Tracing the Pathways of Waters and Protons in Photosystem II and Cytochrome c Oxidase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
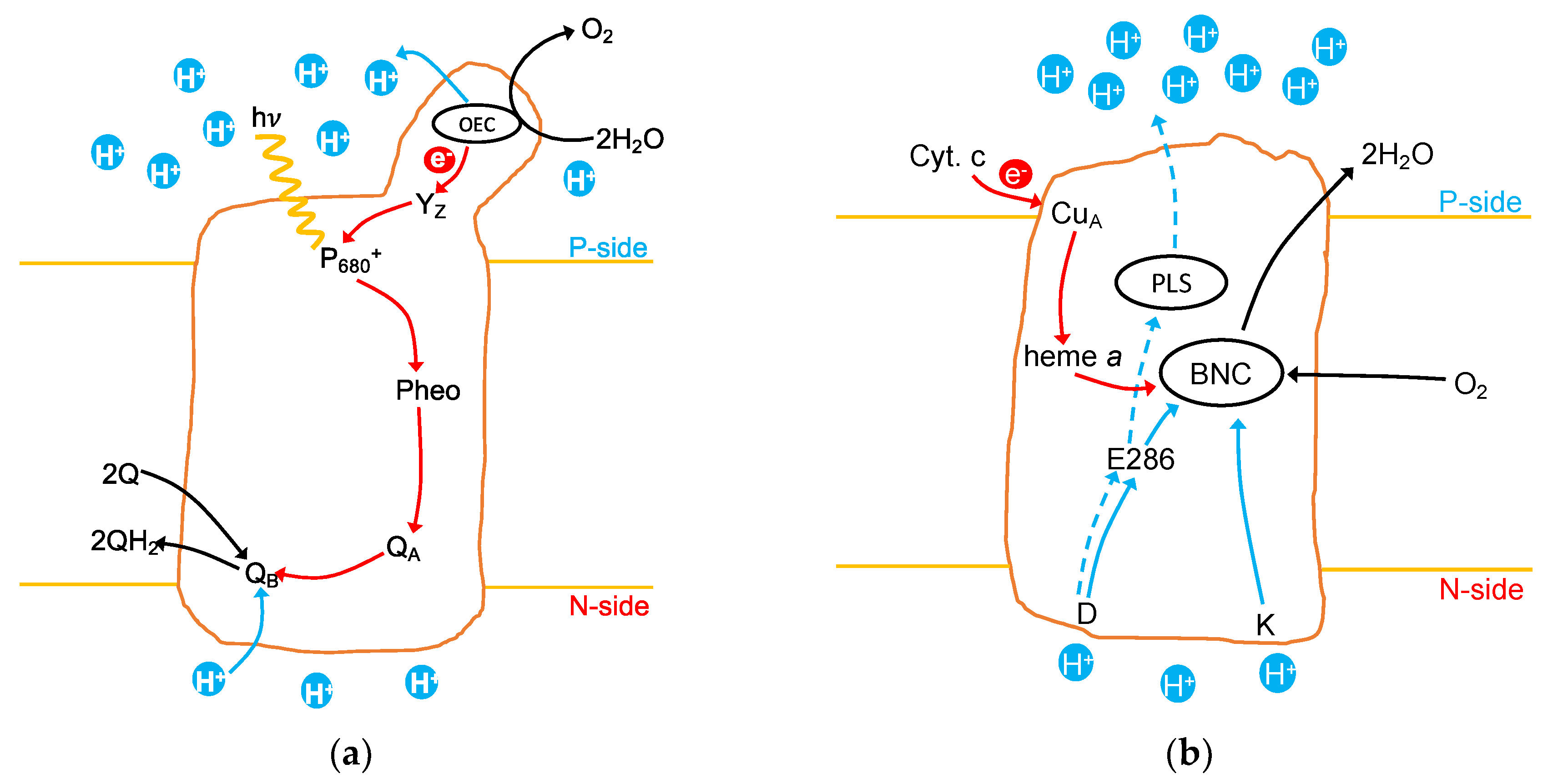
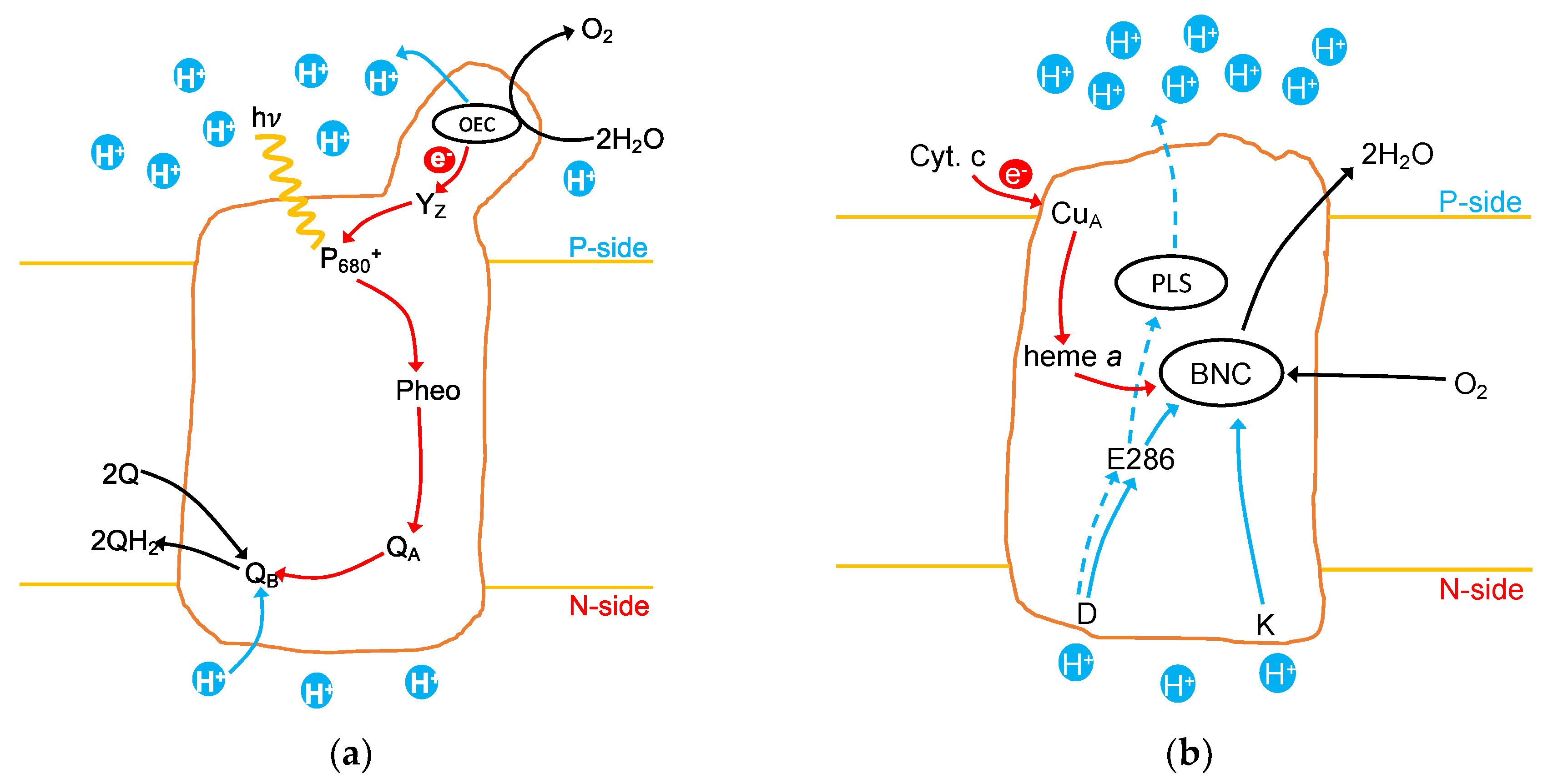
1.1. Photosystem II
1.2. Cytochrome c Oxidase
2. Finding the Protons in the Active Site and the Surrounding Protein
2.1. Methods of Calculation
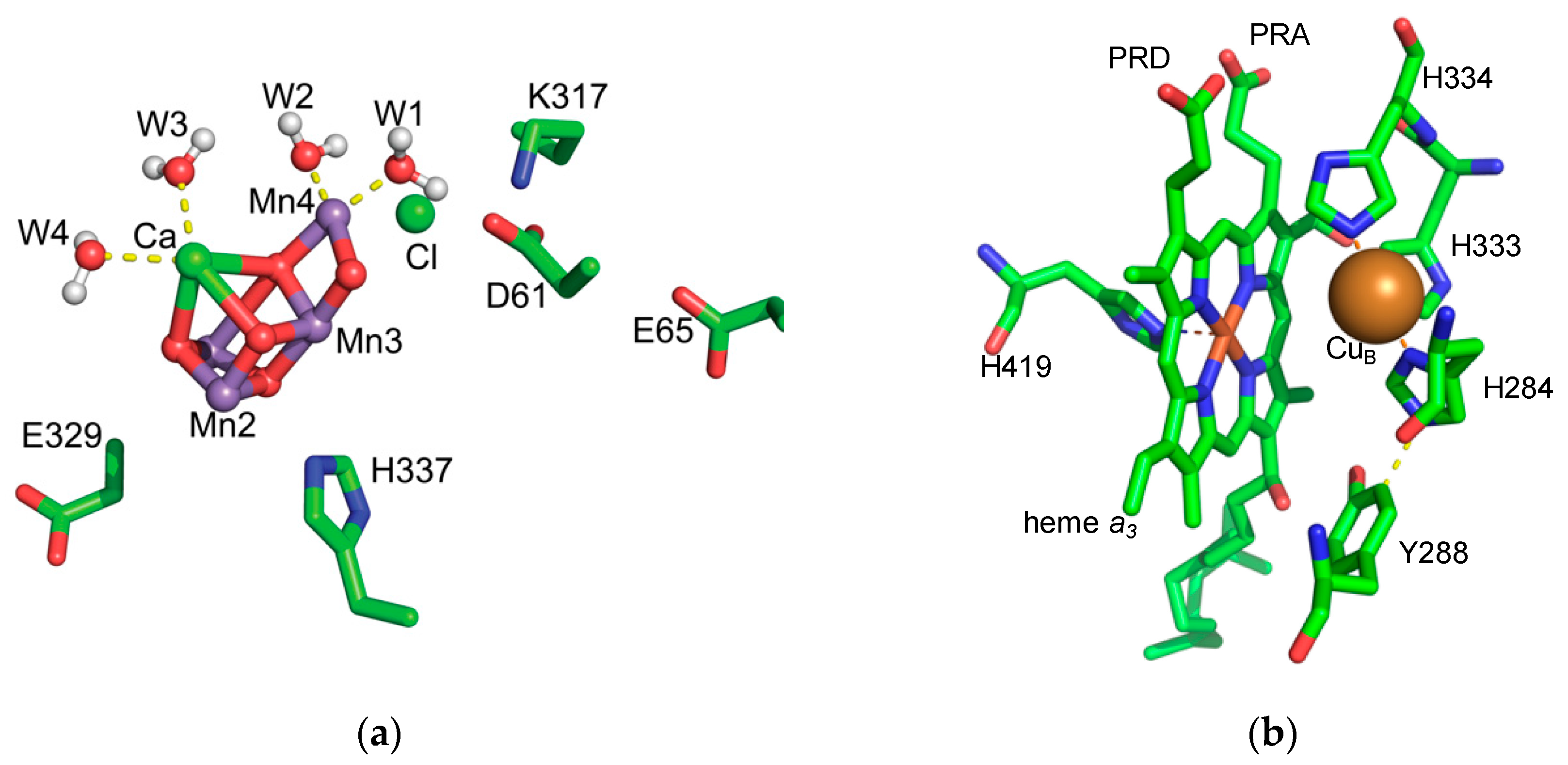
2.2. Changing Protonation States of the OEC through the S-State Cycle
2.3. Changing Protonation States of Residues in the Protein through the Reaction Cycle
2.3.1. PSII Protonation State
2.3.2. CcO Protonation State
3. Finding Water and Proton Transfer Pathways
3.1. Methods to Find Water and Proton Pathways
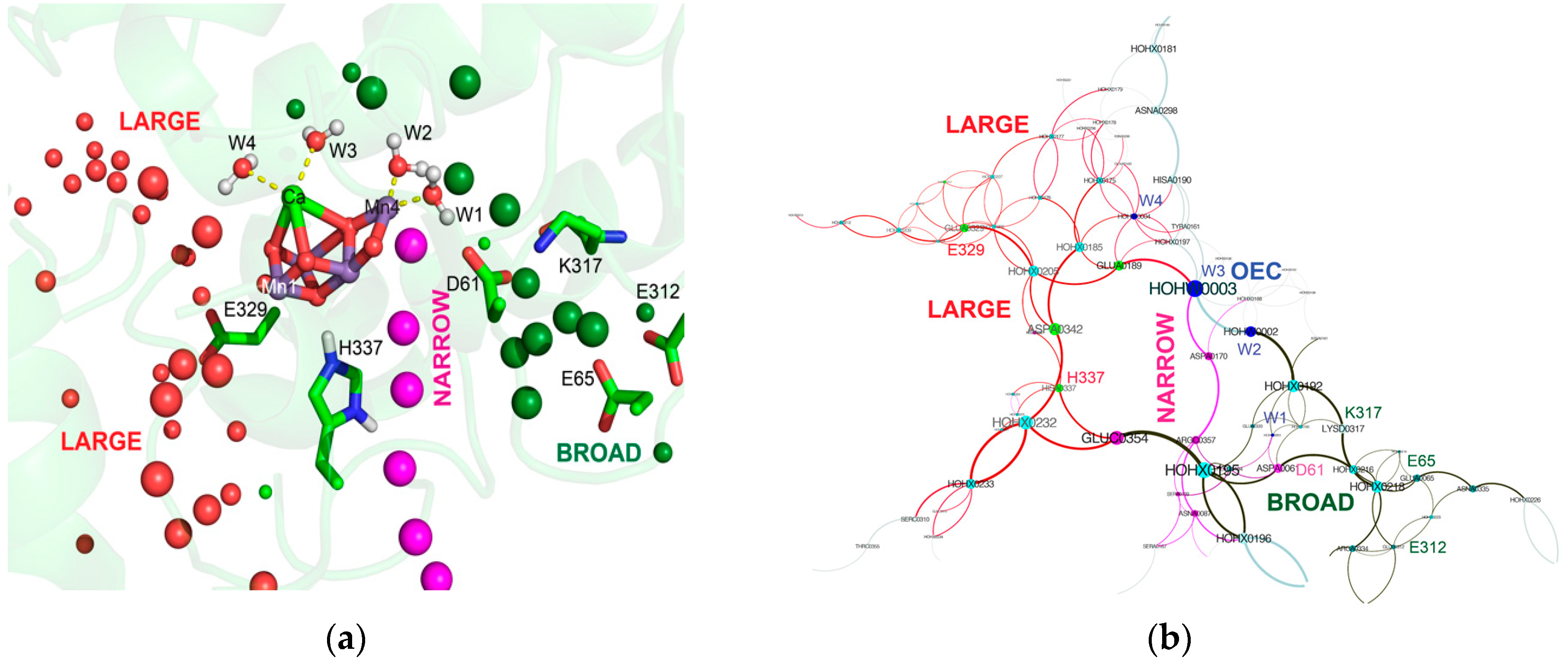
3.2. Water and Proton Transfer Pathways in PSII
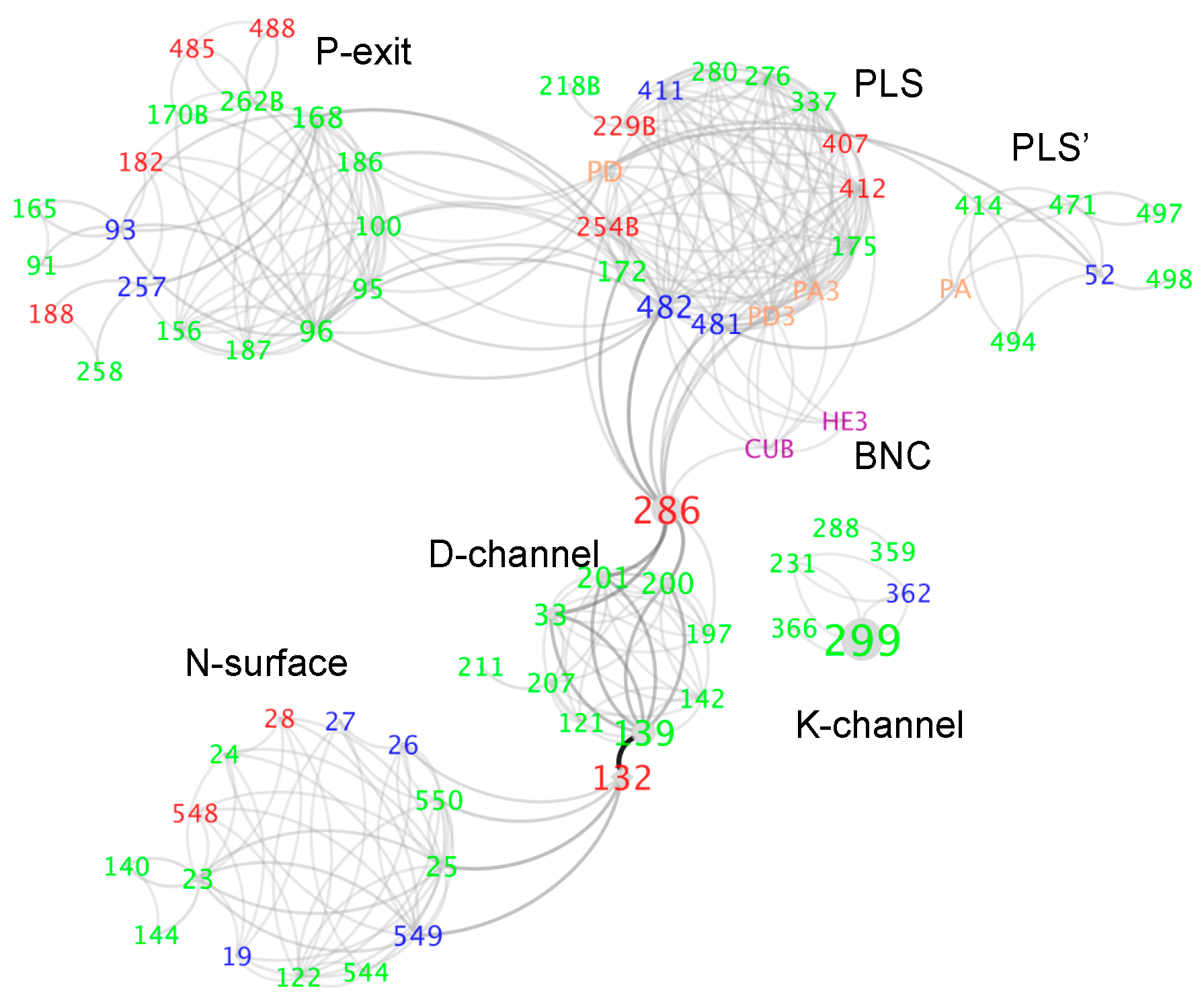
3.3. Water and Proton Transfer Pathways in CcO
3.3.1. The P-Side Exit Path
3.3.2. The Path through the Protein
3.3.3. Testing the Location of Water Channel Experimentally
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Falkowski, P.G.; Godfrey, L.V. Electrons, life and the evolution of Earth’s oxygen cycle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 2705–2716. [Google Scholar] [CrossRef] [PubMed]
- Gunner, M.R.; Amin, M.; Zhu, X.; Lu, J. Molecular mechanisms for generating transmembrane proton gradients. Biochim. Biophys. Acta 2013, 1827, 892–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunner, M.R.; Koder, R. The design features cells use to build their transmembrane proton gradient. Phys. Biol. 2017, 14, 013001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, N.; Messinger, J. Reflections on substrate water and dioxygen formation. Biochim. Biophys. Acta 2013, 1827, 1020–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dau, H.; Zaharieva, I.; Haumann, M. Recent developments in research on water oxidation by photosystem II. Curr. Opin. Chem. Biol. 2012, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Vinyard, D.J.; Brudvig, G.W. Progress toward a molecular mechanism of water oxidation in photosystem II. Annu. Rev. Phys. Chem. 2017, 68, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Styring, S.; Sjöholm, J.; Mamedov, F. Two tyrosines that changed the world: Interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Biochim. Biophys. Acta 2012, 1817, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Joliot, P. Period-four oscillations of the flash-induced oxygen formation in photosynthesis. In Discoveries in Photosynthesis; Govindjee, B.J.T., Gest, H., Allen, J.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 20, pp. 371–378. ISBN 978-1-4020-3323-0. [Google Scholar]
- Kok, B.; Forbush, B.; McGloin, M. Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism. Photochem. Photobiol. 1970, 11, 457–475. [Google Scholar] [CrossRef]
- Nilsson, H.; Cournac, L.; Rappaport, F.; Messinger, J.; Lavergne, J. Estimation of the driving force for dioxygen formation in photosynthesis. Biochim. Biophys. Acta 2016, 1857, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Askerka, M.; Brudvig, G.W.; Batista, V.S. The O2-evolving complex of photosystem II: Recent insights from quantum mechanics/molecular mechanics (QM/MM), extended X-ray absorption fine structure (EXAFS), and femtosecond X-ray crystallography data. Acc. Chem. Res. 2017, 50, 41–48. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Water oxidation mechanism in photosystem II, including oxidations, proton release pathways, O–O bond formation and O2 release. Biochim. Biophys. Acta 2013, 1827, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Kern, J.; Sauer, K.; Latimer, M.J.; Pushkar, Y.; Biesiadka, J.; Loll, B.; Saenger, W.; Messinger, J.; Zouni, A.; et al. Where water is oxidized to dioxygen: Structure of the photosynthetic Mn4Ca cluster. Science 2006, 314, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Ibrahim, M.; Chatterjee, R.; Gul, S.; Fuller, F.; Koroidov, S.; Brewster, A.S.; Tran, R.; Alonso-Mori, R.; Kroll, T.; et al. Structure of photosystem II and substrate binding at room temperature. Nature 2016, 540, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, K.N.; Iverson, T.M.; Maghlaoui, K.; Barber, J.; Iwata, S. Architecture of the photosynthetic oxygen-evolving center. Science 2004, 303, 1831–1838. [Google Scholar] [CrossRef]
- Galstyan, A.; Robertazzi, A.; Knapp, E.W. Oxygen-Evolving Mn Cluster in Photosystem II: The Protonation Pattern and Oxidation State in the High-Resolution Crystal Structure. J. Am. Chem. Soc. 2012, 134, 7442–7449. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Kern, J.; Irrgang, K.-D.; Latimer, M.J.; Bergmann, U.; Glatzel, P.; Pushkar, Y.; Biesiadka, J.; Loll, B.; Sauer, K.; et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA 2005, 102, 12047–12052. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Tran, R.; Alonso-Mori, R.; Koroidov, S.; Echols, N.; Hattne, J.; Ibrahim, M.; Gul, S.; Laksmono, H.; Sierra, R.G.; et al. Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nat. Commun. 2014, 5, 4371. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Alonso-Mori, R.; Tran, R.; Hattne, J.; Gildea, R.J.; Echols, N.; Glöckner, C.; Hellmich, J.; Laksmono, H.; Sierra, R.G.; et al. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science 2013, 340, 491–495. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef]
- Askerka, M.; Vinyard, D.J.; Wang, J.; Brudvig, G.W.; Batista, V.S. Analysis of the radiation-damage-free X-ray structure of photosystem II in light of EXAFS and QM/MM data. Biochemistry 2015, 54, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Ames, W.; Pantazis, D.A.; Krewald, V.; Cox, N.; Messinger, J.; Lubitz, W.; Neese, F. Theoretical evaluation of structural models of the S2 state in the oxygen evolving complex of photosystem II: Protonation states and magnetic interactions. J. Am. Chem. Soc. 2011, 133, 19743–19757. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Sugiura, M.; Noguchi, T. Monitoring proton release during photosynthetic water oxidation in photosystem II by means of isotope-edited infrared spectroscopy. J. Am. Chem. Soc. 2009, 131, 7849–7857. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M. O–O bond formation in the S4 state of the oxygen-evolving complex in photosystem II. Chem. Eur. J. 2006, 12, 9217–9227. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Negre, C.F.; Vogt, L.; Pokhrel, R.; Ertem, M.Z.; Brudvig, G.W.; Batista, V.S. S0-State model of the oxygen-evolving complex of photosystem II. Biochemistry 2013, 52, 7703–7706. [Google Scholar] [CrossRef] [PubMed]
- Luber, S.; Rivalta, I.; Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N.; Brudvig, G.W.; Batista, V.S. S1-state model of the O2-evolving complex of photosystem II. Biochemistry 2011, 50, 6308–6311. [Google Scholar] [CrossRef] [PubMed]
- Askerka, M.; Wang, J.; Brudvig, G.W.; Batista, V.S. Structural changes in the oxygen-evolving complex of photosystem II induced by the S1 to S2 transition: A combined XRD and QM/MM study. Biochemistry 2014, 53, 6860–6862. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Ames, W.; Cox, N.; Lubitz, W.; Neese, F. Two interconvertible structures that explain the spectroscopic properties of the oxygen-evolving complex of photosystem II in the S2 state. Angew. Chem. 2012, 51, 9935–9940. [Google Scholar] [CrossRef]
- Askerka, M.; Wang, J.; Vinyard, D.J.; Brudvig, G.W.; Batista, V.S. S3 state of the O2-evolving complex of photosystem II: Insights from QM/MM, EXAFS, and Femtosecond X-ray Diffraction. Biochemistry 2016, 55, 981–984. [Google Scholar] [CrossRef]
- Retegan, M.; Krewald, V.; Mamedov, F.; Neese, F.; Lubitz, W.; Cox, N.; Pantazis, D.A. A five-coordinate Mn(IV) intermediate in biological water oxidation: Spectroscopic signature and a pivot mechanism for water binding. Chem. Sci. 2015, 7, 72–84. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Substrate water exchange for the oxygen evolving complex in PSII in the S1, S2, and S3 states. J. Am. Chem. Soc. 2013, 135, 9442–9449. [Google Scholar] [CrossRef]
- Cox, N.; Pantazis, D.A.; Neese, F.; Lubitz, W. Biological water oxidation. Acc. Chem. Res. 2013, 46, 1588–1596. [Google Scholar] [CrossRef]
- Bovi, D.; Narzi, D.; Guidoni, L. The S2 state of the oxygen-evolving complex of photosystem II explored by QM/MM dynamics: Spin surfaces and metastable states suggest a reaction path towards the S3 state. Angew. Chem. 2013, 52, 11744–11749. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Orio, M.; Petrenko, T.; Zein, S.; Lubitz, W.; Messinger, J.; Neese, F. Structure of the oxygen-evolving complex of photosystem II: Information on the S2 state through quantum chemical calculation of its magnetic properties. Phys. Chem. Chem. Phys. 2009, 11, 6788–6798. [Google Scholar] [CrossRef]
- Paul, S.; Cox, N.; Pantazis, D.A. What Can We Learn from a Biomimetic Model of Nature’s Oxygen-Evolving Complex? Inorg. Chem. 2017, 56, 3875–3888. [Google Scholar] [CrossRef] [PubMed]
- Boussac, A.; Rutherford, A.W. Comparative study of the g=4.1 EPR signals in the S2 state of photosystem II. Biochim. Biophys. Acta 2000, 1457, 145–156. [Google Scholar] [CrossRef]
- Cox, N.; Retegan, M.; Neese, F.; Pantazis, D.A.; Boussac, A.; Lubitz, W. Electronic structure of the oxygen-evolving complex in photosystem II prior to O–O bond formation. Science 2014, 345, 804–808. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys. Chem. Chem. Phys. 2012, 14, 4849–4856. [Google Scholar] [CrossRef]
- Narzi, D.; Mattioli, G.; Bovi, D.; Guidoni, L. A spotlight on the compatibility between XFEL and ab Initio structures of the oxygen evolving complex in photosystem II. Chem. Eur. J. 2017, 23, 6969–6973. [Google Scholar] [CrossRef]
- Wang, J.; Askerka, M.; Brudvig, G.W.; Batista, V.S. Crystallographic Data Support the Carousel Mechanism of Water Supply to the Oxygen-Evolving Complex of Photosystem II. ACS Energy Lett. 2017, 2, 2299–2306. [Google Scholar] [CrossRef]
- Guo, Y.; Li, H.; He, L.-L.; Zhao, D.-X.; Gong, L.-D.; Yang, Z.-Z. The open-cubane oxo-oxyl coupling mechanism dominates photosynthetic oxygen evolution: A comprehensive DFT investigation on O–O bond formation in the S4 state. Phys. Chem. Chem. Phys. 2017, 19, 13909–13923. [Google Scholar] [PubMed]
- Siegbahn, P.E.M. Nucleophilic water attack is not a possible mechanism for O–O bond formation in photosystem II. Proc. Natl. Acad. Sci. USA 2017, 114, 4966–4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Ugur, I.; Rutherford, A.W.; Kaila, V.R.I. Redox-coupled substrate water reorganization in the active site of Photosystem II—The role of calcium in substrate water delivery. Biochim. Biophys. Acta 2016, 1857, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Popović, D.M. Current advances in research of cytochrome c oxidase. Amino Acids 2013, 45, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Rich, P.R. Mitochondrial cytochrome c oxidase: Catalysis, coupling and controversies. Biochem. Soc. Trans. 2017, 45, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Sharma, V. Proton pumping by cytochrome c oxidase—A 40 year anniversary. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Shimada, A. Reaction mechanism of cytochrome c oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef]
- Sousa, F.L.; Alves, R.J.; Ribeiro, M.A.; Pereira-Leal, J.B.; Teixeira, M.; Pereira, M.M. The superfamily of heme–copper oxygen reductases: Types and evolutionary considerations. Biochim. Biophys. Acta 2012, 1817, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Kaila, V.R.I.; Verkhovsky, M.I.; Wikström, M. Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 2010, 110, 7062–7081. [Google Scholar] [CrossRef]
- Brändén, G.; Gennis, R.B.; Brzezinski, P. Transmembrane proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta 2006, 1757, 1052–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faxén, K.; Gilderson, G.; Adelroth, P.; Brzezinski, P. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature 2005, 437, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Proshlyakov, D.A.; Pressler, M.A.; DeMaso, C.; Leykam, J.F.; DeWitt, D.L.; Babcock, G.T. Oxygen activation and reduction in respiration: Involvement of redox-active tyrosine 244. Science 2000, 290, 1588–1591. [Google Scholar] [CrossRef]
- Gorbikova, E.A.; Belevich, I.; Wikström, M.; Verkhovsky, M.I. The proton donor for O–O bond scission by cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2008, 105, 10733–10737. [Google Scholar] [CrossRef] [PubMed]
- Adelroth, P.; Ek, M.; Brzezinski, P. Factors determining electron-transfer rates in cytochrome c oxidase: Investigation of the oxygen reaction in the R. sphaeroides enzyme. Biochim. Biophys. Acta 1998, 1367, 107–117. [Google Scholar] [CrossRef]
- Verkhovsky, M.I.; Morgan, J.E.; Wikström, M. Oxygen binding and activation: Early steps in the reaction of oxygen with cytochrome c oxidase. Biochemistry 1994, 33, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Belevich, I.; Bloch, D.A.; Belevich, N.; Wikström, M.; Verkhovsky, M.I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. USA 2007, 104, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.A. Mechanism of Oxygen Reduction in Cytochrome c Oxidase and the Role of the Active Site Tyrosine. Biochemistry 2016, 55, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Han Du, W.-G.; Götz, A.W.; Noodleman, L. A water dimer shift activates a proton pumping pathway in the Pr→F Transition of ba3 cytochrome c oxidase. Inorg. Chem. 2018, 57, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Gunner, M.R.; Madeo, J.; Zhu, Z. Modification of quinone electrochemistry by the proteins in the biological electron transfer chains: Examples from photosynthetic reaction centers. J. Bioenerg. Biomembr. 2008, 40, 509–519. [Google Scholar] [CrossRef]
- Limburg, J.; Vrettos, J.S.; Liable-Sands, L.M.; Rheingold, A.L.; Crabtree, R.H.; Brudvig, G.W. A functional model for O–O bond formation by the O2-evolving complex in photosystem II. Science 1999, 283, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tagore, R.; Das, S.; Incarvito, C.; Faller, J.W.; Crabtree, R.H.; Brudvig, G.W. General synthesis of di-μ-oxo dimanganese complexes as functional models for the oxygen evolving complex of photosystem II. Inorg. Chem. 2005, 44, 7661–7670. [Google Scholar] [CrossRef]
- Amin, M.; Vogt, L.; Vassiliev, S.; Rivalta, I.; Sultan, M.M.; Bruce, D.; Brudvig, G.W.; Batista, V.S.; Gunner, M.R. Electrostatic effects on proton coupled electron transfer in oxomanganese complexes inspired by the oxygen-evolving complex of photosystem II. J. Phys. Chem. B 2013, 117, 6217–6226. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Mao, J.; Gunner, M.R. Electrostatic environment of hemes in proteins: pKas of hydroxyl ligands. Biochemistry 2006, 45, 7949–7958. [Google Scholar] [CrossRef] [PubMed]
- Gunner, M.R.; Baker, N.A. Continuum electrostatics approaches to calculating pKas and Ems in proteins. Meth. Enzymol. 2016, 578, 1–20. [Google Scholar] [PubMed]
- Nielsen, J.E.; Gunner, M.R.; García-Moreno, B.E. The pKa Cooperative: A collaborative effort to advance structure-based calculations of pKa values and electrostatic effects in proteins. Proteins 2011, 79, 3249–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexov, E.; Mehler, E.L.; Baker, N.; Baptista, A.M.; Huang, Y.; Milletti, F.; Nielsen, J.E.; Farrell, D.; Carstensen, T.; Olsson, M.H.M.; et al. Progress in the prediction of pKa values in proteins. Proteins 2011, 79, 3260–3275. [Google Scholar] [CrossRef] [Green Version]
- Kieseritzky, G.; Knapp, E.-W. Optimizing pKa computation in proteins with pH adapted conformations. Proteins 2008, 71, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Mao, J.; Gunner, M.R. MCCE2: Improving protein pKa calculations with extensive side chain rotamer sampling. J. Comput. Chem. 2009, 30, 2231–2247. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, B. Karlsberg Online Manual; Freie Universität: Berlin, Germany, 1999. [Google Scholar]
- Gunner, M.R.; Alexov, E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim. Biophys. Acta 2000, 1458, 63–87. [Google Scholar] [CrossRef] [Green Version]
- Di Russo, N.V.; Estrin, D.A.; Martí, M.A.; Roitberg, A.E. pH-Dependent conformational changes in proteins and their effect on experimental pKas: The case of Nitrophorin 4. PLoS Comput. Biol. 2012, 8, e1002761. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cui, Q. Microscopic mechanisms that govern the titration response and pKa values of buried residues in Staphylococcal nuclease mutants. Proteins 2017, 85, 268–281. [Google Scholar] [PubMed]
- Cruzeiro, V.W.D.; Amaral, M.S.; Roitberg, A.E. Redox potential replica exchange molecular dynamics at constant pH in AMBER: Implementation and validation. J. Chem. Phys. 2018, 149, 072338. [Google Scholar] [CrossRef] [PubMed]
- Damjanovic, A.; Miller, B.T.; Okur, A.; Brooks, B.R. Reservoir pH replica exchange. J. Chem. Phys. 2018, 149, 072321. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Miller, B.T.; Brooks, B.R. Computational scheme for pH-dependent binding free energy calculation with explicit solvent. Protein Sci. 2016, 25, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Machuqueiro, M.; Baptista, A.M. Is the prediction of pKa values by constant-pH molecular dynamics being hindered by inherited problems? Proteins 2011, 79, 3437–3447. [Google Scholar] [CrossRef] [PubMed]
- Machuqueiro, M.; Baptista, A.M. Molecular dynamics at constant pH and reduction potential: Application to cytochrome c(3). J. Am. Chem. Soc. 2009, 131, 12586–12594. [Google Scholar] [CrossRef]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Correction to constant-pH molecular dynamics simulations for large biomolecular systems. J. Chem. Theory Comput. 2018, 14, 6748–6749. [Google Scholar] [CrossRef]
- Leontyev, I.V.; Stuchebrukhov, A.A. Polarizable molecular interactions in condensed phase and their equivalent nonpolarizable models. J. Chem. Phys. 2014, 141, 014103. [Google Scholar] [CrossRef] [Green Version]
- Blomberg, M.R.A.; Borowski, T.; Himo, F.; Liao, R.-Z.; Siegbahn, P.E.M. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Thiel, W. Semiempirical quantum-chemical methods: Semiempirical quantum-chemical methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Blomberg, M.R.A.; Siegbahn, P.E.M. The mechanism for proton pumping in cytochrome c oxidase from an electrostatic and quantum chemical perspective. Biochim. Biophys. Acta 2012, 1817, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, M.R.A.; Siegbahn, P.E.M. Quantum chemistry applied to the mechanisms of transition metal containing enzymes—Cytochrome c oxidase, a particularly challenging case. J. Comput. Chem. 2006, 27, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Haumann, M. The manganese complex of photosystem II in its reaction cycle—Basic framework and possible realization at the atomic level. Coord. Chem. Rev. 2008, 252, 273–295. [Google Scholar] [CrossRef]
- Debus, R.J. The manganese and calcium ions of photosynthetic oxygen evolution. Biochim. Biophys. Acta 1992, 1102, 269–352. [Google Scholar] [CrossRef]
- Brudvig, G.W. Water oxidation chemistry of photosystem II. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Brettel, K.; Schlodder, E.; Witt, H.T. Nanosecond reduction kinetics of photooxidized chlorophyll-aII (P-680) in single flashes as a probe for the electron pathway, H+-release and charge accumulation in the O2-evolving complex. Biochim. Biophys. Acta 1984, 766, 403–415. [Google Scholar] [CrossRef]
- Rappaport, F.; Lavergne, J. Proton release during successive oxidation steps of the photosynthetic water oxidation process: Stoichiometries and pH dependence. Biochemistry 1991, 30, 10004–10012. [Google Scholar] [CrossRef]
- Jahns, P.; Junge, W. Proton release during the four steps of photosynthetic water oxidation: Induction of 1:1:1:1 pattern due to lack of chlorophyll a/b binding proteins. Biochemistry 1992, 31, 7398–7403. [Google Scholar] [CrossRef]
- Lavergne, J.; Junge, W. Proton release during the redox cycle of the water oxidase. Photosynth. Res. 1993, 38, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T. FTIR detection of water reactions in the oxygen-evolving centre of photosystem II. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 1189–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokhrel, R.; Brudvig, G.W. Oxygen-evolving complex of photosystem II: Correlating structure with spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 11812–11821. [Google Scholar] [CrossRef] [PubMed]
- Koua, F.H.M.; Umena, Y.; Kawakami, K.; Shen, J.-R. Structure of Sr-substituted photosystem II at 2.1 A resolution and its implications in the mechanism of water oxidation. Proc. Natl. Acad. Sci. USA 2013, 110, 3889–3894. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Sugiura, M. Structure of an active water molecule in the water-oxidizing complex of photosystem II as studied by FTIR spectroscopy. Biochemistry 2000, 39, 10943–10949. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E. Recent theoretical studies of water oxidation in photosystem II. J. Photochem. Photobiol. B 2011, 104, 94–99. [Google Scholar] [CrossRef]
- Davis, K.M.; Pushkar, Y.N. Structure of the oxygen evolving complex of photosystem II at room temperature. J. Phys. Chem. B 2015, 119, 3492–3498. [Google Scholar] [CrossRef]
- Robertazzi, A.; Galstyan, A.; Knapp, E.W. PSII manganese cluster: Protonation of W2, O5, O4 and His337 in the S1 state explored by combined quantum chemical and electrostatic energy computations. Biochim. Biophys. Acta 2014, 1837, 1316–1321. [Google Scholar] [CrossRef]
- Kaur, D.; Szejgis, W.; Mao, J.; Amin, M.; Reiss, K.M.; Askerka, M.; Cai, X.; Khaniya, U.; Zhang, Y.; Brudvig, G.W.; et al. Relative Stability of the S2 isomers of the oxygen evolving complex of photosystem II. 2018. revision. [Google Scholar]
- Lee, H.J.; Reimann, J.; Huang, Y.; Adelroth, P. Functional proton transfer pathways in the heme–copper oxidase superfamily. Biochim. Biophys. Acta 2012, 1817, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, M.; Vogt, L.; Szejgis, W.; Vassiliev, S.; Brudvig, G.W.; Bruce, D.; Gunner, M.R. Proton-Coupled Electron Transfer During the S-State Transitions of the Oxygen-Evolving Complex of Photosystem II. J Phys. Chem. B 2015, 119, 7366–7377. [Google Scholar] [CrossRef] [PubMed]
- Namslauer, A.; Aagaard, A.; Katsonouri, A.; Brzezinski, P. Intramolecular proton-transfer reactions in a membrane-bound proton pump: The effect of pH on the peroxy to ferryl transition in cytochrome c oxidase. Biochemistry 2003, 42, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belevich, I.; Verkhovsky, M.I.; Wikström, M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature 2006, 440, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Lu, J.; Yang, S.; Gunner, M.R.; Cui, Q. Changing hydration level in an internal cavity modulates the proton affinity of a key glutamate in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 18886–18891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quenneville, J.; Popović, D.M.; Stuchebrukhov, A.A. Combined DFT and electrostatics study of the proton pumping mechanism in cytochrome c oxidase. Biochim. Biophys. Acta 2006, 1757, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Sugitani, R.; Medvedev, E.S.; Stuchebrukhov, A.A. Theoretical and computational analysis of the membrane potential generated by cytochrome c oxidase upon single electron injection into the enzyme. Biochim. Biophys. Acta 2008, 1777, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Gunner, M.R. Characterizing the proton loading site in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2014, 111, 12414–12419. [Google Scholar] [CrossRef] [Green Version]
- Petřek, M.; Otyepka, M.; Banáš, P.; Košinová, P.; Koča, J.; Damborský, J. CAVER: A new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinform. 2006, 7, 316. [Google Scholar] [CrossRef]
- Smart, O.S.; Neduvelil, J.G.; Wang, X.; Wallace, B.A.; Sansom, M.S.P. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. [Google Scholar] [CrossRef]
- Morozenko, A.; Stuchebrukhov, A.A. Dowser++, a new method of hydrating protein structures. Proteins 2016, 84, 1347–1357. [Google Scholar] [PubMed] [Green Version]
- Ge, X.; Gunner, M.R. Unraveling the mechanism of proton translocation in the extracellular half-channel of bacteriorhodopsin. Proteins 2016, 84, 639–654. [Google Scholar] [PubMed]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Xu, J.; Voth, G.A. Free energy profiles for H+ conduction in the D-pathway of Cytochrome c Oxidase: A study of the wild type and N98D mutant enzymes. Biochim. Biophys. Acta 2006, 1757, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Son, C.Y.; Yethiraj, A.; Cui, Q. Cavity hydration dynamics in cytochrome c oxidase and functional implications. Proc. Natl. Acad. Sci. USA 2017, 114, E8830–E8836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Haider, K.; Lu, J.; Radic, S.; Son, C.Y.; Cui, Q.; Gunner, M.R. Network analysis of a proposed exit pathway for protons to the P-side of cytochrome c oxidase. Biochim. Biophys. Acta 2018, 1859, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In the Third International AAAI Conference on Weblogs and Social Media. 2009. Available online: https://www.aaai.org/ocs/index.php/ICWSM/09/paper/view/154 (accessed on 10 December 2018).
- Vogt, L.; Vinyard, D.J.; Khan, S.; Brudvig, G.W. Oxygen-evolving complex of Photosystem II: An analysis of second-shell residues and hydrogen-bonding networks. Curr. Opin. Chem. Biol. 2015, 25, 152–158. [Google Scholar] [CrossRef]
- Ho, F.M.; Styring, S. Access channels and methanol binding site to the CaMn4 cluster in photosystem II based on solvent accessibility simulations, with implications for substrate water access. Biochim. Biophys. Acta 2008, 1777, 140–153. [Google Scholar] [CrossRef]
- Takaoka, T.; Sakashita, N.; Saito, K.; Ishikita, H. pKa of a Proton-Conducting Water Chain in Photosystem II. J. Phys. Chem. Lett. 2016, 7, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Bondar, A.-N.; Dau, H. Extended protein/water H-bond networks in photosynthetic water oxidation. Biochim. Biophys. Acta 2012, 1817, 1177–1190. [Google Scholar] [CrossRef] [Green Version]
- Ishikita, H.; Saenger, W.; Loll, B.; Biesiadka, J.; Knapp, E.-W. Energetics of a possible proton exit pathway for water oxidation in photosystem II. Biochemistry 2006, 45, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Rutherford, A.W.; Ishikita, H. Energetics of proton release on the first oxidation step in the water-oxidizing enzyme. Nat. Commun. 2015, 6, 8488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabdulkhakov, A.; Guskov, A.; Broser, M.; Kern, J.; Müh, F.; Saenger, W.; Zouni, A. Probing the accessibility of the Mn4Ca cluster in photosystem II: Channels calculation, noble gas derivatization, and cocrystallization with DMSO. Structure 2009, 17, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Linke, K.; Ho, F.M. Water in Photosystem II: Structural, functional and mechanistic considerations. Biochim. Biophys. Acta 2014, 1837, 14–32. [Google Scholar] [CrossRef] [Green Version]
- Loll, B.; Kern, J.; Saenger, W.; Zouni, A.; Biesiadka, J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature 2005, 438, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.W.; Barber, J. Structural characteristics of channels and pathways in photosystem II including the identification of an oxygen channel. J. Struct. Biol. 2007, 159, 228–237. [Google Scholar] [CrossRef]
- Vassiliev, S.; Zaraiskaya, T.; Bruce, D. Exploring the energetics of water permeation in photosystem II by multiple steered molecular dynamics simulations. Biochim. Biophys. Acta 2012, 1817, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Su, X.; Cao, P.; Liu, X.; Chang, W.; Li, M.; Zhang, X.; Liu, Z. Structure of spinach photosystem II–LHCII supercomplex at 3.2 Å resolution. Nature 2016, 534, 69–74. [Google Scholar] [CrossRef]
- Sakashita, N.; Watanabe, H.C.; Ikeda, T.; Ishikita, H. Structurally conserved channels in cyanobacterial and plant photosystem II. Photosynth. Res. 2017, 133, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, N.; Watanabe, H.C.; Ikeda, T.; Saito, K.; Ishikita, H. Origins of water molecules in the photosystem II crystal structure. Biochemistry 2017, 56, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Bommer, M.; Bondar, A.-N.; Zouni, A.; Dobbek, H.; Dau, H. Crystallographic and computational analysis of the barrel part of the PsbO protein of photosystem II: Carboxylate–water clusters as putative proton transfer relays and structural switches. Biochemistry 2016, 55, 4626–4635. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Zimmermann, J.L.; Inoue, Y.; Rutherford, A.W. EPR evidence for a modified S-state transition in chloride-depleted Photosystem II. Biochim.Biophys. Acta 1986, 851, 193–201. [Google Scholar] [CrossRef]
- Amin, M.; Pokhrel, R.; Brudvig, G.W.; Badawi, A.; Obayya, S.S.A. Effect of chloride depletion on the magnetic properties and the redox leveling of the oxygen-evolving complex in photosystem II. J. Phys. Chem. B 2016, 120, 4243–4248. [Google Scholar] [CrossRef] [PubMed]
- Rivalta, I.; Amin, M.; Luber, S.; Vassiliev, S.; Pokhrel, R.; Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N.; Bruce, D.; et al. Structural–functional role of chloride in photosystem II. Biochemistry 2011, 50, 6312–6315. [Google Scholar] [CrossRef] [PubMed]
- Bricker, T.M.; Mummadisetti, M.P.; Frankel, L.K. Recent advances in the use of mass spectrometry to examine structure/function relationships in photosystem II. J. Photochem. Photobiol. B 2015, 152, 227–246. [Google Scholar] [CrossRef] [Green Version]
- Weisz, D.A.; Gross, M.L.; Pakrasi, H.B. The use of advanced mass spectrometry to dissect the life-cycle of photosystem II. Front. Plant Sci. 2016, 7, 617. [Google Scholar] [CrossRef]
- Weisz, D.A.; Gross, M.L.; Pakrasi, H.B. Reactive oxygen species leave a damage trail that reveals water channels in Photosystem II. Sci. Adv. 2017, 3, eaao3013. [Google Scholar] [CrossRef]
- Dilbeck, P.L.; Bao, H.; Neveu, C.L.; Burnap, R.L. Perturbing the water cavity surrounding the manganese cluster by mutating the residue D1-Valine 185 has a strong effect on the water oxidation mechanism of photosystem II. Biochemistry 2013, 52, 6824–6833. [Google Scholar] [CrossRef]
- Debus, R.J. Evidence from FTIR difference spectroscopy that D1-Asp61 influences the water reactions of the oxygen-evolving Mn4CaO5 cluster of photosystem II. Biochemistry 2014, 53, 2941–2955. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, R.; Service, R.J.; Debus, R.J.; Brudvig, G.W. Mutation of lysine 317 in the D2 subunit of photosystem II alters chloride binding and proton transport. Biochemistry 2013, 52, 4758–4773. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Sharma, V.; Kaila, V.R.I.; Hosler, J.P.; Hummer, G. New perspectives on proton pumping in cellular respiration. Chem. Rev. 2015, 115, 2196–2221. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, A.A.; Siletsky, S.; Mitchell, D.; Kaulen, A.; Gennis, R.B. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc. Natl. Acad. Sci. USA 1997, 94, 9085–9090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikström, M.; Jasaitis, A.; Backgren, C.; Puustinen, A.; Verkhovsky, M.I. The role of the D- and K-pathways of proton transfer in the function of the haem–copper oxidases. Biochim. Biophys. Acta 2000, 1459, 514–520. [Google Scholar] [CrossRef] [Green Version]
- Wikström, M.; Verkhovsky, M.I. The D-channel of cytochrome oxidase: An alternative view. Biochim. Biophys. Acta 2011, 1807, 1273–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popović, D.M.; Stuchebrukhov, A.A. Proton exit channels in bovine cytochrome c oxidase. J. Phys. Chem. B 2005, 109, 1999–2006. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K. The O2 reduction and proton pumping gate mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 2011, 1807, 1279–1286. [Google Scholar] [CrossRef]
- Lee, H.M.; Das, T.K.; Rousseau, D.L.; Mills, D.; Ferguson-Miller, S.; Gennis, R.B. Mutations in the putative H-channel in the cytochrome c oxidase from Rhodobacter sphaeroides show that this channel is not important for proton conduction but reveal modulation of the properties of heme a. Biochemistry 2000, 39, 2989–2996. [Google Scholar] [CrossRef]
- Sharma, V.; Jambrina, P.G.; Kaukonen, M.; Rosta, E.; Rich, P.R. Insights into functions of the H channel of cytochrome c oxidase from atomistic molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2017, 114, E10339–E10348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supekar, S.; Kaila, V.R.I. Dewetting transitions coupled to K-channel activation in cytochrome c oxidase. Chem. Sci. 2018, 9, 6703–6710. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Swanson, J.M.J.; Peng, Y.; Wikström, M.; Voth, G.A. Multiscale simulations reveal key features of the proton-pumping mechanism in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2016, 113, 7420–7425. [Google Scholar] [CrossRef] [PubMed]
- Wikström, M.; Verkhovsky, M.I.; Hummer, G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta 2003, 1604, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef] [PubMed]
- Busenlehner, L.S.; Salomonsson, L.; Brzezinski, P.; Armstrong, R.N. Mapping protein dynamics in catalytic intermediates of the redox-driven proton pump cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2006, 103, 15398–15403. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, D.; Cai, X.; Khaniya, U.; Zhang, Y.; Mao, J.; Mandal, M.; Gunner, M.R. Tracing the Pathways of Waters and Protons in Photosystem II and Cytochrome c Oxidase. Inorganics 2019, 7, 14. https://doi.org/10.3390/inorganics7020014
Kaur D, Cai X, Khaniya U, Zhang Y, Mao J, Mandal M, Gunner MR. Tracing the Pathways of Waters and Protons in Photosystem II and Cytochrome c Oxidase. Inorganics. 2019; 7(2):14. https://doi.org/10.3390/inorganics7020014
Chicago/Turabian StyleKaur, Divya, Xiuhong Cai, Umesh Khaniya, Yingying Zhang, Junjun Mao, Manoj Mandal, and Marilyn R. Gunner. 2019. "Tracing the Pathways of Waters and Protons in Photosystem II and Cytochrome c Oxidase" Inorganics 7, no. 2: 14. https://doi.org/10.3390/inorganics7020014