L. Electrochemical and spectroscopic characterization of the novel charge-transfer ground state in diimine complexes of ytterbocene. Inorg. Chem. 2003, 42, 5551–5559. [Google Scholar] [CrossRef]
- Schubert, E.M. Utilizing the Evans method with a superconducting NMR spectrometer in the undergraduate laboratory. J. Chem. Educ. 1992, 69, 62. [Google Scholar] [CrossRef]
- Evans, D.F. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003. [Google Scholar] [CrossRef]
- Awaga, K.; Maruyama, Y. Ferromagnetic and antiferromagnetic intermolecular interactions of organic radicals, α-nitronyl nitroxides. II. Phys. Chem. Chem. Phys. 1989, 91, 2743–2747. [Google Scholar] [CrossRef]
- Fujita, W.; Awaga, K. Room-Temperature Magnetic Bistability in Organic Radical Crystals. Science 1999, 286, 261–263. [Google Scholar] [CrossRef]
- Eusterwiemann, S.; Doerenkamp, C.; Dresselhaus, T.; Janka, O.; de Oliveira, M.; Daniliuc, C.G.; Eckert, H.; Neugebauer, J.; Pöttgen, R.; Studer, A. Strong intermolecular antiferromagnetic verdazyl-verdazyl coupling in the solid state. Phys. Chem. Chem. Phys. PCCP 2017, 19, 15681–15685. [Google Scholar] [CrossRef]
- Roffia, S.; Marcaccio, M.; Paradisi, C.; Paolucci, F.; Balzani, V.; Denti, G.; Serroni, S.; Campagna, S. Electrochemical reduction of (2,2′-bipyridine)- and bis ((2-pyridyl) pyrazine) ruthenium(II) complexes used as building blocks for supramolecular species. Redox series made of 8, 10, and 12 redox steps. Inorg. Chem. 1993, 32, 3003–3009. [Google Scholar] [CrossRef]
- Margel, S. Electrochemistry of 2,2′-Bipyridine Complexes of Cobalt in the Presence of Acrylonitrile. J. Electrochem. Soc. 1978, 125, 241. [Google Scholar] [CrossRef]
- Braterman, P.S.; Song, J.I.; Peacock, R.D. Electronic absorption spectra of the iron(II) complexes of 2,2′-bipyridine, 2,2′-bipyrimidine, 1,10-phenanthroline, and 2,2′:6′,2′′-terpyridine and their reduction products. Inorg. Chem. 1992, 31, 555–559. [Google Scholar] [CrossRef]
- De Bruin, B.; Bill, E.; Bothe, E.; Weyhermüller, T.; Wieghardt, K. Molecular and Electronic Structures of Bis (pyridine-2,6-diimine) metal Complexes [ML2](PF6)n (n = 0, 1, 2, 3; M = Mn, Fe, Co, Ni, Cu, Zn). Inorg. Chem. 2000, 39, 2936–2947. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.P.; Bolinger, C.M.; Conrad, D.; Vining, W.J.; Meyer, T.J. One- and two-electron pathways in the electrocatalytic reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine). Chem. Commun. 1985, 1414–1416. [Google Scholar] [CrossRef]
- La Mar, G.N.; Horrocks, W.D.; Holm, R.H. NMR of Paramagnetic Molecules: Principles and Applications; Elsevier Science: Burlington, NJ, USA, 1973. [Google Scholar]
- Bertini, I.; Luchinat, C.; Parigi, G.; Pierattelli, R. NMR spectroscopy of paramagnetic metalloproteins. Chembiochem 2005, 6, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Bürger, H.; Wannagat, U. Silylamido-Derivate von Eisen und Kobalt. Monatsh. Chem. 1963, 94, 1007–1012. [Google Scholar] [CrossRef]
- Bürger, H.; Wannagat, U. Silylamido-Verbindungen von Chrom, Mangan, Nickel und Kupfer. Monatsh. Chem. 1964, 95, 1099–1102. [Google Scholar] [CrossRef]
- Conley, M.P.; Delley, M.F.; Siddiqi, G.; Lapadula, G.; Norsic, S.; Monteil, V.; Safonova, O.V.; Copéret, C. Polymerization of Ethylene by Silica-Supported Dinuclear CrIII Sites through an Initiation Step Involving C–H Bond Activation. Angew. Chem. Int. Ed. 2014, 53, 1872–1876. [Google Scholar] [CrossRef]
- Dalvit, C.; Invernizzi, C.; Vulpetti, A. Fluorine as a hydrogen-bond acceptor: Experimental evidence and computational calculations. Chem. Eur. J. 2014, 20, 11058–11068. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- International Union of Crystallography. International Tables for Crystallography, 1st ed.; Springer: Chester, UK; New York, NY, USA, 2006. [Google Scholar]
- Bruker AXS Inc. SADABS; 2016/2; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- STOE&Cie GmbH. X-Area; X.-R. 1.6.1.0; STOE&Cie GmbH: Darmstadt, Germany, 2016. [Google Scholar]











© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).