A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit?



Abstract
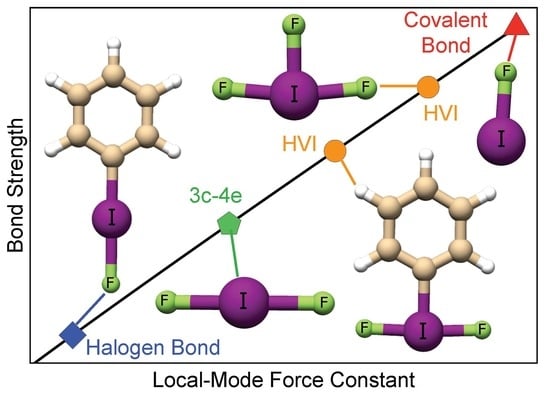
1. Introduction
2. Results and Discussion
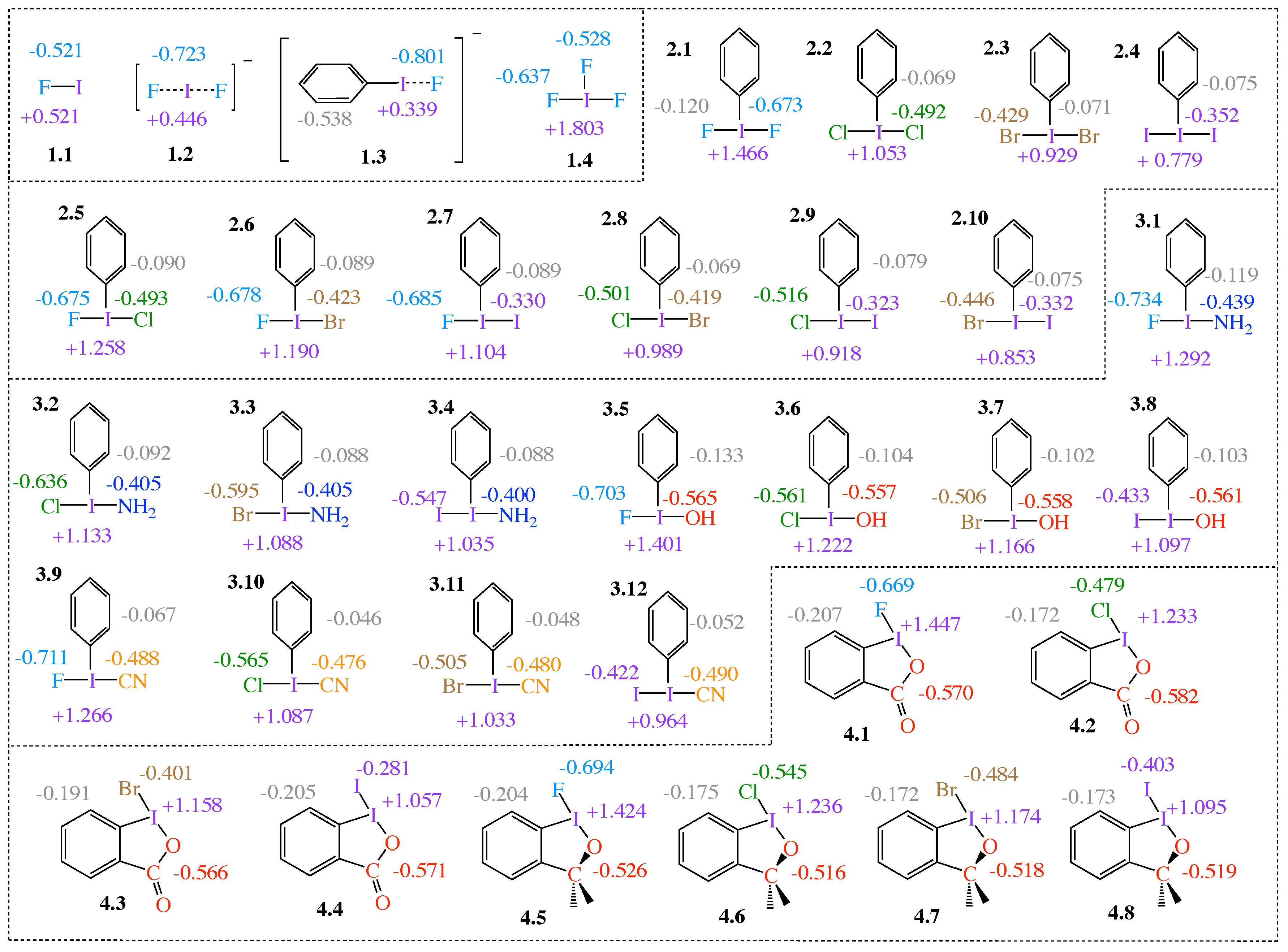
2.1. NBO Charge Analysis
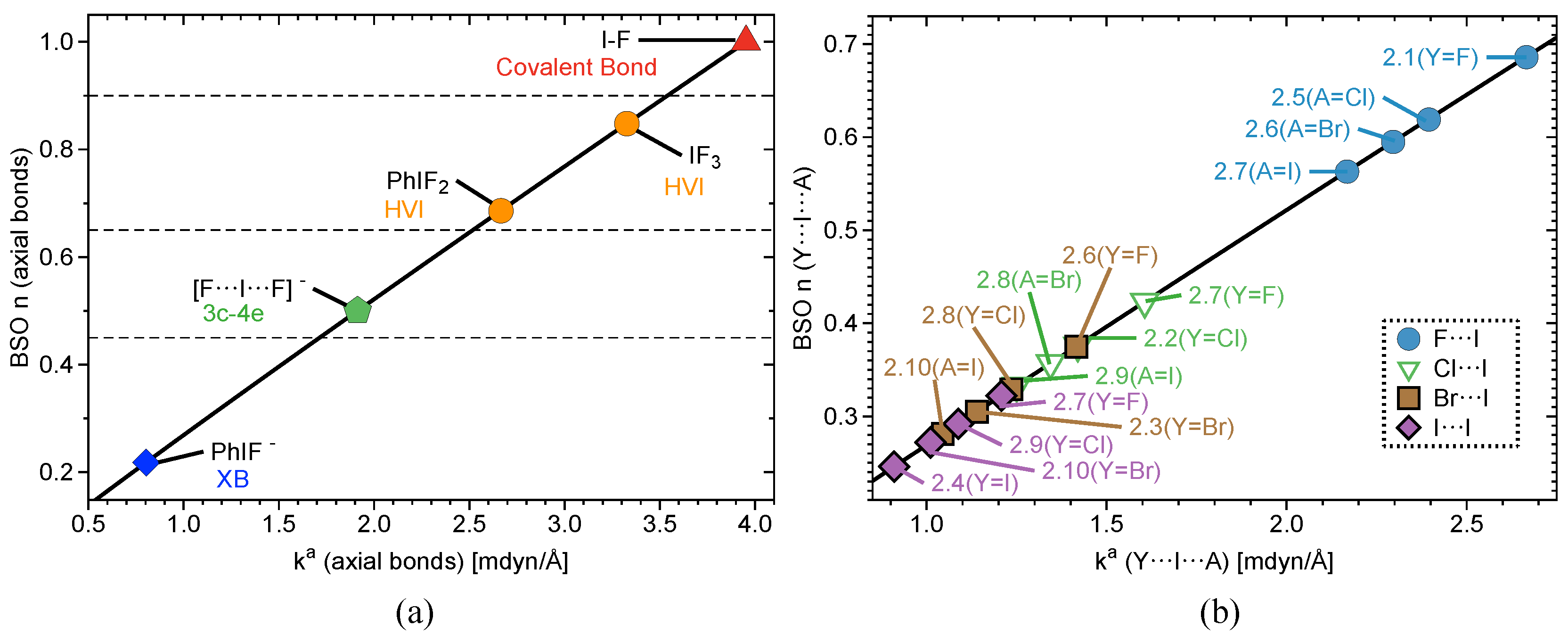
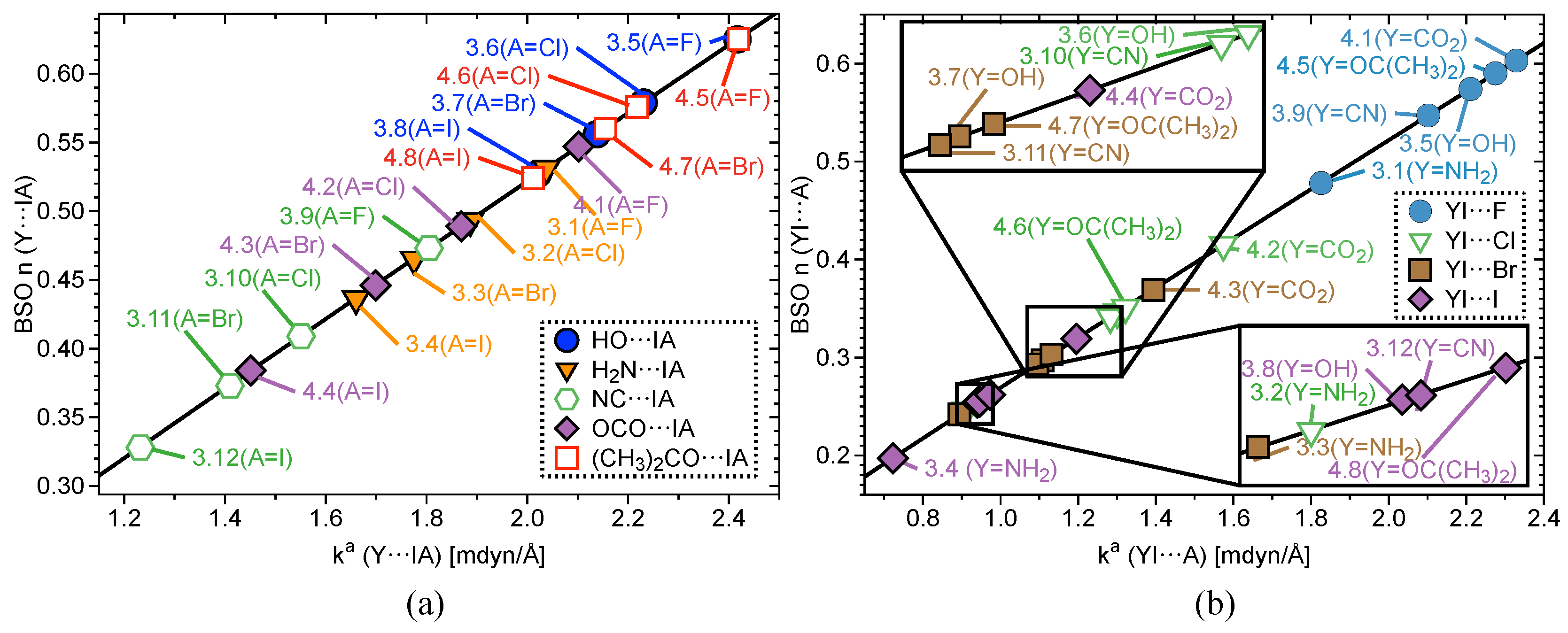
2.2. Bond Strength Order
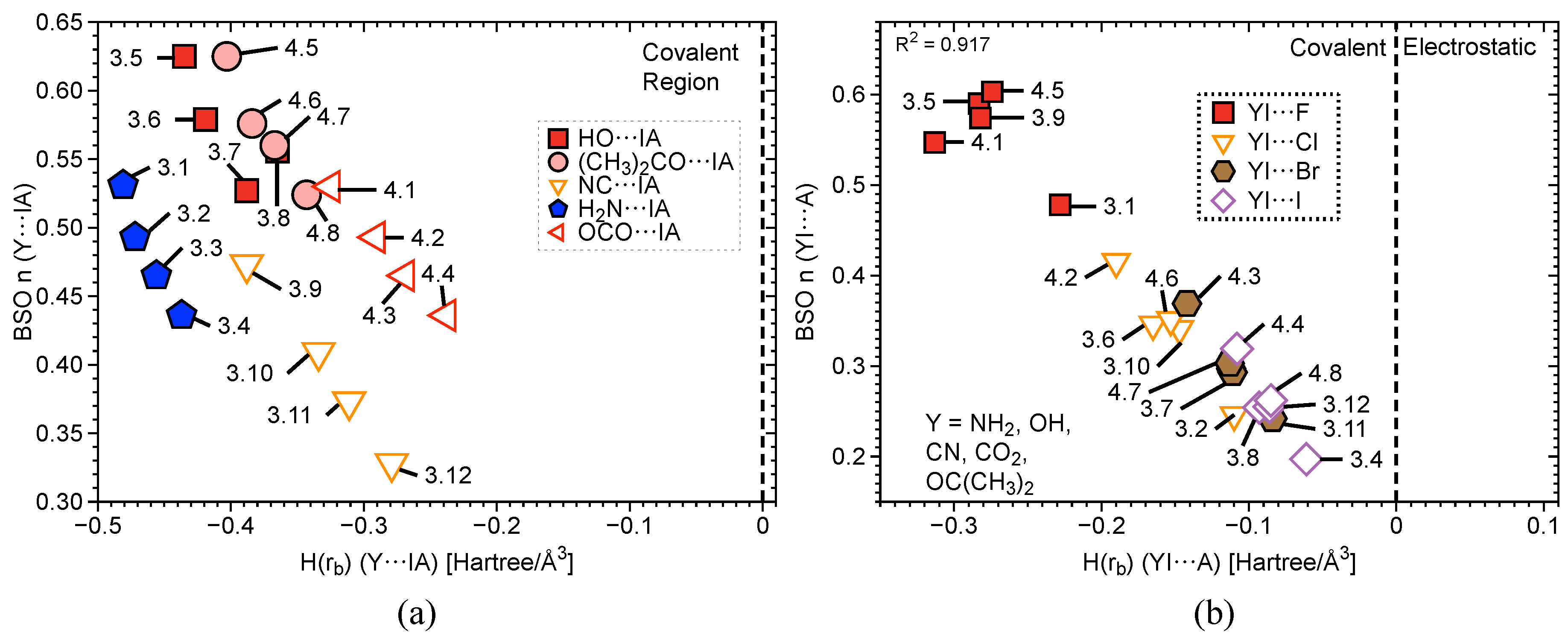
2.3. Covalent/Electrostatic Contributions
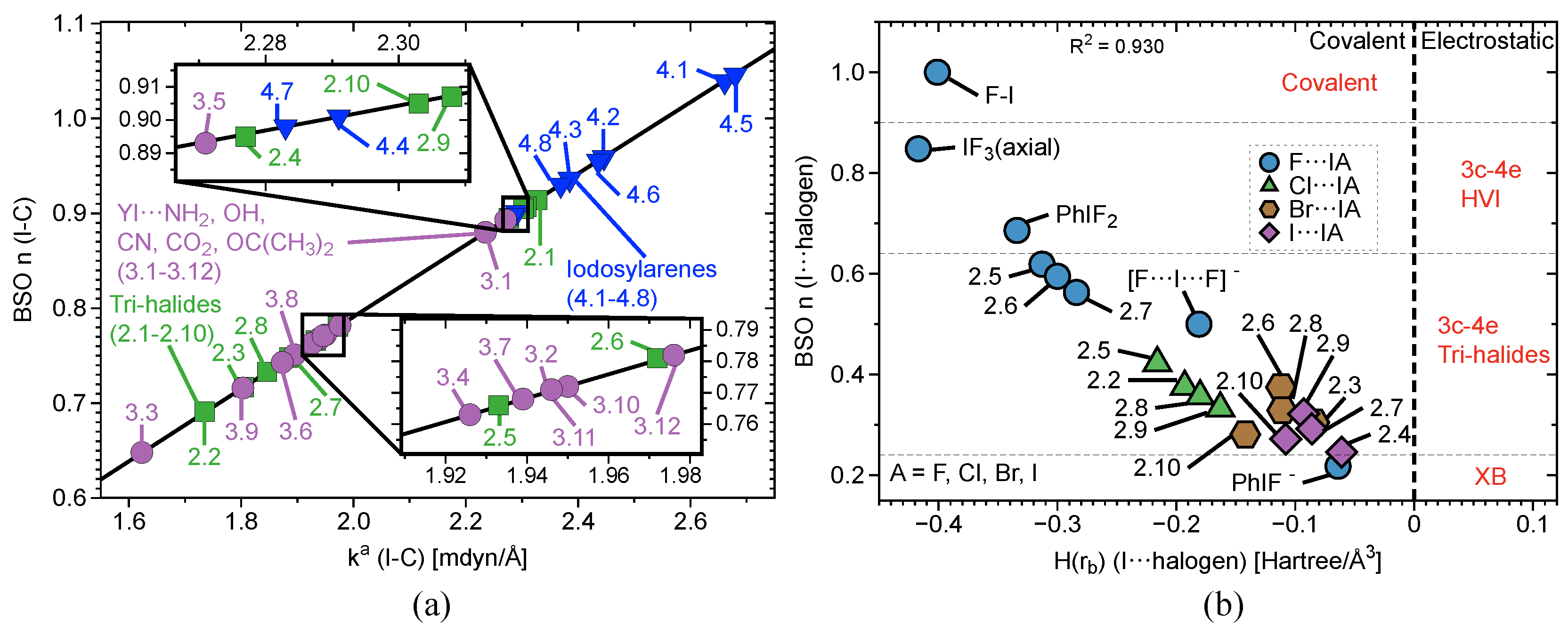
2.4. 3c–4e Bonding
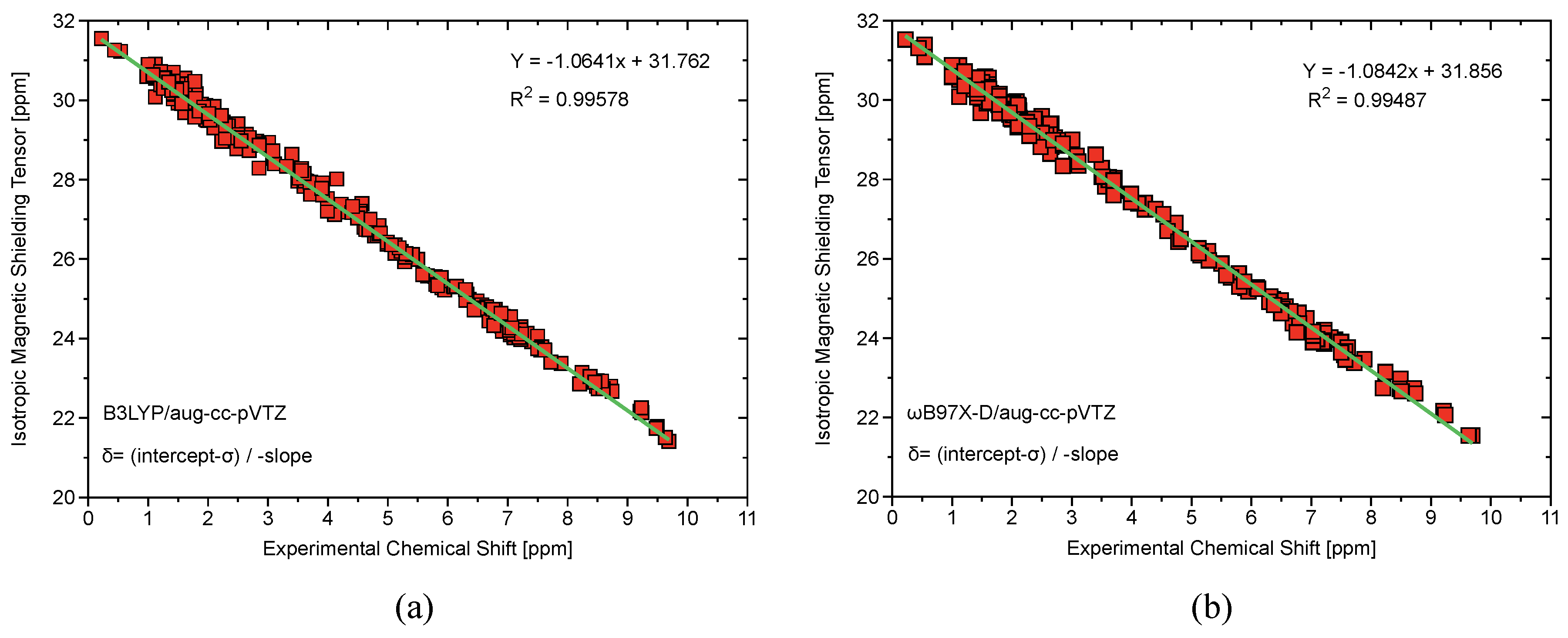
3. Computational Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | r (I–Cl, F) | % | r (I–Cl, F) | % | % | |
|---|---|---|---|---|---|---|
| Basis Set | [Å] | Error | [Å] | Error | Degrees | Error |
| (ICl)B2PLYP | ||||||
| cc-pVDZ | 2.432 | 1.97 | 2.770 | 1.83 | N/A | N/A |
| aug-cc-pVTZ | 2.397 | 0.50 | 2.733 | 0.48 | N/A | N/A |
| Def2TZP | 2.448 | 2.64 | 2.773 | 1.95 | N/A | N/A |
| B3LYP | ||||||
| aug-cc-pVTZ | 2.412 | 1.13 | 2.758 | 1.40 | N/A | N/A |
| B97X-D | ||||||
| aug-cc-pVTZ | 2.364 | 0.88 | 2.744 | 0.88 | N/A | N/A |
| MP2 | ||||||
| cc-pVDZ | 2.420 | 1.47 | 2.746 | 0.96 | N/A | N/A |
| Experiment | 2.385 | N/A | 2.720 | N/A | N/A | N/A |
| IF | ||||||
| B2PLYP | ||||||
| cc-pVDZ | 1.900 | 3.04 | 1.941 | 3.85 | N/A | N/A |
| aug-cc-pVTZ | 1.847 | 0.16 | 1.908 | 2.09 | N/A | N/A |
| Def2TZP | 1.859 | 0.81 | 1.919 | 2.68 | N/A | N/A |
| B3LYP | ||||||
| aug-cc-pVTZ | 1.857 | 0.70 | 1.918 | 2.62 | N/A | N/A |
| B97X-D | ||||||
| aug-cc-pVTZ | 1.840 | 0.22 | 1.901 | 1.71 | N/A | N/A |
| MP2 | ||||||
| cc-pVDZ | 1.895 | 2.77 | 1.933 | 3.42 | N/A | N/A |
| Experiment | 1.844 | N/A | 1.869 | N/A | N/A | N/A |
| IF | ||||||
| B2PLYP | ||||||
| cc-pVDZ | 1.931 | 3.15 | 1.984 | 0.05 | 169.2 | 5.55 |
| aug-cc-pVTZ | 1.885 | 0.69 | 1.960 | 1.16 | 167.7 | 4.61 |
| Def2TZP | 1.900 | 1.50 | 1.975 | 0.40 | 168.6 | 5.18 |
| B3LYP | ||||||
| aug-cc-pVTZ | 1.900 | 1.50 | 1.975 | 0.40 | 168.6 | 5.18 |
| B97X-D | ||||||
| aug-cc-pVTZ | 1.876 | 0.21 | 1.951 | 1.61 | 167.5 | 4.49 |
| MP2 | ||||||
| cc-pVDZ | 1.925 | 2.83 | 1.977 | 0.30 | 168.2 | 4.93 |
| Experiment | 1.872 | N/A | 1.983 | N/A | 160.3 | N/A |
| Method | r (I–Cl, O) | % | r (I–C) | % | % | |
|---|---|---|---|---|---|---|
| Basis Set | [Å] | Error | [Å] | Error | Degrees | Error |
| PhICl | ||||||
| B2PLYP | ||||||
| cc-pVDZ | 2.536 | 2.26 | N/A | N/A | 90.5 | 1.46 |
| aug-cc-pVTZ | 2.505 | 1.01 | N/A | N/A | 89.6 | 0.45 |
| Def2TZP | 2.558 | 3.15 | N/A | N/A | 89.8 | 0.67 |
| B3LYP | ||||||
| aug-cc-pVTZ | 2.524 | 1.77 | N/A | N/A | 90.3 | 1.23 |
| B97X-D | ||||||
| aug-cc-pVTZ | 2.481 | 0.04 | N/A | N/A | 89.2 | 0.00 |
| MP2 | ||||||
| cc-pVDZ | 2.517 | 1.49 | N/A | N/A | 88.9 | 0.34 |
| Experiment | 2.480 | N/A | N/A | N/A | 89.2 | N/A |
| PhI(OAc) | ||||||
| B2PLYP | ||||||
| cc-pVDZ | 2.212 | 2.60 | 2.150 | 2.87 | 164.5 | 0.30 |
| aug-cc-pVTZ | 2.178 | 1.02 | 2.081 | 0.43 | 162.8 | 0.73 |
| Def2TZP | 2.179 | 1.07 | 2.111 | 1.00 | 163.1 | 0.55 |
| B3LYP | ||||||
| aug-cc-pVTZ | 2.194 | 1.76 | 2.124 | 1.63 | 163.7 | 0.18 |
| B97X-D | ||||||
| aug-cc-pVTZ | 2.149 | 0.32 | 2.104 | 0.67 | 162.9 | 0.66 |
| MP2 | ||||||
| cc-pVDZ | 2.187 | 1.44 | 2.122 | 1.53 | 162.7 | 0.79 |
| Experiment | 2.156 | N/A | 2.090 | N/A | 164.0 | N/A |
| Molecule | r (FI) Equatorial | (FI) Equatorial | r (FI) Axial | (FI) Axial |
|---|---|---|---|---|
| Level of Theory | (Å) | (mdyn/Å) | (Å) | (mdyn/Å) |
| FI | ||||
| B97X-D/aug-cc-pVTZ | 1.921 | 3.953 | - | - |
| CCSD(T)/aug-cc-pVTZ | 1.931 | 3.705 | - | - |
| [F⋯I⋯F] | ||||
| B97X-D/aug-cc-pVTZ | 2.089 | 1.913 | - | - |
| CCSD(T)/aug-cc-pVTZ | 2.085 | 1.746 | - | - |
| IF | ||||
| B97X-D/aug-cc-pVTZ | 1.876 | 4.087 | 1.951 | 3.327 |
| CCSD(T)/aug-cc-pVTZ | 1.878 | 4.278 | 1.950 | 3.325 |
| IF | ||||
| B97X-D/aug-cc-pVTZ | 1.840 | 4.529 | 1.901 | 3.634 |
| CCSD(T)/aug-cc-pVTZ | 1.840 | 4.706 | 1.895 | 3.834 |
| B97X-D | B3LYP | |||||
|---|---|---|---|---|---|---|
| Magnetic Isotropic | -Calculated | -Experimental | % | Magnetic Isotropic | -Calculated | % |
| Shielding Tensor | (ppm) | (ppm) | Error | Shielding Tensor | (ppm) | Error |
| PhICl | ||||||
| 23.64 | 7.58 | 7.16 | 5.51 | 23.77 | 7.51 | 4.65 |
| 23.57 | 7.65 | 7.40 | 3.24 | 23.71 | 7.57 | 2.19 |
| 23.41 | 7.79 | 7.68 | 1.42 | 23.57 | 7.70 | 0.24 |
| PhIOAc | ||||||
| 23.42 | 7.79 | 8.24 | 5.81 | 23.56 | 7.71 | 6.91 |
| 23.50 | 7.70 | 7.68 | 0.37 | 23.67 | 7.61 | 0.91 |
| 23.59 | 7.62 | 7.58 | 0.59 | 23.74 | 7.54 | 0.44 |
| 29.68 | 2.01 | 1.92 | 4.72 | 29.64 | 2.00 | 4.02 |
| 1-Hydroxy-1,2 | ||||||
| -benziodoxol-3(1H)-one | ||||||
| 23.45 | 7.76 | 7.71 | 0.58 | 23.61 | 7.66 | 0.65 |
| 22.91 | 8.26 | 8.02 | 2.85 | 23.04 | 8.20 | 2.19 |
| 23.20 | 7.99 | 7.97 | 0.19 | 23.36 | 7.90 | 0.90 |
| 23.30 | 7.89 | 7.85 | 0.50 | 23.44 | 7.82 | 0.35 |

References
- Kieltsch, I.; Eisenberger, P.; Togni, A. Mild Electrophilic Trifluoromethylation of Carbon- and Sulfur-Centered Nucleophiles by a Hypervalent Iodine(III)–CF3 Reagent. Angew. Chem. Int. 2007, 46, 754–757. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.C.S.; Tierno, A.F.; Wengryniuk, S.E. Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry. Molecules 2017, 22, 780. [Google Scholar] [CrossRef]
- Charpentier, J.; Früh, N.; Togni, A. Electrophilic Trifluoromethylation by Use of Hypervalent Iodine Reagents. Chem. Rev. 2014, 115, 650–682. [Google Scholar] [CrossRef] [PubMed]
- Maddox, V.H.; Godefroi, E.F.; Parcell, R.F. The Synthesis of Phencyclidine and Other 1-Arylcyclohexylamines. J. Med. Chem. 1965, 8, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, R.M.; Enache, L.A.; Zhao, L.; Gilardi, R.; Mattson, M.V.; Prakash, O. Rigid Phencyclidine Analogues. Binding to the Phencyclidine and σ1 Receptors. J.Med. Chem. 1998, 41, 468–477. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; Meyer, F. Halogen bonding in polymer science: From crystal engineering to functional supramolecular polymers and materials. Polym. Chem. 2015, 6, 3559–3580. [Google Scholar] [CrossRef]
- Murphy, G.K.; Racicot, L.; Carle, M.S. The Chemistry between Hypervalent Iodine(III) Reagents and Organophosphorus Compounds. Asian J. Org. Chem. 2018, 7, 837–851. [Google Scholar] [CrossRef]
- Ghosh, S.; Pradhan, S.; Chatterjee, I. A survey of chiral hypervalent iodine reagents in asymmetric synthesis. Beilstein J. Org. Chem 2018, 14, 1244–1262. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Wulfsberg, G. Inorganic Chemistry; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]
- Crabtree, R.H. Hypervalency, secondary bonding and hydrogen bonding: Siblings under the skin. Chem. Soc. Rev. 2017, 46, 1720–1729. [Google Scholar] [CrossRef]
- Berger, G.; Soubhye, J.; van der Lee, A.; Vande Velde, C.; Wintjens, R.; Dubois, P.; Clément, S.; Meyer, F. Interplay between Halogen Bonding and Lone Pair-π Interactions: A Computational and Crystal Packing Study. ChemPlusChem 2014, 79, 552–558. [Google Scholar] [CrossRef]
- Labattut, A.; Tremblay, P.L.; Moutounet, O.; Legault, C.Y. Experimental and Theoretical Quantification of the Lewis Acidity of Iodine(III) Species. J. Org. Chem. 2017, 82, 11891–11896. [Google Scholar] [CrossRef] [PubMed]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Quantitative Assessment of the Multiplicity of Carbon-Halogen Bonds: Carbenium and Halonium Ions with F, Cl, Br, and I. J. Phys. Chem. A 2014, 118, 1948–1963. [Google Scholar] [CrossRef] [PubMed]
- Kalescky, R.; Kraka, E.; Cremer, D. Are carbon–halogen double and triple bonds possible? Int. J. Quant. Chem. 2014, 114, 1060–1072. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed]
- Archer, E.M.; van Schalkwyk, T.G. The crystal structure of benzene iododichloride. Acta. Crystallogr. A 1953, 6, 88–92. [Google Scholar] [CrossRef]
- Landrum, G.A.; Goldberg, N.; Hoffmann, R.; Minyaev, R.M. Intermolecular interactions between hypervalent molecules: Ph2IX and XF3 (X = Cl, Br, I) dimers. New J. Chem. 1998, 22, 883–890. [Google Scholar] [CrossRef]
- Bersuker, I.B. Modern Aspects of the Jahn–Teller Effect Theory and Applications To Molecular Problems. Chem. Rev. 2001, 101, 1067–1114. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Nyburg, S.C.; Faerman, C.H.; Prasad, L. A revision of van der Waals atomic radii for molecular crystals. II: Hydrogen bonded to carbon. Acta Crystallogr. B 1987, 43, 106–110. [Google Scholar] [CrossRef]
- Nyburg, S.C.; Faerman, C.H. A revision of van der Waals atomic radii for molecular crystals: N, O, F, S, Cl, Se, Br and I bonded to carbon. Acta Crystallogr. B 1985, 41, 274–279. [Google Scholar] [CrossRef]
- Ishikawa, M.; Ikuta, S.; Katada, M.; Sano, H. Anisotropy of van der Waals radii of atoms in molecules: Alkali-metal and halogen atoms. Acta Crystallogr. B 1990, 46, 592–598. [Google Scholar] [CrossRef]
- Badenhoop, J.K.; Weinhold, F. Natural steric analysis: Ab initio van der Waals radii of atoms and ions. J. Chem. Phys. 1997, 107, 5422–5432. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Henneker, W.H.; Cade, P.E. Molecular Charge Distributions and Chemical Binding. J. Chem. Phys. 1967, 46, 3341–3363. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Bandrauk, A.D. Relaxation of the Molecular Charge Distribution and the Vibrational Force Constant. J. Chem. Phys. 1968, 49, 1666–1675. [Google Scholar] [CrossRef]
- Molina, J.M.; Dobado, J.A. The three-center-four-electron (3c–4e) bond nature revisited. An atoms-in-molecules theory (AIM) and ELF study. Theor. Chim. Acta. 2001, 105, 328–337. [Google Scholar] [CrossRef]
- Musher, J.I. The Chemistry of Hypervalent Molecules. Angew. Chem. Ed. 1969, 8, 54–68. [Google Scholar] [CrossRef]
- Reed, A.E.; von Ragué Schleyer, P. Chemical Bonding in Hypervalent Molecules. The Dominance of Ionic Bonding and Negative Hyperconjugation over d-orbital Participation. J. Am. Chem. Soc. 1990, 112, 1434–1445. [Google Scholar] [CrossRef]
- Magnusson, E. Hypercoordinate Molecules of Second-Row Elements: d Functions or d Orbitals? J. Am. Chem. Soc. 1990, 112, 7940–7951. [Google Scholar] [CrossRef]
- Landrum, G.A.; Goldberg, N.; Hoffmann, R. Bonding in the trihalides (X3−), mixed trihalides (X2Y−) and hydrogen bihalides (X2H−). The connection between hypervalent, electron-rich three-center, donor-acceptor and strong hydrogen bonding. Dalton Trans. 1997, 0, 3605–3613. [Google Scholar] [CrossRef]
- Yang, L.M.; Ganz, E.; Chen, Z.; Wang, Z.X.; von Ragué Schleyer, P. Four Decades of the Chemistry of Planar Hypercoordinate Compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The intrinsic strength of the halogen bond: Electrostatic and covalent contributions described by coupled cluster theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen Bonding versus Hydrogen Bonding: A Molecular Orbital Perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Elder, P.J.; Vargas-Baca, I. A survey of tellurium-centered secondary-bonding supramolecular synthons. Coord. Chem. Rev. 2011, 255, 1426–1438. [Google Scholar] [CrossRef]
- Freindorf, M.; Kraka, E.; Cremer, D. A comprehensive analysis of hydrogen bond interactions based on local vibrational modes. Int. J. Quant. Chem. 2012, 112, 3174–3187. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int. J. Quantum Chem. 2012, 113, 1609–1620. [Google Scholar] [CrossRef]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2012, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2016, 56, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Cremer, D. Transition from metal–ligand bonding to halogen bonding involving a metal as halogen acceptor a study of Cu, Ag, Au, Pt, and Hg complexes. Chem. Phys. Lett. 2017, 681, 56–63. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121, 9544–9556. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of pnicogen bonding with the help of vibrational spectroscopy—The missing link between theory and experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the Pnicogen Bond in Complexes Involving Group VA Elements N, P, and As. J. Phys. Chem. A 2014, 119, 1642–1656. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Tian, C.; Verma, N.; Zou, W.; Wang, C.; Cremer, D.; Kraka, E. Recovering Intrinsic Fragmental Vibrations Using the Generalized Subsystem Vibrational Analysis. J. Chem. Theory Comput. 2018, 14, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Cremer, D. Dieter Cremer’s Contribution to the Field of Theoretical Chemistry. Int. J. Quantum Chem. 2019, 119, e25849. [Google Scholar] [CrossRef]
- Bene, J.D.; Alkorta, I.; Elguero, J. Halogen Bonding Involving CO and CS with Carbon as the Electron Donor. Molecules 2017, 22, 1955. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Bene, J.E.D. Boron as an Electron-Pair Donor for B⋯Cl Halogen Bonds. ChemPhysChem. 2016, 17, 3112–3119. [Google Scholar] [CrossRef]
- Oliveira, V.; Cremer, D.; Kraka, E. The Many Facets of Chalcogen Bonding: Described by Vibrational Spectroscopy. J. Phys. Chem. A 2017, 121, 6845–6862. [Google Scholar] [CrossRef]
- Bene, J.E.D.; Alkorta, I.; Elguero, J. Hydrogen and Halogen Bonding in Cyclic FH(4-n):FCln Complexes, for n = 0–4. J. Phys. Chem. A 2018, 122, 2587–2597. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Murray, J.S.; Politzer, P.; Pilati, T.; Ursini, M.; Resnati, G. Halogen bonding in hypervalent iodine and bromine derivatives: Halonium salts. IUCrJ 2017, 4, 411–419. [Google Scholar] [CrossRef]
- Heinen, F.; Engelage, E.; Dreger, A.; Weiss, R.; Huber, S.M. Iodine(III) Derivatives as Halogen Bonding Organocatalysts. Angew. Chem. Int. Ed. 2018, 57, 3830–3833. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; John Wiley and Sons Ltd.: West Sussex, UK, 2014. [Google Scholar]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croatica Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Cremer, D. New Ways of Analyzing Chemical Bonding. In Modelling of Structure and Properties of Molecules; Maksic, Z.B., Ed.; Ellis Horwood: Chichester, UK, 1987; p. 125. [Google Scholar]
- Kraka, E.; Cremer, D. Chemical Implication of Local Features of the Electron Density Distribution. In Theoretical Models of Chemical Bonding. The Concept of the Chemical Bond; Maksic, Z.B., Ed.; Springer: Heidelberg, Germany, 1990; Volume 2, p. 453. [Google Scholar]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On The Nature of the Halogen Bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef]
- Wang, C.; Guan, L.; Danovich, D.; Shaik, S.; Mo, Y. The origins of the directionality of noncovalent intermolecular interactions. J. Comput. Chem. 2015, 37, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Konkoli, Z.; Cremer, D. A new way of analyzing vibrational spectra. I. Derivation of adiabatic internal modes. Int. J. Quant. Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A new way of analyzing vibrational spectra. IV. Application and testing of adiabatic modes within the concept of the characterization of normal modes. Int. J. Quant. Chem. 1998, 67, 41–55. [Google Scholar] [CrossRef]
- Cremer, D.; Larsson, J.A.; Kraka, E. New developments in the analysis of vibrational spectra On the use of adiabatic internal vibrational modes. In Theoretical and Computational Chemistry; Parkanyi, C., Ed.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 259–327. [Google Scholar]
- Alcock, N.W.; Countryman, R.M. Secondary bonding. Part 4. The crystal and molecular structure of μ-oxo-bis[nitrato(phenyl)iodine(III)]. J. Chem. Soc., Dalton Trans. 1979, 0, 851–853. [Google Scholar] [CrossRef]
- Hoyer, S.; Seppelt, K. The structure of IF3. Angew. Chem. Int. Ed. 2000, 39, 1448–1449. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Zhdankin, V.V.; Simons, J.; Stang, P.J. Macrocyclic, square planar, tetraalkynyl tetraiodonium salts: Structures, stabilities, and vibrational frequencies via ab initio calculations. J. Am. Chem. Soc. 1992, 114, 10569–10572. [Google Scholar] [CrossRef]
- Boswijk, K.H.; Wiebenga, E.H. The crystal structure of I2Cl6(ICl3). Acta Crystallogr. 1954, 7, 417–423. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first? Row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. 1994, 100, 2975–2988. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Cremer, D. Møller-Plesset Perturbation Theory. In Encyclopedia of Computational Chemistry; Schleyer, P., Allinger, N., Clark, T., Gasteiger, J., Kollman, P., Schaefer, H., III, Schreiner, P., Eds.; John Wiley & Sons: New York, NY, USA, 1998; pp. 1706–1735. [Google Scholar]
- Cremer, D. Møller-Plesset perturbation theory: From small molecule methods to methods for thousands of atoms. WIREs Comput. Mol. Sci. 2011, 1, 509–530. [Google Scholar] [CrossRef]
- Görling, A.; Levy, M. Correlation-energy functional and its high-density limit obtained from a coupling-constant perturbation expansion. Phys. Rev. B 1993, 47, 13105–13113. [Google Scholar] [CrossRef]
- Görling, A.; Levy, M. Exact Kohn-Sham scheme based on perturbation theory. Phys. Rev. A 1994, 50, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Mori-Sánchez, P.; Wu, Q.; Yang, W. Orbital-dependent correlation energy in density-functional theory based on a second-order perturbation approach: Success and failure. J. Chem. Phys. 2005, 123, 062204. [Google Scholar] [CrossRef] [PubMed]
- Biczysko, M.; Panek, P.; Scalmani, G.; Bloino, J.; Barone, V. Harmonic and Anharmonic Vibrational Frequency Calculations with the Double-Hybrid B2PLYP Method: Analytic Second Derivatives and Benchmark Studies. J. Chem. Theory Comput. 2010, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, D.; Brémond, E.; Sancho-García, J.C.; Ciofini, I.; Adamo, C. Is There Still Room for Parameter Free Double Hybrids? Performances of PBE0-DH and B2PLYP over Extended Benchmark Sets. J. Chem. Theory Comput. 2013, 9, 3444–3452. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Sherrill, C.D. High-Accuracy Quantum Mechanical Studies of π–π Interactions in Benzene Dimers. J. Phys. Chem. A 2006, 110, 10656–10668. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, K.; Fujii, A.; Mikami, N.; Tsuzuki, S. Magnitude of the CH/π Interaction in the Gas Phase: Experimental and Theoretical Determination of the Accurate Interaction Energy in Benzene-methane. J. Phys. Chem. A 2006, 110, 4397–4404. [Google Scholar] [CrossRef]
- Janowski, T.; Pulay, P. High accuracy benchmark calculations on the benzene dimer potential energy surface. Chem. Phys. Lett. 2007, 447, 27–32. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16-18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- London, F. The quantic theory of inter-atomic currents in aromatic combinations. J. Phys. Radium 1937, 8, 397–409. [Google Scholar] [CrossRef]
- McWeeny, R. Perturbation Theory for the Fock-Dirac Density Matrix. Phys. Rev. 1962, 126, 1028–1034. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Rablen, P.R.; Pearlman, S.A.; Finkbiner, J. A Comparison of Density Functional Methods for the Estimation of Proton Chemical Shifts with Chemical Accuracy. J. Phys. Chem. A 1999, 103, 7357–7363. [Google Scholar] [CrossRef]
- Jain, R.; Bally, T.; Rablen, P.R. Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets. J. Org. Chem. 2009, 74, 4017–4023. [Google Scholar] [CrossRef]
- Bally, T.; Rablen, P.R. Quantum-Chemical Simulation of 1H NMR Spectra. 2.† Comparison of DFT-Based Procedures for Computing Proton-Proton Coupling Constants in Organic Molecules. J. Org. Chem. 2011, 76, 4818–4830. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2011, 112, 1839–1862. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Soldi, C.; Jones, P.B.; Olmstead, M.M.; Rita, J.; Shaw, J.T.; Tantillo, D.J. The Correct Structure of Aquatolide-Experimental Validation of a Theoretically-Predicted Structural Revision. J. Am. Chem. Soc. 2012, 134, 18550–18553. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Tantillo, D.J. Prediction of the Structure of Nobilisitine A Using Computed NMR Chemical Shifts. J. Nat. Prod. 2011, 74, 1339–1343. [Google Scholar] [CrossRef]
- Brand, J.; Charpentier, J.; Waser, J. Direct Alkynylation of Indole and Pyrrole Heterocycles. Angew. Chem. Int. Ed. 2009, 48, 9346–9349. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E. From Molecular Vibrations to Bonding, Chemical Reactions, and Reaction Mechanism. Curr. Org. Chem. 2010, 14, 1524–1560. [Google Scholar] [CrossRef]
- Kraka, E.; Larsson, J.A.; Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Computational Spectroscopy; Grunenberg, J., Ed.; Wiley: New York, NY, USA, 2010; pp. 105–149. [Google Scholar]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A new way of analyzing vibrational spectra. II. Comparison of internal mode frequencies. Int. J. Quant. Chem. 1998, 67, 11–27. [Google Scholar] [CrossRef]
- Zou, W.; Kalescky, R.; Kraka, E.; Cremer, D. Relating Normal Vibrational Modes to Local Vibrational Modes with the Help of an Adiabatic Connection Scheme. J. Chem. Phys. 2012, 137, 084114. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Local vibrational modes of the water dimer—Comparison of theory and experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Zou, W.; Kalescky, R.; Kraka, E.; Cremer, D. Relating normal vibrational modes to local vibrational modes: benzene and naphthalene. J. Mol. Model. 2012, 19, 2865–2877. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Local vibrational modes of the formic acid dimer—The strength of the double hydrogen bond. Mol. Phys. 2013, 111, 1497–1510. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. New Approach to Tolman’s Electronic Parameter Based on Local Vibrational Modes. Inorg. Chem. 2013, 53, 478–495. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A new way of analyzing vibrational spectra. III. Characterization of normal vibrational modes in terms of internal vibrational modes. Int. J. Quant. Chem. 1998, 67, 29–40. [Google Scholar] [CrossRef]
- Zou, W.; Cremer, D. C2 in a Box: Determining its Intrinsic Bond Strength for the X1Σ+g Ground State. Chem. Eur. J. 2016, 22, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, D.; Kraka, E.; Cremer, D. Hidden Bond Anomalies: The Peculiar Case of the Fluorinated Amine Chalcogenides. J. Phys. Chem. A 2015, 119, 9541–9556. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Setiawan, D.; Cremer, D. Re-evaluation of the bond length-bond strength rule: The stronger bond is not always the shorter bond. J. Comp. Chem. 2015, 37, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, D.; Sethio, D.; Cremer, D.; Kraka, E. From Strong to Weak NF Bonds: On the Design of a New Class of Fluorinating Agents. Phys. Chem. Chem. Phys. 2018, 20, 23913–23927. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zou, W.; Jia, J.; Li, W.; Cremer, D. Different Ways of Hydrogen Bonding in Water—Why Does Warm Water Freeze Faster than Cold Water? J. Chem. Theory Comput. 2016, 13, 55–76. [Google Scholar] [CrossRef]
- Tao, Y.; Zou, W.; Kraka, E. Strengthening of hydrogen bonding with the push-pull effect. Chem. Phys. Lett. 2017, 685, 251–258. [Google Scholar] [CrossRef]
- Li, Y.; Oliveira, V.; Tang, C.; Cremer, D.; Liu, C.; Ma, J. The Peculiar Role of the Au3 Unit in Aum Clusters: σ-Aromaticity of the Au5Zn+ Ion. Inorg. Chem. 2017, 56, 5793–5803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dai, H.; Yan, H.; Zou, W.; Cremer, D. B–H π Interaction: A New Type of Nonclassical Hydrogen Bonding. J. Am. Chem. Soc. 2016, 138, 4334–4337. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhang, X.; Dai, H.; Yan, H.; Cremer, D.; Kraka, E. Description of an unusual hydrogen bond between carborane and a phenyl group. J. Organometal. Chem. 2018, 865, 114–127. [Google Scholar] [CrossRef]
- Setiawan, D.; Kalescky, R.; Kraka, E.; Cremer, D. Direct Measure of Metal–Ligand Bonding Replacing the Tolman Electronic Parameter. Inorg. Chem. 2016, 55, 2332–2344. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Generalization of the Tolman electronic parameter: The metal–ligand electronic parameter and the intrinsic strength of the metal–ligand bond. Dalton Trans. 2017, 46, 8323–8338. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Quantitative Assessment of Aromaticity and Antiaromaticity Utilizing Vibrational Spectroscopy. J. Org. Chem. 2016, 81, 9669–9686. [Google Scholar] [CrossRef] [PubMed]
- Badger, R.M. A Relation Between Internuclear Distances and Bond Force Constants. J. Chem. Phys. 1934, 2, 128–131. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence indices: A personal account. J. Comput. Chem. 2006, 28, 204–221. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density? Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Electron density shift description of non-bonding intramolecular interactions. Comput. Theor. Chem. 2012, 991, 124–133. [Google Scholar] [CrossRef]
- Kraka, E.; Zou, W.; Filatov, M.; Tao, Y.; Grafenstein, J.; Izotov, D.; Gauss, J.; He, Y.; Wu, A.; Konkoli, Z.; et al. COLOGNE2018. 2018. Available online: http://www.smu.edu/catco (accessed on 15 April 2018).
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Sosa, G.L.; Peruchena, N.M.; Contreras, R.H.; Castro, E.A. Topological and NBO analysis of hydrogen bonding interactions involving C–H⋯O bonds. J. Mol. Struct. THEOCHEM 2002, 577, 219–228. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll (Version 17.11.14). 2017. Available online: aim.tkgristmill.com (accessed on 2 April 2018).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Clark, T.; Murray, J.S.; Politzer, P. The σ-Hole Coulombic Interpretation of Trihalide Anion Formation. ChemPhysChem 2018, 19, 3044–3049. [Google Scholar] [CrossRef]
- de Magalhães, H.P.; Lüthi, H.P.; Bultinck, P. Exploring the role of the 3-center-4-electron bond in hypervalent λ3-iodanes using the methodology of domain averaged Fermi holes. Phys. Chem. Chem. Phys. 2016, 18, 846–856. [Google Scholar] [CrossRef]
- Ruedenberg, K. The Physical Nature of the Chemical Bond. Rev. Mod. Phys. 1962, 34, 326–376. [Google Scholar] [CrossRef]
- Bacskay, G.B.; Nordholm, S.; Ruedenberg, K. The Virial Theorem and Covalent Bonding. J. Phys. Chem. A 2018, 122, 7880–7893. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Sexton, T.; Kraka, E.; Freindorf, M.; Cremer, D. A New Method for Describing the Mechanism of a Chemical Reaction Based on the Unified Reaction Valley Approach. J. Chem. Theory Comput. 2016, 12, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E. Reaction path Hamiltonian and the unified reaction valley approach. WIREs: Comput. Mol. Sci. 2011, 1, 531–556. [Google Scholar] [CrossRef]
- Kraka, E.; Cremer, D. Computational Analysis of the Mechanism of Chemical Reactions in Terms of Reaction Phases: Hidden Intermediates and Hidden Transition States. Acc. Chem. Res. 2010, 43, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Vaish, A.; Runčevski, T.; Tsarevsky, N.V. Hypervalent Iodine Compounds with Tetrazole Ligands. J. Org. Chem. 2018, 83, 12496–12506. [Google Scholar] [CrossRef]
- Vaish, A.; Tsarevsky, N. Hypervalent Iodine Compounds in Polymer Science and Technology. In Main Group Strategies towards Functional Hybrid Materials; Baumgartner, T., Jaekle, F., Eds.; Wiley: New York, NY, USA, 2018; Chapter 19; pp. 483–514. [Google Scholar]





| # | Bond Analyzed | r | BSO n | r | BSO n | %3c–4e | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Y–I; I–A) | (Y–I) | (Y–I) | (Y–I) | (Y–I) | (Y–I) | (Y–I) | (I–A) | (I–A) | (I–A) | (I–A) | (I–A) | (I–A) | |||
| Group1 | 1.1 | F–I | 1.921 | 3.953 | 1.000 | 637 | 0.900 | −0.401 | - | - | - | - | - | - | - |
| 1.2 | F–I; I–F | 2.089 | 1.913 | 0.500 | 443 | 0.615 | −0.181 | 2.089 | 1.913 | 0.500 | 443 | 0.615 | −0.181 | 100 | |
| 1.3 | C–I; I–F | 2.173 | 1.843 | 0.482 | 534 | 0.761 | −0.359 | 2.282 | 0.803 | 0.218 | 287 | 0.428 | −0.064 | 45 | |
| 1.4 | F–I; I–F | 1.951 | 3.327 | 0.848 | 585 | 0.875 | −0.417 | 1.951 | 3.327 | 0.848 | 585 | 0.875 | −0.417 | 100 | |
Group2 | 2.1 | F–I; I F | 1.997 | 2.666 | 0.686 | 523 | 0.783 | −0.334 | 1.997 | 2.666 | 0.686 | 523 | 0.783 | −0.334 | 100 |
| 2.2 | Cl–I; I–Cl | 2.481 | 1.420 | 0.376 | 297 | 0.542 | −0.193 | 2.481 | 1.420 | 0.376 | 297 | 0.542 | −0.193 | 100 | |
| 2.3 | Br–I; I–Br | 2.651 | 1.140 | 0.305 | 155 | 0.465 | −0.136 | 2.651 | 1.140 | 0.305 | 155 | 0.465 | −0.136 | 100 | |
| 2.4 | I–I; I–I | 2.892 | 0.910 | 0.246 | 199 | 0.380 | −0.092 | 2.892 | 0.904 | 0.244 | 156 | 0.380 | −0.092 | 100 | |
| 2.5 | F–I; I–Cl | 2.014 | 2.395 | 0.619 | 496 | 0.759 | −0.313 | 2.454 | 1.606 | 0.423 | 315 | 0.567 | −0.216 | 68 | |
| 2.6 | F–I; I–Br | 2.025 | 2.296 | 0.595 | 486 | 0.745 | −0.300 | 2.606 | 1.416 | 0.375 | 222 | 0.503 | −0.164 | 63 | |
| 2.7 | F–I; I–I) | 2.039 | 2.168 | 0.563 | 472 | 0.725 | −0.284 | 2.821 | 1.208 | 0.322 | 180 | 0.431 | −0.123 | 57 | |
| 2.8 | Cl–I; I–Br | 2.498 | 1.344 | 0.357 | 288 | 0.525 | −0.180 | 2.632 | 1.235 | 0.329 | 208 | 0.481 | −0.147 | 92 | |
| 2.9 | Cl–I; I–I | 2.522 | 1.250 | 0.333 | 278 | 0.503 | −0.163 | 2.847 | 1.088 | 0.292 | 171 | 0.412 | −0.111 | 88 | |
| 2.10 | Br–I; I–I | 2.677 | 1.045 | 0.281 | 191 | 0.443 | −0.121 | 2.865 | 1.011 | 0.272 | 164 | 0.399 | −0.103 | 97 | |
Group 3 | 3.1 | N–I; I–F | 2.080 | 2.038 | 0.531 | 524 | 0.854 | −0.481 | 2.084 | 1.826 | 0.478 | 433 | 0.654 | −0.228 | 90 |
| 3.2 | N–I; I–Cl | 2.087 | 1.887 | 0.493 | 504 | 0.851 | −0.472 | 2.611 | 0.911 | 0.246 | 238 | 0.419 | −0.110 | 50 | |
| 3.3 | N–I; I–Br | 2.100 | 1.775 | 0.465 | 489 | 0.835 | −0.456 | 2.767 | 0.894 | 0.242 | 177 | 0.371 | −0.084 | 52 | |
| 3.4 | N–I; I–I | 2.113 | 1.660 | 0.436 | 473 | 0.817 | −0.437 | 2.998 | 0.723 | 0.197 | 139 | 0.315 | −0.061 | 45 | |
| 3.5 | O–I; I–F | 2.031 | 2.417 | 0.625 | 537 | 0.836 | −0.435 | 2.036 | 2.275 | 0.590 | 483 | 0.722 | −0.283 | 94 | |
| 3.6 | O–I; I–Cl | 2.042 | 2.231 | 0.579 | 516 | 0.822 | −0.419 | 2.520 | 1.301 | 0.346 | 284 | 0.500 | −0.165 | 60 | |
| 3.7 | O–I; I–Br | 2.051 | 2.138 | 0.556 | 505 | 0.761 | −0.365 | 2.676 | 1.108 | 0.297 | 197 | 0.425 | −0.112 | 53 | |
| 3.8 | O–I; I–I | 2.064 | 2.021 | 0.527 | 491 | 0.790 | −0.388 | 2.895 | 0.940 | 0.254 | 159 | 0.378 | −0.093 | 48 | |
| 3.9 | F–I; I–C | 2.038 | 2.211 | 0.574 | 476 | 0.722 | −0.282 | 2.154 | 1.805 | 0.473 | 529 | 0.763 | −0.388 | 82 | |
| 3.10 | C–I; I–Cl | 2.174 | 1.551 | 0.409 | 490 | 0.705 | −0.334 | 2.519 | 1.283 | 0.341 | 282 | 0.474 | −0.147 | 83 | |
| 3.11 | C–I; I–Br | 2.189 | 1.410 | 0.373 | 467 | 0.681 | −0.311 | 2.672 | 1.095 | 0.293 | 195 | 0.421 | −0.111 | 79 | |
| 3.12 | C–I; I–I | 2.213 | 1.232 | 0.328 | 437 | 0.644 | −0.279 | 2.887 | 0.946 | 0.255 | 159 | 0.365 | −0.086 | 78 | |
Group 4 | 4.1 | F–I; I–O | 1.993 | 2.622 | 0.675 | 519 | 0.757 | −0.313 | 2.079 | 2.102 | 0.547 | 501 | 0.719 | −0.325 | 81 |
| 4.2 | O–I; I–Cl | 2.102 | 1.869 | 0.489 | 473 | 0.682 | −0.291 | 2.448 | 1.574 | 0.415 | 312 | 0.536 | −0.190 | 85 | |
| 4.3 | O–I; I–Br | 2.117 | 1.699 | 0.446 | 451 | 0.657 | −0.269 | 2.598 | 1.394 | 0.369 | 221 | 0.473 | −0.142 | 83 | |
| 4.4 | O–I; I–I | 2.141 | 1.451 | 0.384 | 416 | 0.623 | −0.238 | 2.809 | 1.195 | 0.319 | 179 | 0.408 | −0.108 | 83 | |
| 4.5 | O–I; I–F | 2.032 | 2.419 | 0.625 | 538 | 0.800 | −0.403 | 2.024 | 2.329 | 0.603 | 489 | 0.710 | −0.274 | 96 | |
| 4.6 | O–I; I–Cl | 2.041 | 2.218 | 0.576 | 515 | 0.781 | −0.384 | 2.503 | 1.322 | 0.351 | 286 | 0.482 | −0.153 | 61 | |
| 4.7 | O–I; I–Br | 2.050 | 2.155 | 0.560 | 507 | 0.763 | −0.367 | 2.658 | 1.131 | 0.303 | 199 | 0.424 | −0.113 | 54 | |
| 4.8 | O–I; I–I | 2.063 | 2.011 | 0.524 | 490 | 0.739 | −0.343 | 2.875 | 0.973 | 0.262 | 161 | 0.364 | −0.085 | 50 |
| # | Bond Analyzed | r | BSO n (Scaled) | |||||
|---|---|---|---|---|---|---|---|---|
| Group 1 | 1.4 | I–F | 1.876 | 1.037 | −0.569 | 4.087 | 1.565 | 648 |
Group 2 | 2.1 | I–C | 2.123 | 0.895 | −0.476 | 2.327 | 0.914 | 600 |
| 2.2 | I–C | 2.116 | 0.895 | −0.476 | 1.735 | 0.691 | 518 | |
| 2.3 | I–C | 2.107 | 0.905 | −0.488 | 2.277 | 0.717 | 594 | |
| 2.4 | I–C | 2.107 | 0.901 | −0.484 | 1.805 | 0.895 | 529 | |
| 2.5 | I–C | 2.129 | 0.880 | −0.461 | 1.933 | 0.766 | 547 | |
| 2.6 | I–C | 2.125 | 0.884 | −0.466 | 1.972 | 0.781 | 553 | |
| 2.7 | I–C | 2.120 | 0.891 | −0.473 | 1.886 | 0.748 | 540 | |
| 2.8 | I–C | 2.112 | 0.900 | −0.482 | 1.846 | 0.733 | 535 | |
| 2.9 | I–C | 2.103 | 0.912 | −0.495 | 2.308 | 0.907 | 598 | |
| 2.10 | I–C | 2.105 | 0.907 | −0.490 | 2.303 | 0.905 | 597 | |
Group 3 | 3.1 | I–C | 2.139 | 0.869 | −0.452 | 2.235 | 0.880 | 588 |
| 3.2 | I–C | 2.157 | 0.836 | −0.418 | 1.950 | 0.772 | 549 | |
| 3.3 | I–C | 2.142 | 0.858 | −0.44 0 | 1.623 | 0.648 | 501 | |
| 3.4 | I–C | 2.132 | 0.872 | −0.455 | 1.926 | 0.763 | 546 | |
| 3.5 | I–C | 2.128 | 0.888 | −0.471 | 2.271 | 0.893 | 593 | |
| 3.6 | I–C | 2.133 | 0.874 | −0.457 | 1.895 | 0.751 | 542 | |
| 3.7 | I–C | 2.130 | 0.834 | −0.424 | 1.939 | 0.768 | 548 | |
| 3.8 | I–C | 2.125 | 0.885 | −0.468 | 1.873 | 0.743 | 538 | |
| 3.9 | I–C | 2.138 | 0.865 | −0.445 | 1.803 | 0.716 | 528 | |
| 3.10 | I–C | 2.130 | 0.822 | −0.402 | 1.926 | 0.763 | 546 | |
| 3.11 | I–C | 2.127 | 0.829 | −0.409 | 1.946 | 0.771 | 549 | |
| 3.12 | I–C | 2.123 | 0.841 | −0.421 | 1.976 | 0.782 | 553 | |
Group 4 | 4.1 | I–C | 2.090 | 0.907 | −0.490 | 2.661 | 1.039 | 642 |
| 4.2 | I–C | 2.110 | 0.863 | −0.445 | 2.446 | 0.959 | 615 | |
| 4.3 | I–C | 2.115 | 0.852 | −0.434 | 2.385 | 0.936 | 608 | |
| 4.4 | I–C | 2.123 | 0.838 | −0.421 | 2.291 | 0.901 | 596 | |
| 4.5 | I–C | 2.094 | 0.901 | −0.485 | 2.680 | 1.046 | 644 | |
| 4.6 | I–C | 2.113 | 0.860 | −0.444 | 2.436 | 0.955 | 614 | |
| 4.7 | I–C | 2.118 | 0.851 | −0.434 | 2.369 | 0.930 | 606 | |
| 4.8 | I–C | 2.124 | 0.838 | −0.422 | 2.283 | 0.898 | 594 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. https://doi.org/10.3390/inorganics7040047
Yannacone S, Oliveira V, Verma N, Kraka E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics. 2019; 7(4):47. https://doi.org/10.3390/inorganics7040047
Chicago/Turabian StyleYannacone, Seth, Vytor Oliveira, Niraj Verma, and Elfi Kraka. 2019. "A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit?" Inorganics 7, no. 4: 47. https://doi.org/10.3390/inorganics7040047
APA StyleYannacone, S., Oliveira, V., Verma, N., & Kraka, E. (2019). A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics, 7(4), 47. https://doi.org/10.3390/inorganics7040047