pH Dependent Reversible Formation of a Binuclear Ni2 Metal-Center within a Peptide Scaffold



Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Considerations
2.2. Preparation of S-triphenylmethyl-thioglycolic Acid
2.3. Preparation of {Ni2(SODmds)}
2.4. Determination of Ni–S(H+)–Cys pKa
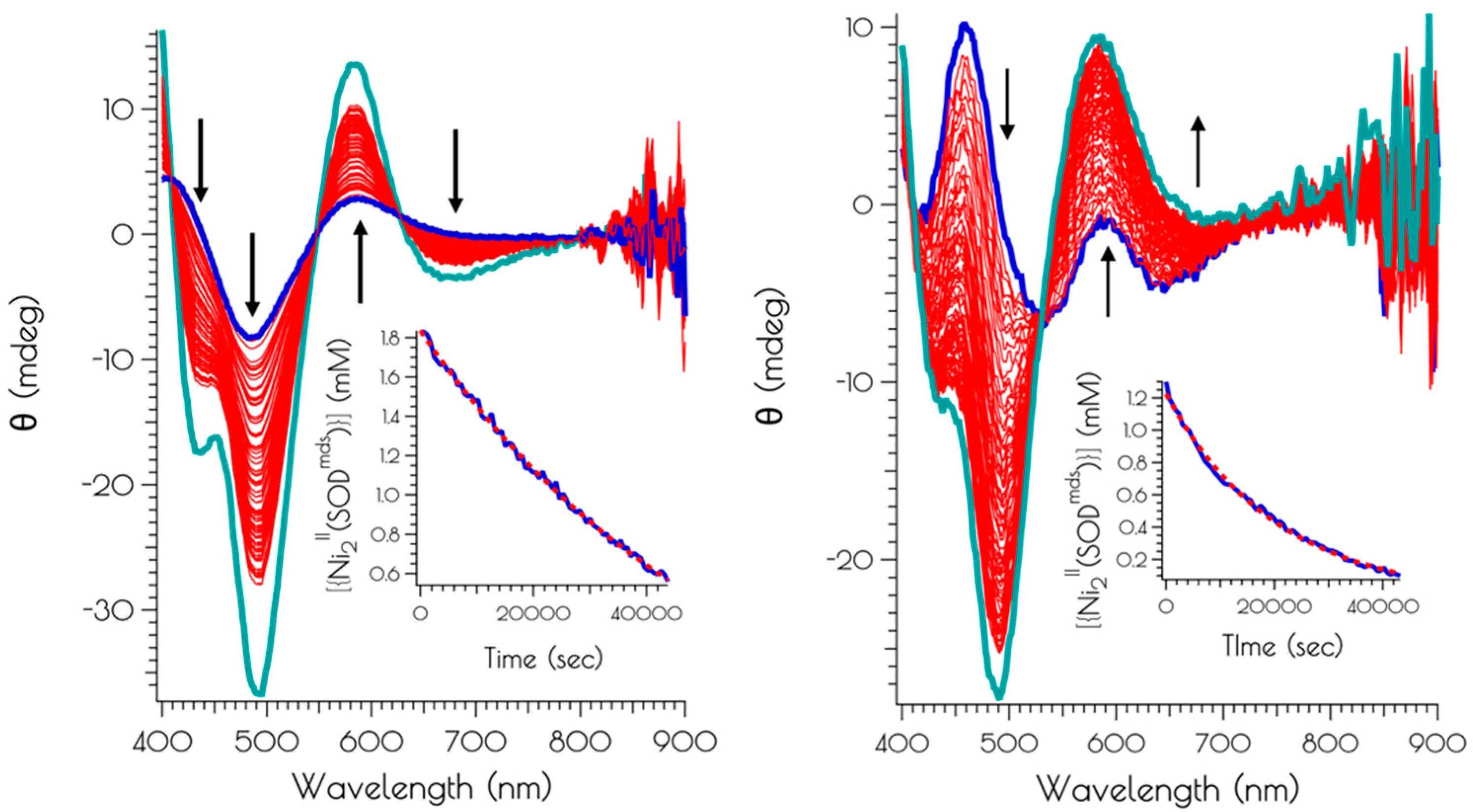
2.5. Kinetics of the Air Oxidation of {Ni2II(m1S3)2}
2.6. Nickel K-Edge X-ray Absorption Spectroscopy
2.7. Sulfur K-Edge X-ray Absorption Spectroscopy
2.8. Electronic Structure Calculation
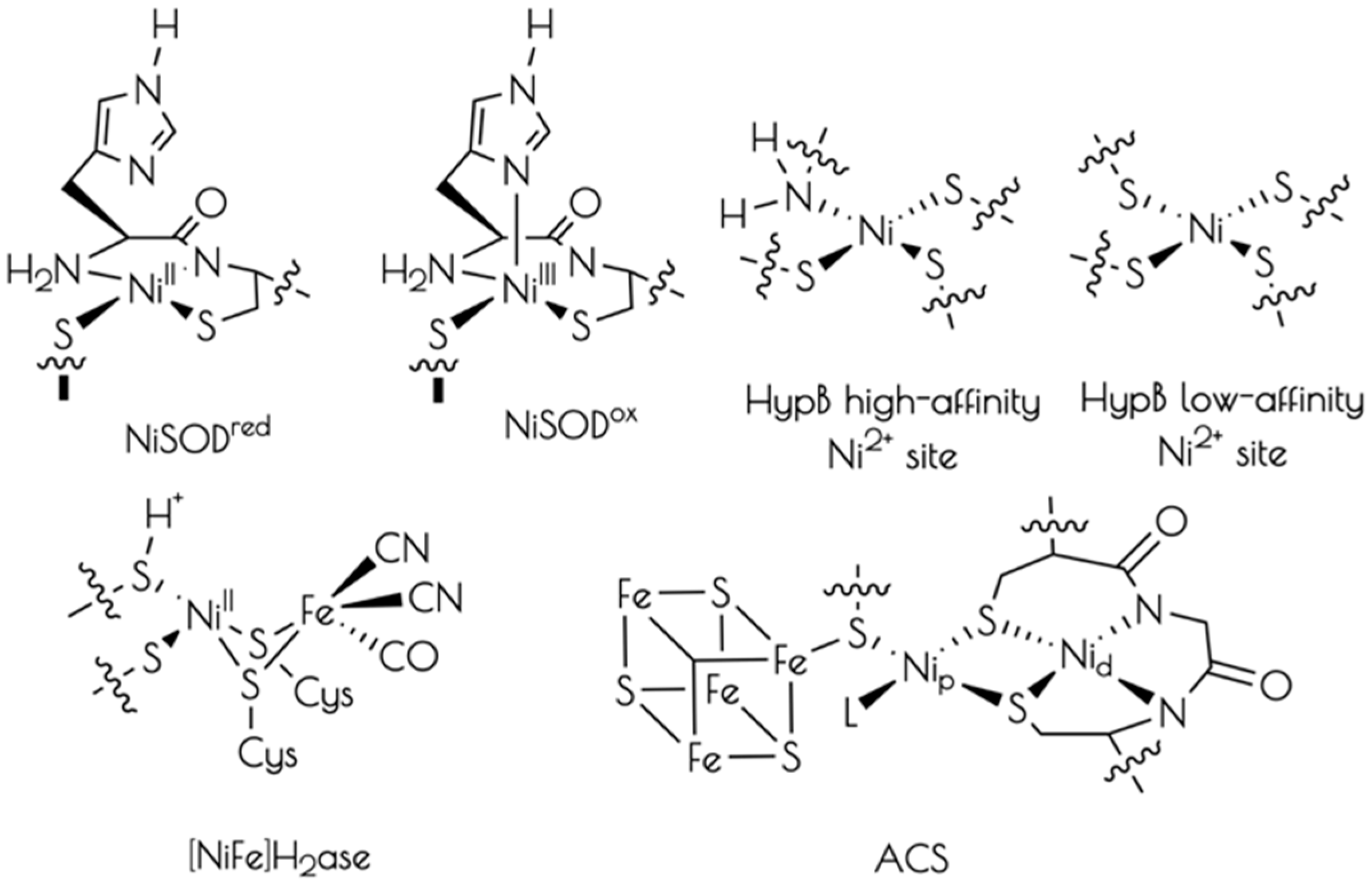
3. Results
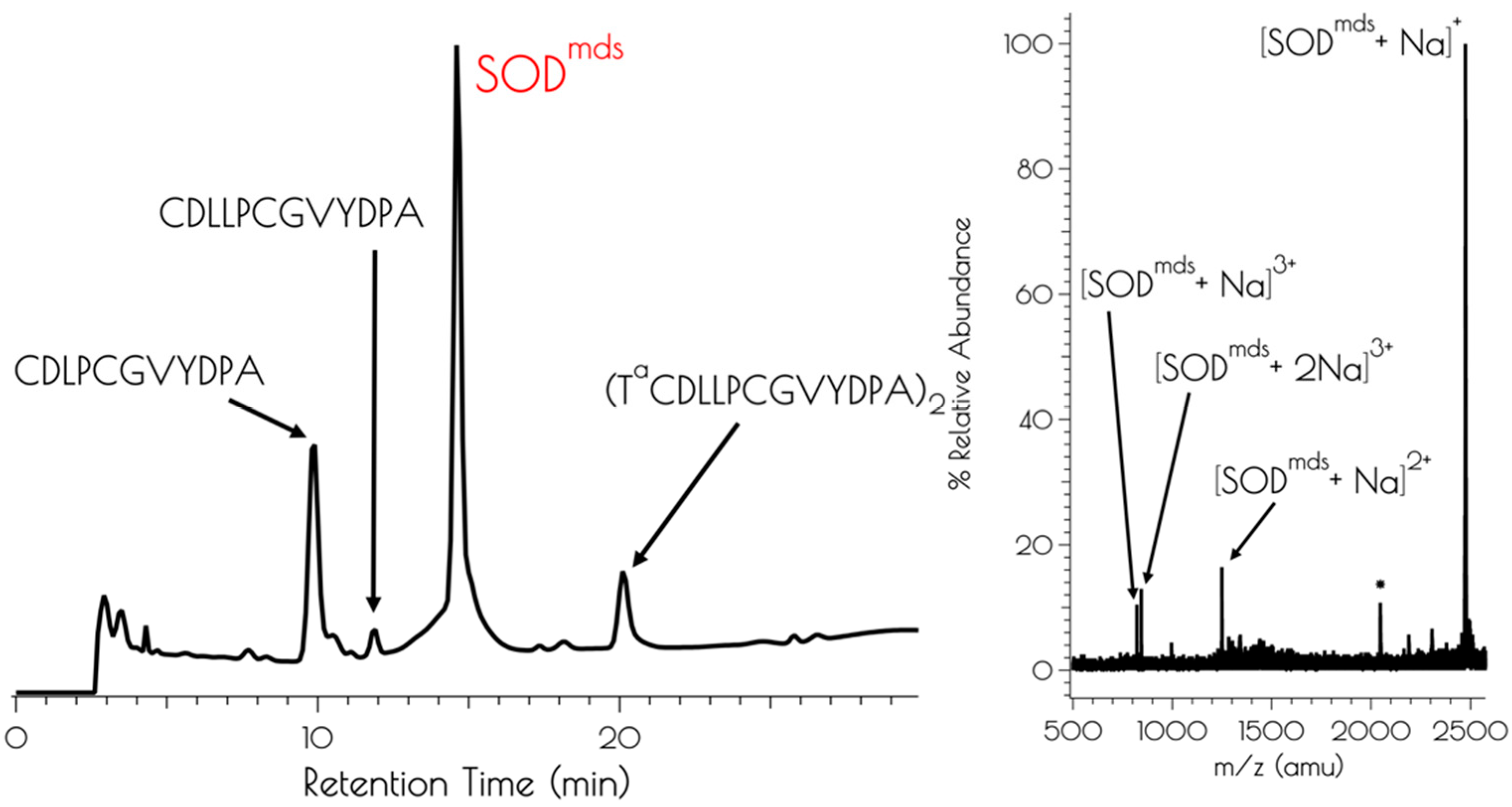
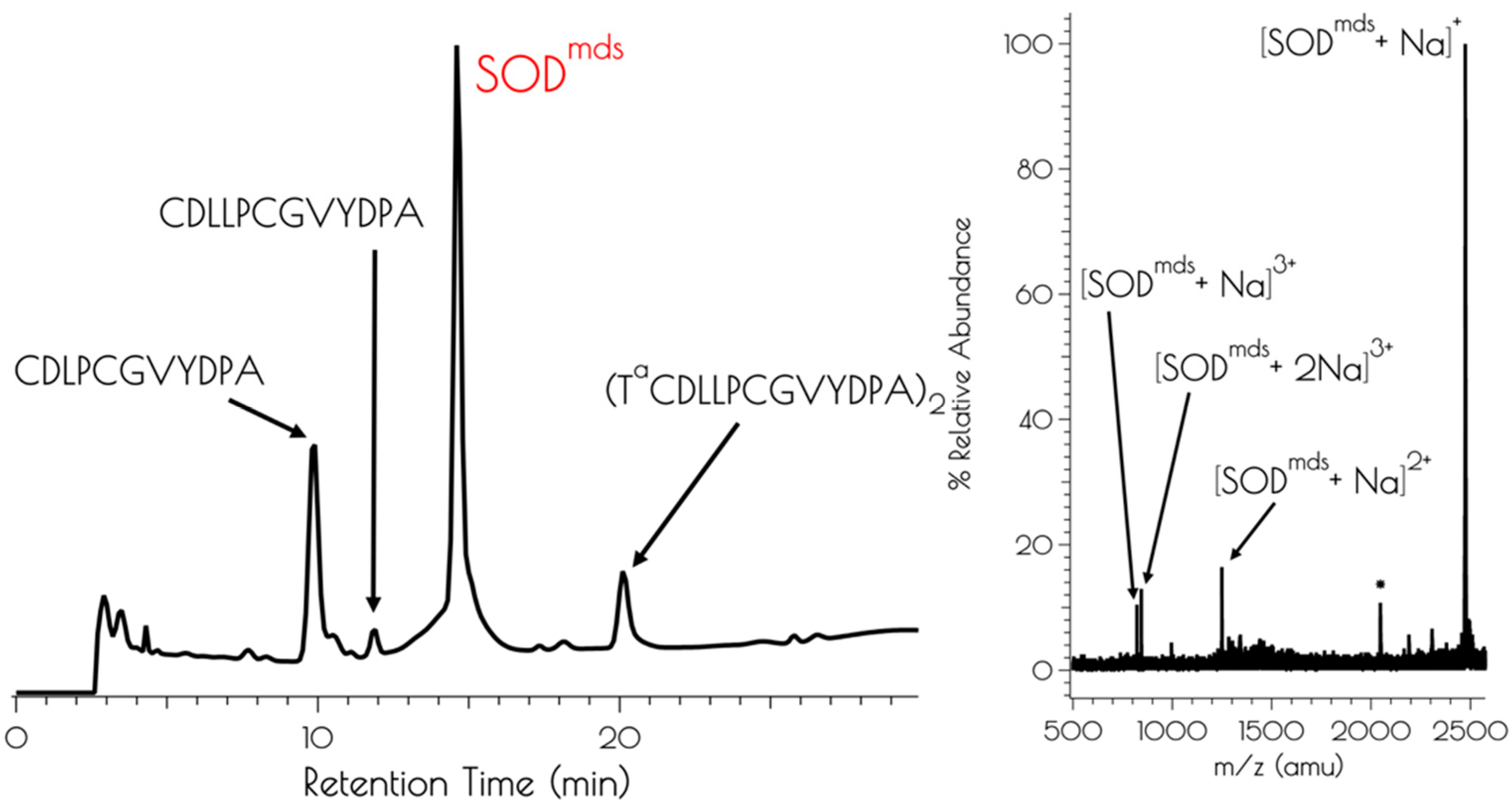
3.1. Generation of {Ni2II(SODmds)}
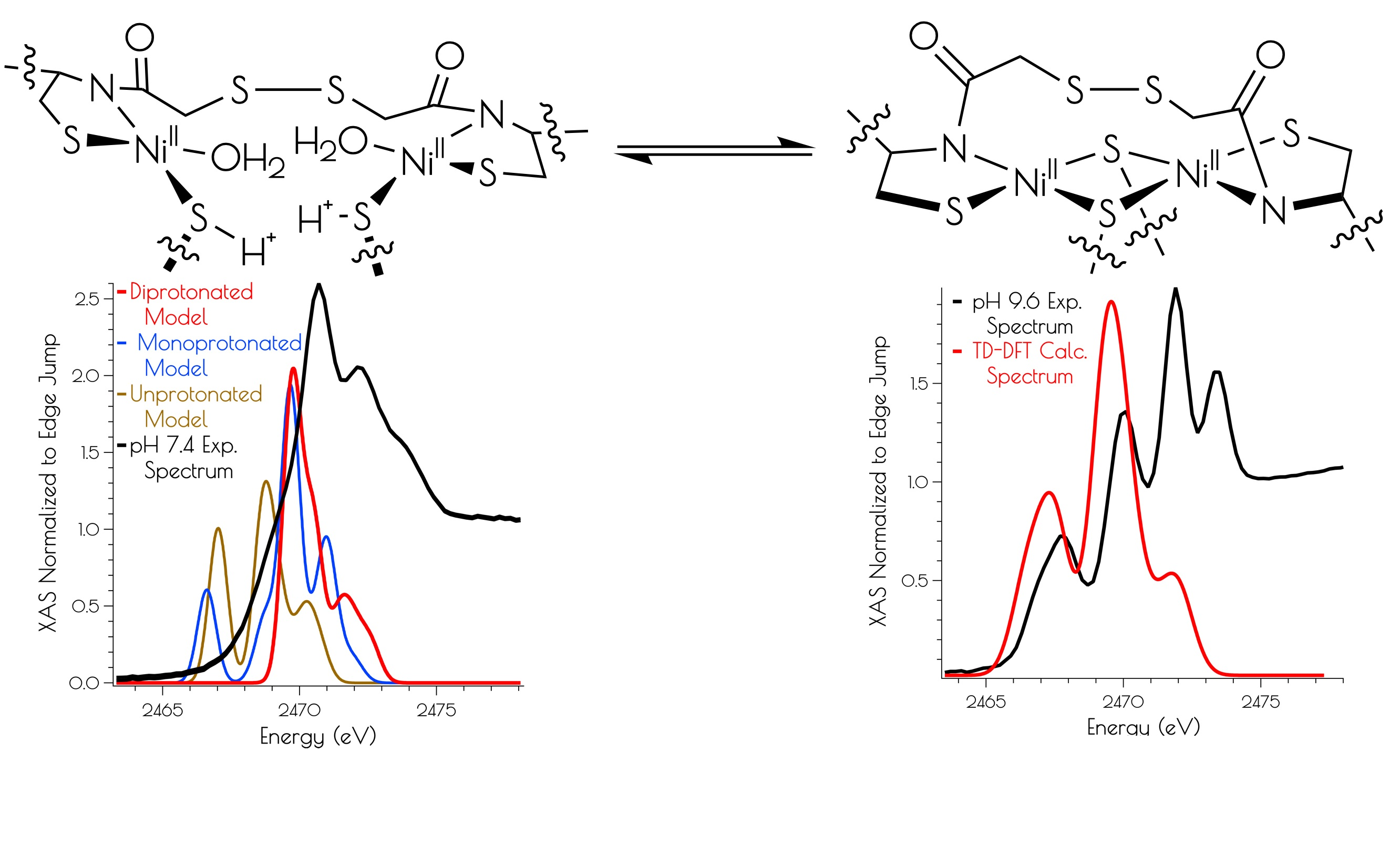
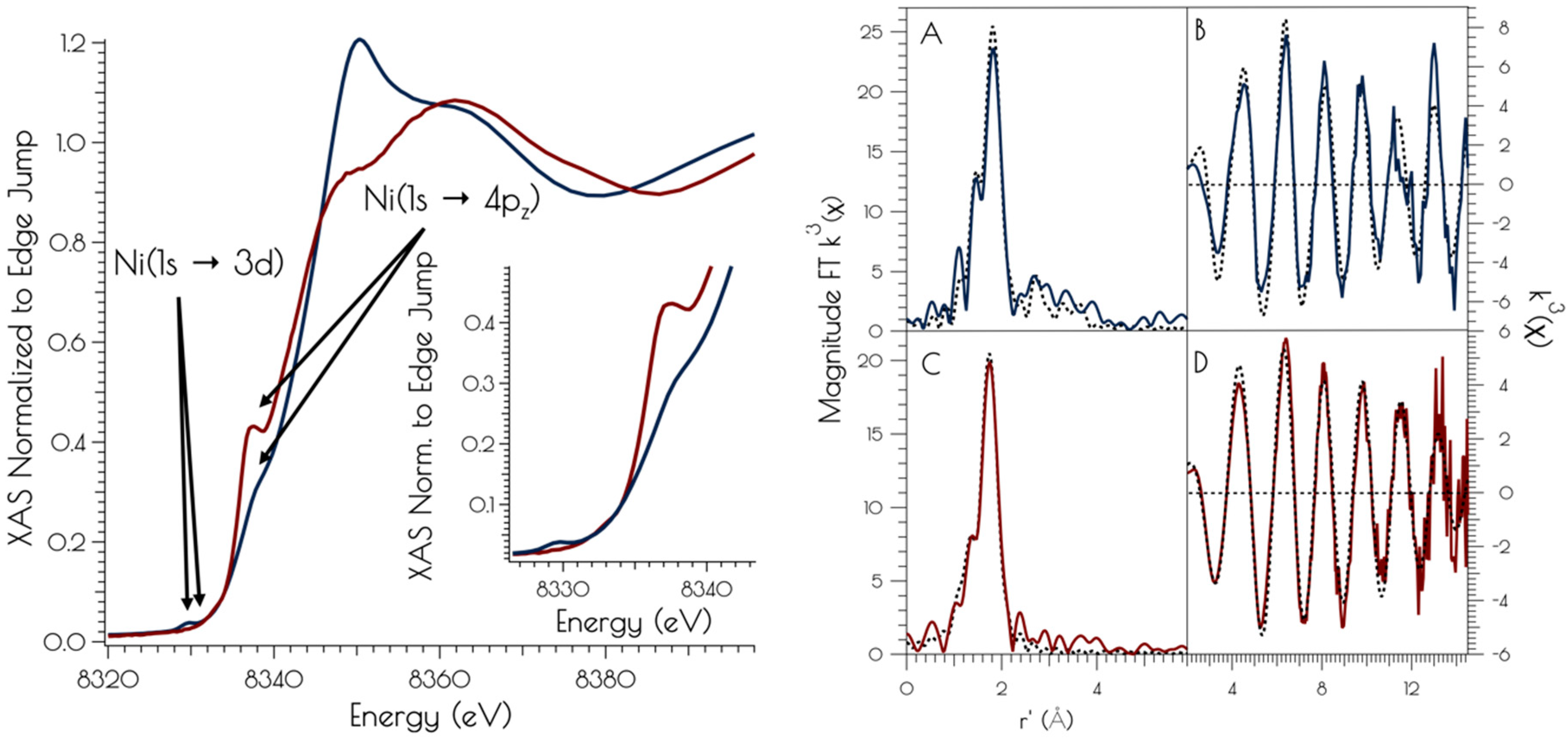
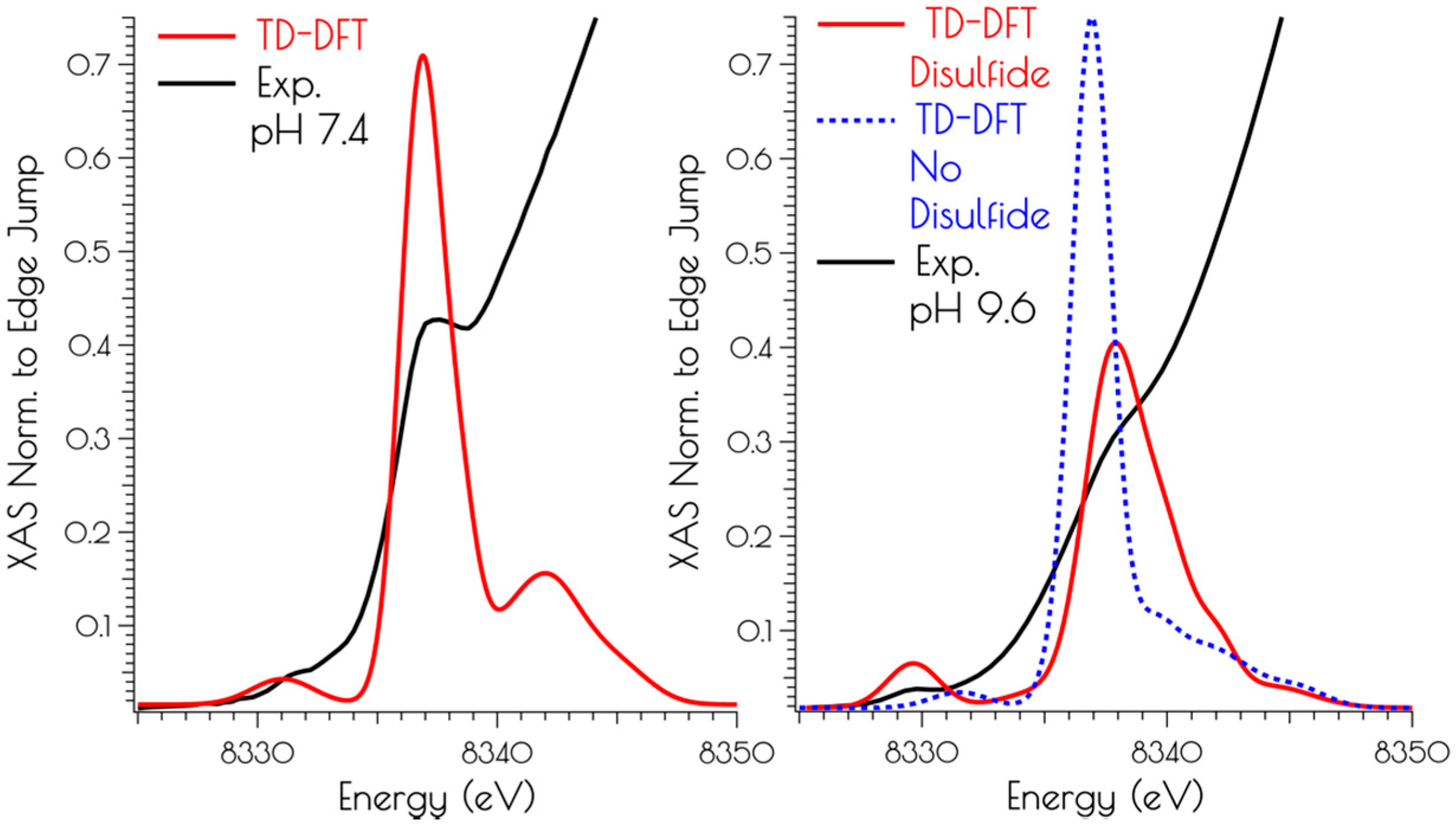
3.2. Nickel K-Edge X-ray Absorption Spectroscopy
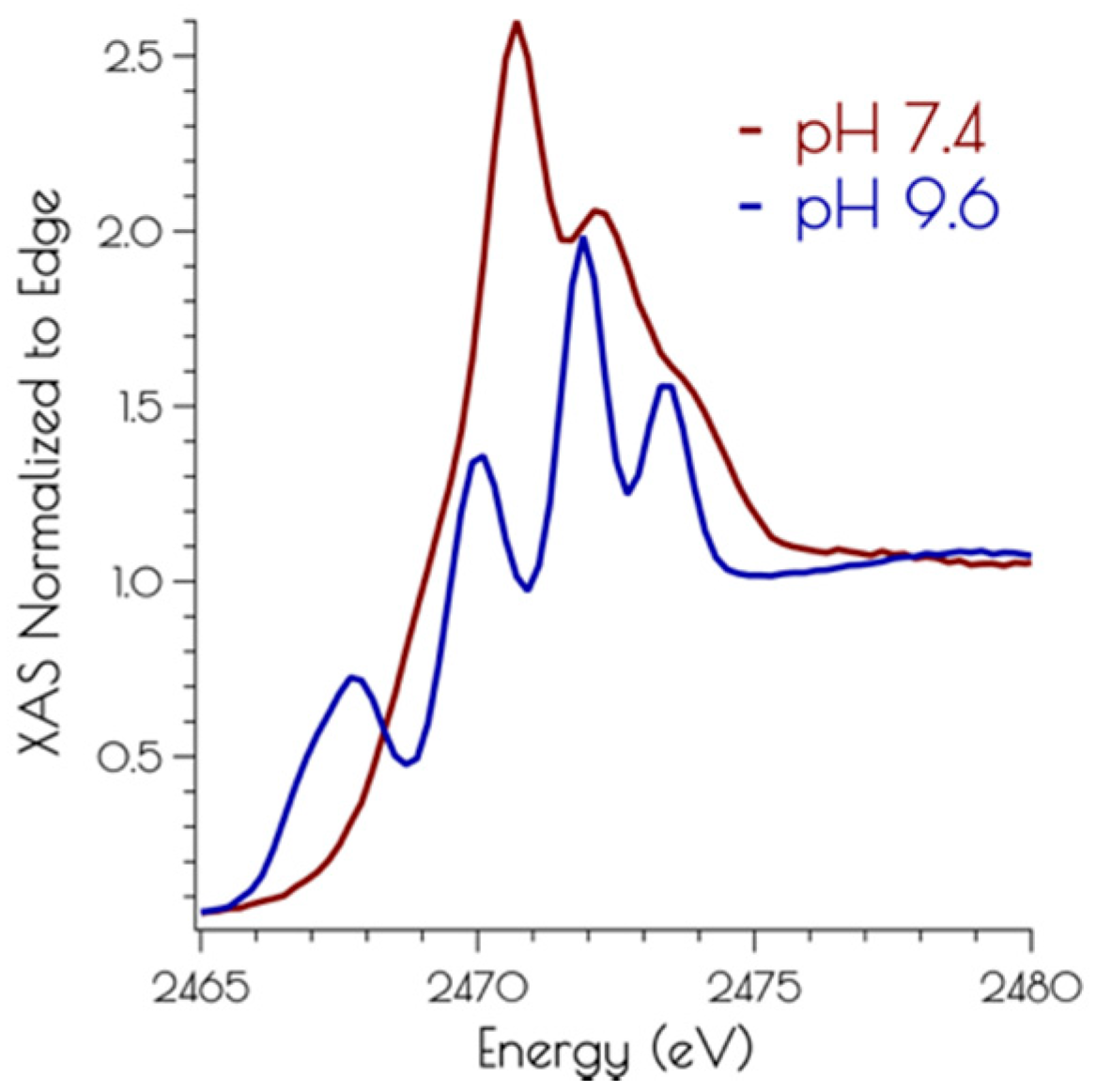
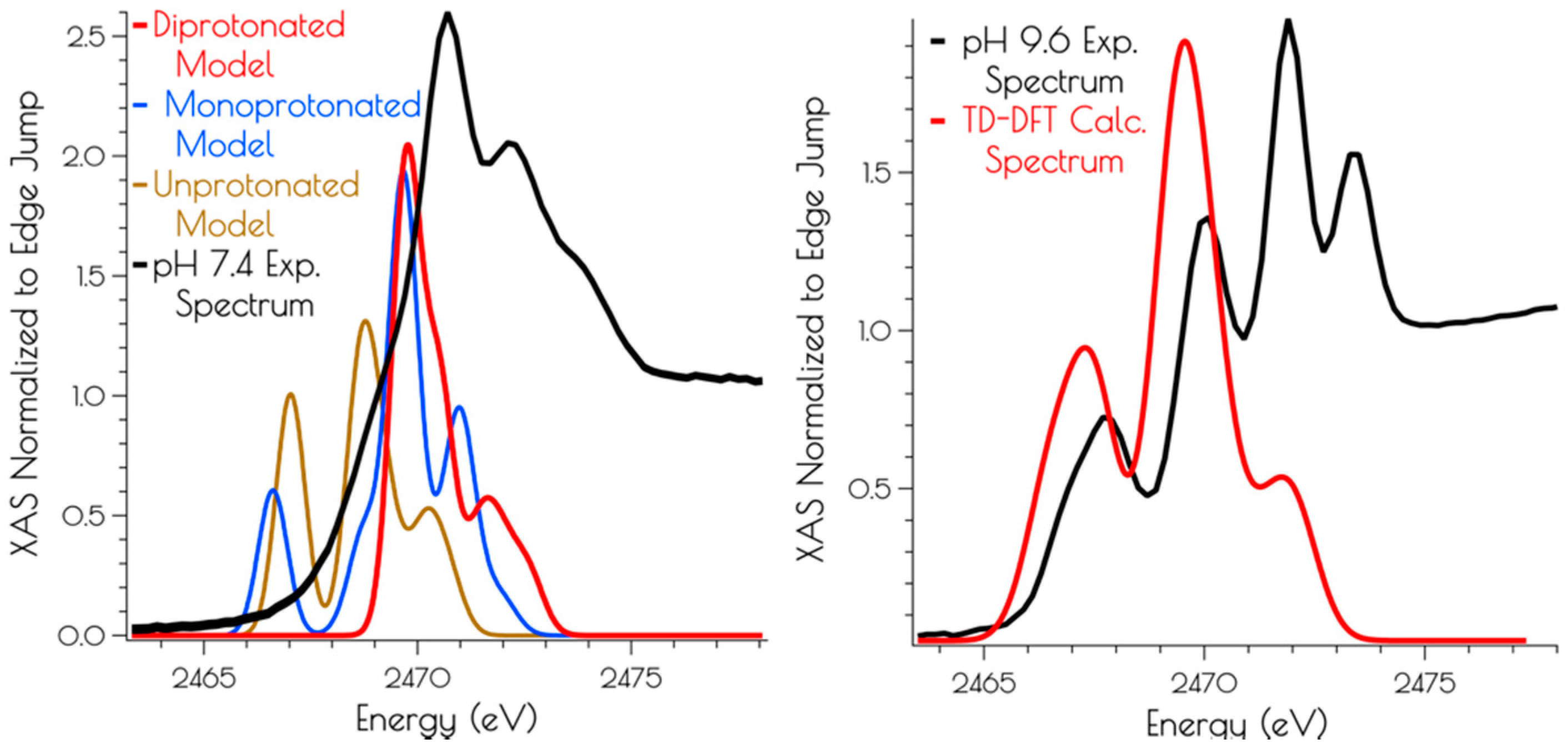
3.3. Sulfur K-Edge X-ray Absorption Spectroscopy of {Ni2II(SODmds)}
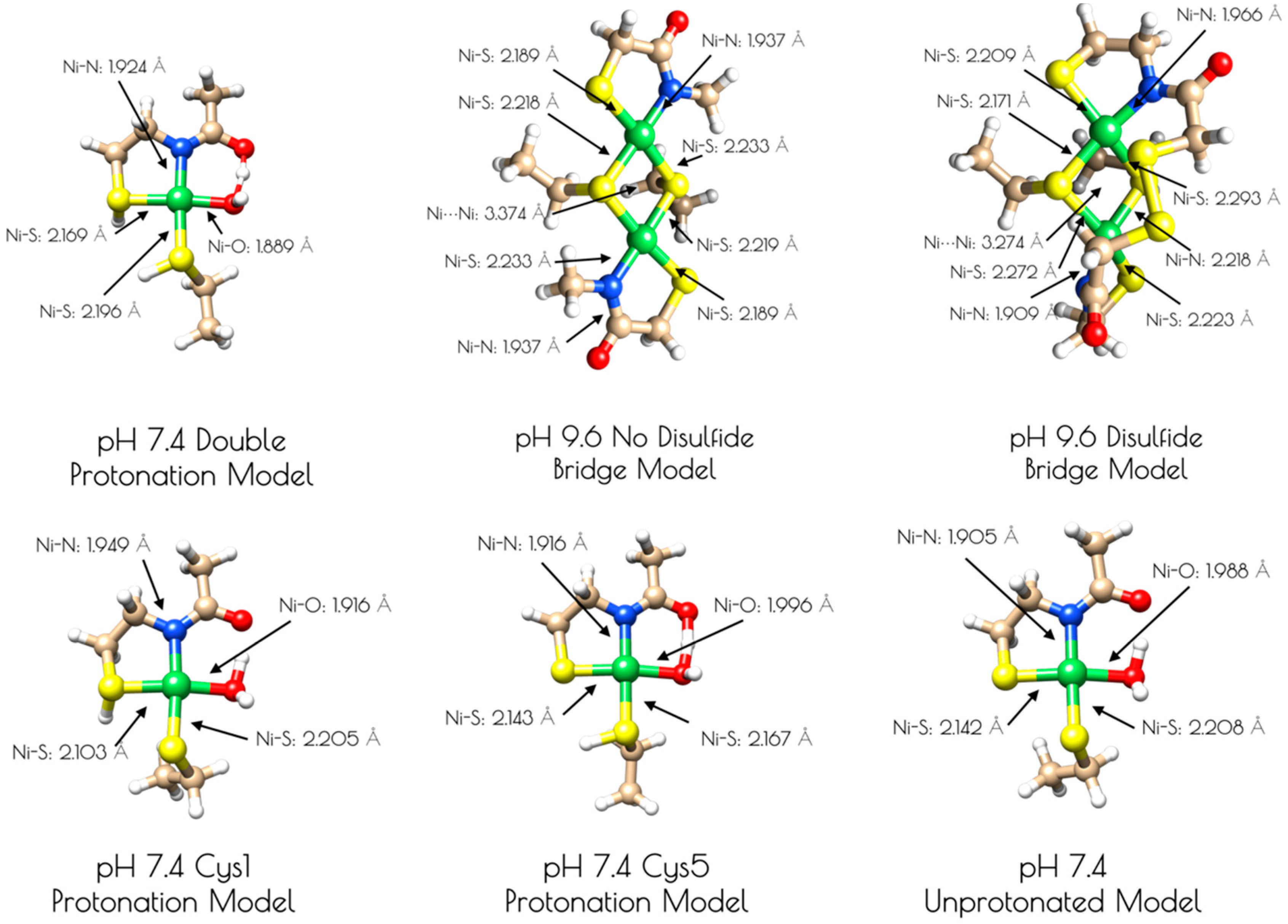
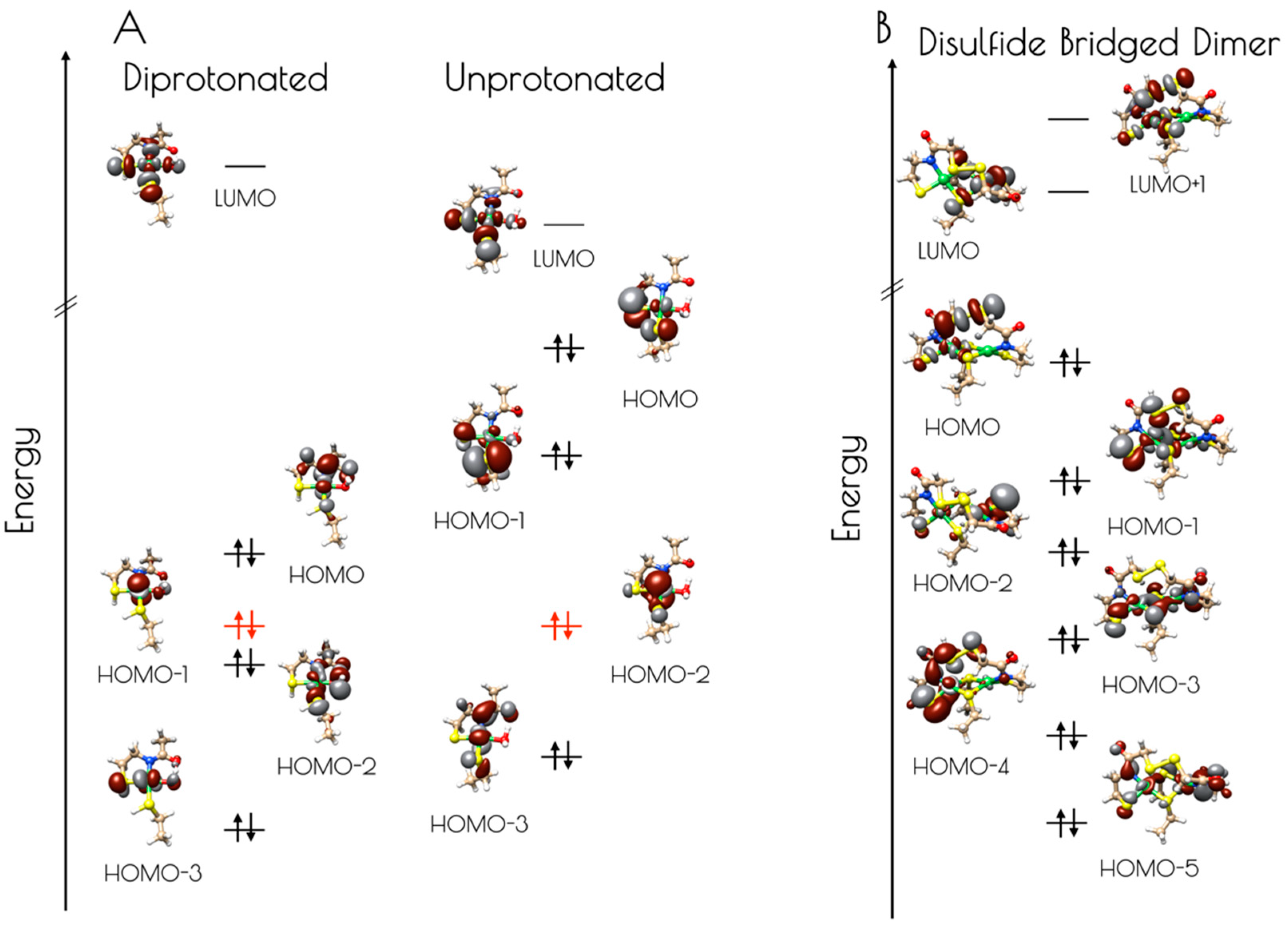
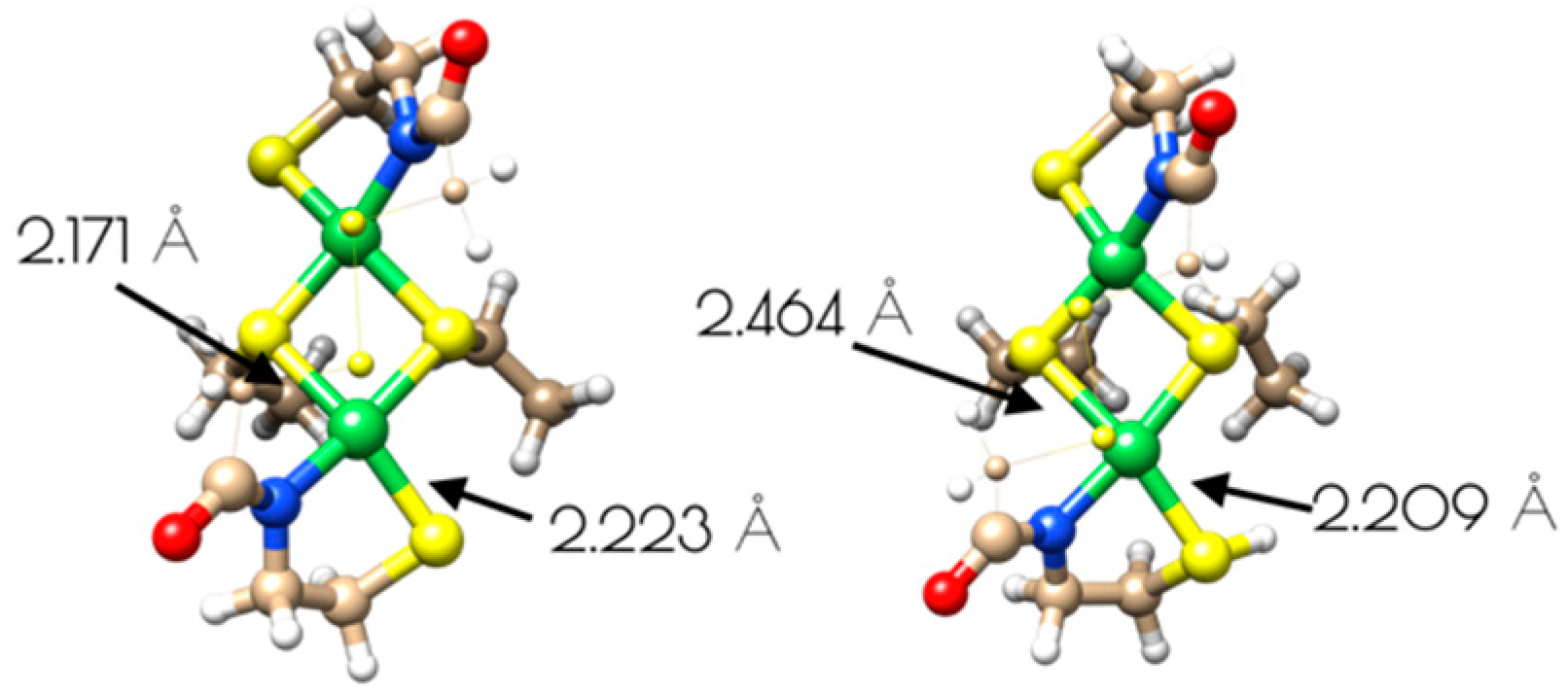
3.4. DFT Generated Structure for {Ni2II(SODmds)} and TD-DFT Calculations of Ni and S K-edge XAS Transitions
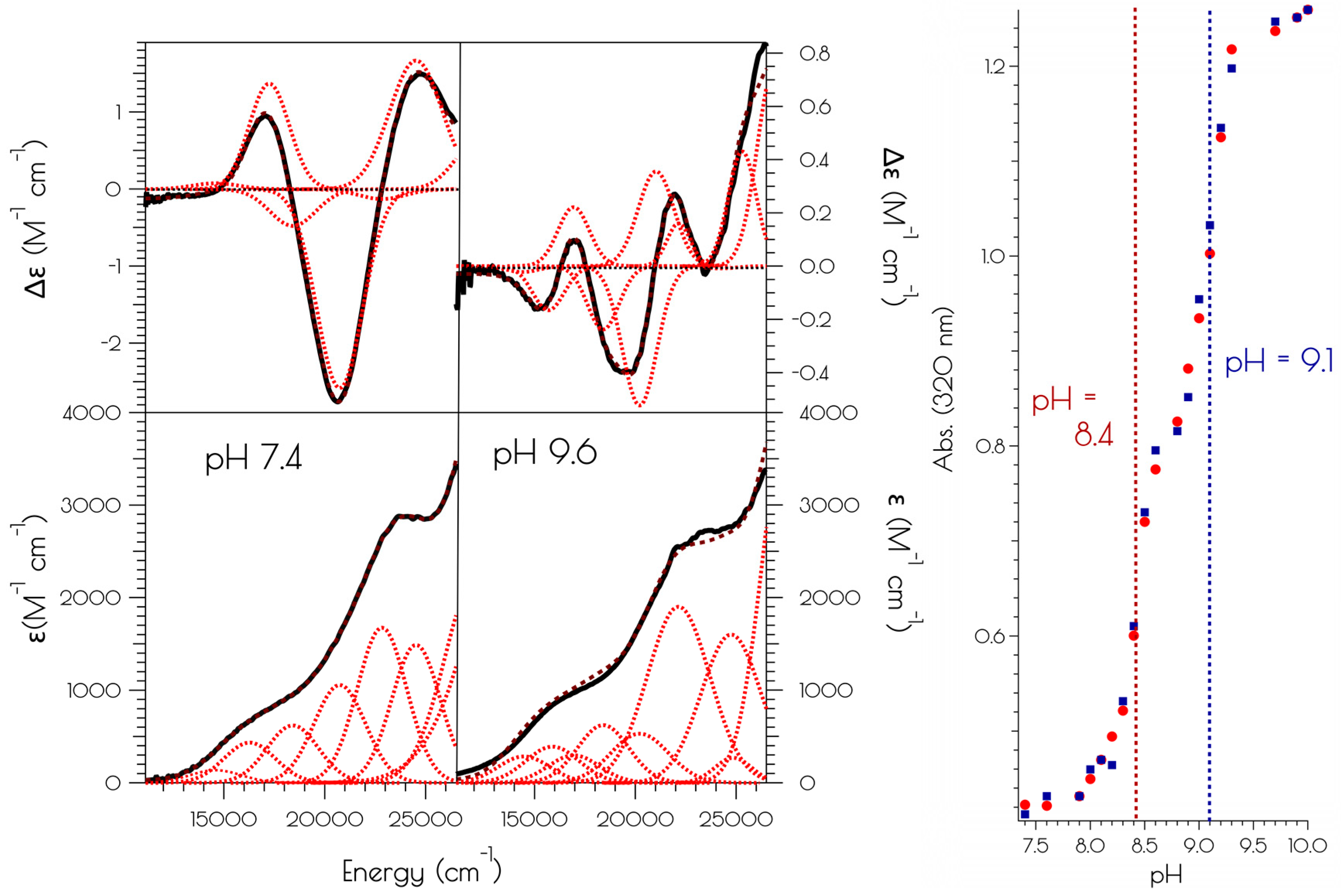
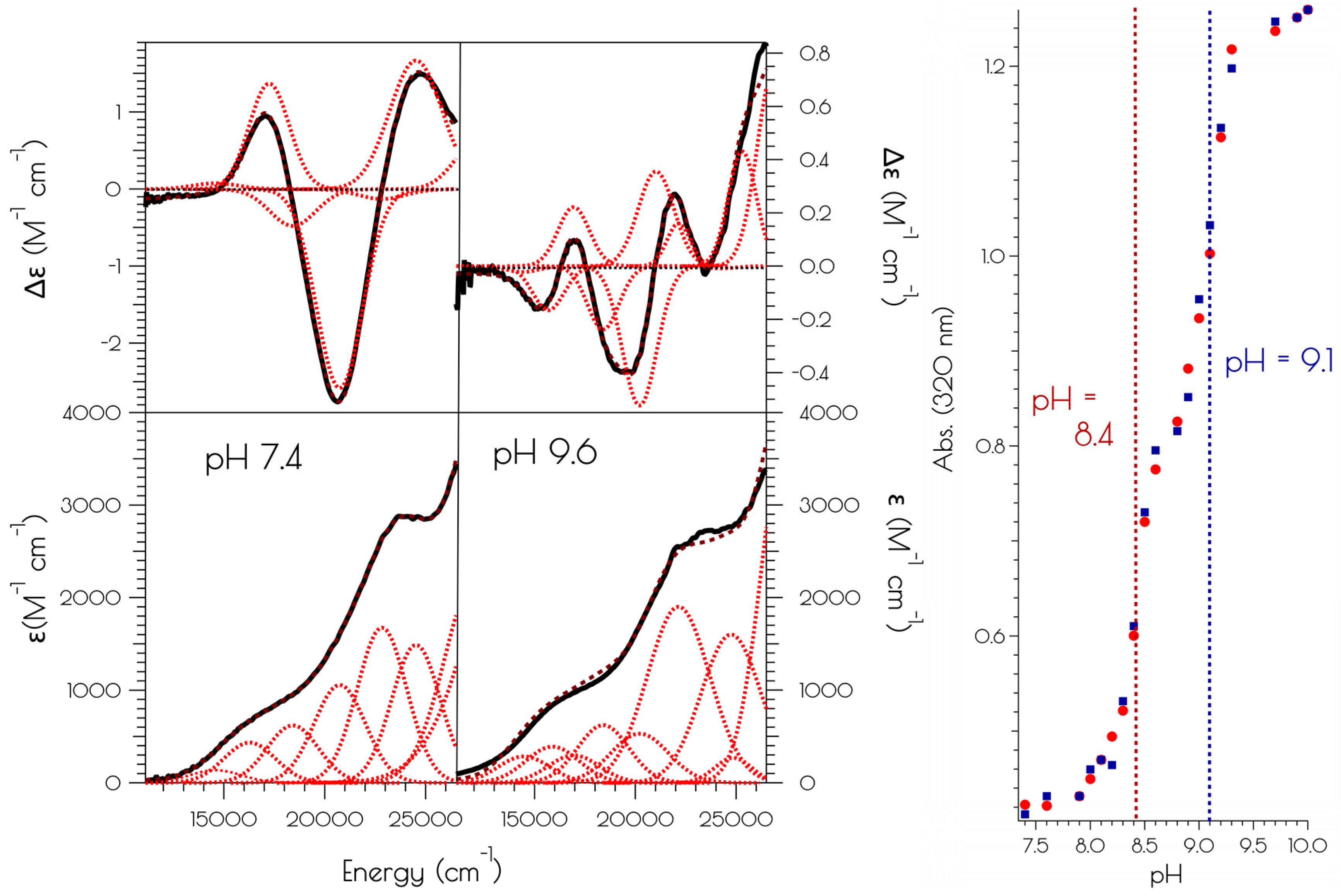
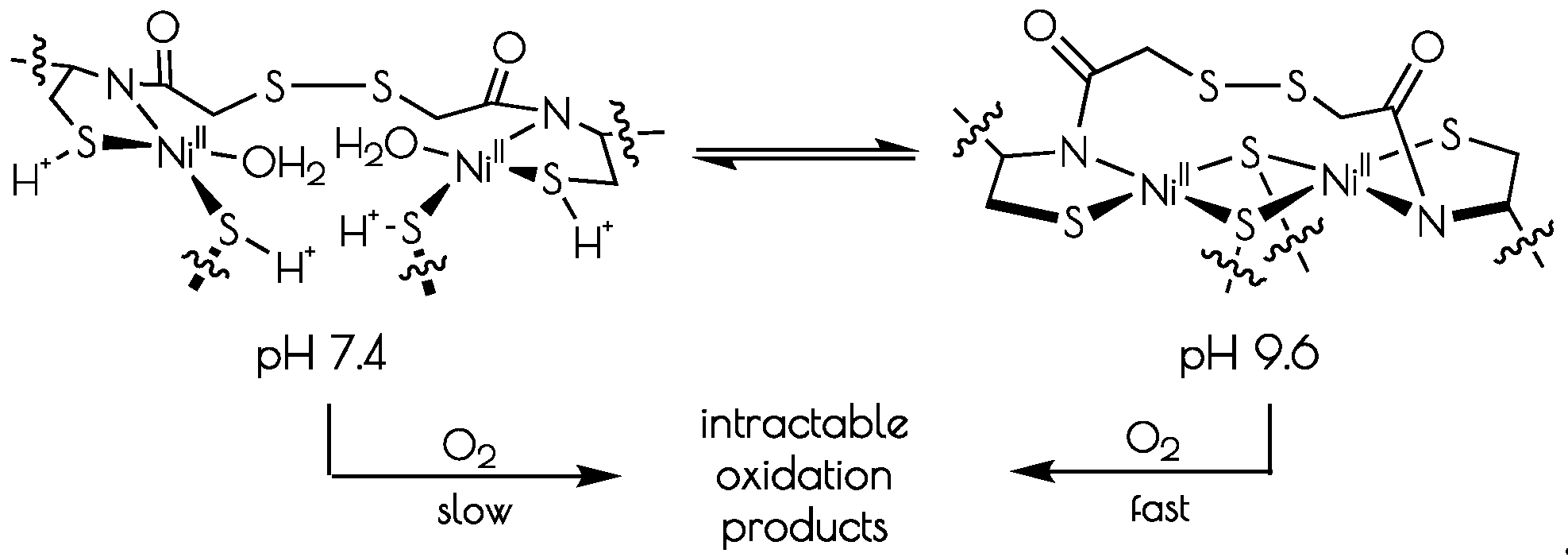
3.5. Oxidation of {Ni2II(SODmds)} at pH 7.4 and 9.6
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase. Chem. Rev. 2014, 114, 4149–4174. [Google Scholar] [CrossRef] [PubMed]
- Maroney, M.J.; Ciurli, S. Nonredox nickel enzymes. Chem. Rev. 2014, 114, 4206–4228. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Biochemistry of Methyl-Coenzyme M Reductase: The Nickel Metalloenzyme That Catalyzes the Final Step in Synthesis and the First Step in Anaerobic Oxidation of the Greenhouse Gas Methane; Springer: Dordrecht, The Netherlands, 2014; pp. 125–145. [Google Scholar]
- Ragsdale, S.W. Nickel biochemistry. Curr. Opin. Chem. Biol. 1998, 2, 208–215. [Google Scholar] [CrossRef]
- Ragsdale, S.W. Nickel-based enzyme systems. J. Biol. Chem. 2009, 284, 18571–18575. [Google Scholar] [CrossRef] [PubMed]
- Higgins, K.A.; Carr, C.E.; Maroney, M.J. Specific metal recognition in nickel trafficking. Biochemistry 2012, 51, 7816–7832. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.C.C.; Cao, L.; Dias, A.V.; Pickering, I.J.; George, G.N.; Zamble, D.B. A High-Affinity Metal-Binding Peptide from Escherichia coli HypB. J. Am. Chem. Soc. 2008, 130, 14056–14057. [Google Scholar] [CrossRef]
- Dias, A.V.; Mulvihill, C.M.; Leach, M.R.; Pickering, I.J.; George, G.N.; Zamble, D.B. Structural and Biological Analysis of the Metal Sites of Escherichia coli Hydrogenase Accessory Protein HypB. Biochemistry 2008, 47, 11981–11991. [Google Scholar] [CrossRef]
- Douglas, C.D.; Ngu, T.T.; Kaluarachchi, H.; Zamble, D.B. Metal Transfer within the Escherichia coli HypB-HypA Complex of Hydrogenase Accessory Proteins. Biochemistry 2013, 52, 6030–6039. [Google Scholar] [CrossRef]
- Lacasse, M.J.; Douglas, C.D.; Zamble, D.B. Mechanism of Selective Nickel Transfer from HypB to HypA, Escherichia coli [NiFe]-Hydrogenase Accessory Proteins. Biochemistry 2016, 55, 6821–6831. [Google Scholar] [CrossRef]
- Schreiter, E.R.; Sintchak, M.D.; Guo, Y.; Chivers, P.T.; Sauer, R.T.; Drennan, C.L. Crystal structure of the nickel-responsive transcription factor NikR. Nat. Struct. Mol. Biol. 2003, 10, 794–799. [Google Scholar] [CrossRef]
- Clegg, W.; Henderson, R.A. Kinetic Evidence for Intramolecular Proton Transfer Between Nickel and Coordinated Thiolate. Inorg. Chem. 2002, 41, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Autissier, V.; Zarza, P.M.; Petrou, A.; Henderson, R.A.; Harrington, R.W.; Clegg, W.C. Proton Transfer to Nickel-Thiolate Complexes. 2. Rate-Limiting Intramolecular Proton Transfer in the Reactions of [Ni(SC6H4R-4)(PhP{CHCH2PPh2}2)]+ (R = NO2, Cl, H, Me, or MeO). Inorg. Chem. 2004, 43, 3106–3115. [Google Scholar] [CrossRef] [PubMed]
- Alwaaly, A.; Henderson, R.A. Sterics level the rates of proton transfer to [Ni(XPh){PhP(CH2CH2PPh2)2}]+ (X = O, S or Se). Chem. Commun. 2014, 50, 9669–9671. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, R.K.; Bryngelson, P.A.; Maroney, M.J.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. S K-Edge X-ray Absorption Spectroscopic Investigation of the Ni-Containing Superoxide Dismutase Active Site: New Structural Insight into the Mechanism. J. Am. Chem. Soc. 2004, 126, 3018–3019. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571. [Google Scholar] [CrossRef] [PubMed]
- Shearer, J. Insight into the structure and mechanism of nickel-containing superoxide dismutase derived from peptide-based mimics. Acc. Chem. Res. 2014, 47, 2332–2341. [Google Scholar] [CrossRef]
- Shearer, J.; Peck, K.L.; Schmitt, J.C.; Neupane, K.P. Cysteinate protonation and water hydrogen bonding at the active-site of a nickel superoxide dismutase metallopeptide-based mimic: Implications for the mechanism of superoxide reduction. J. Am. Chem. Soc. 2014, 136, 16009–16022. [Google Scholar] [CrossRef]
- Shearer, J.; Schmitt, J.C.; Clewett, H.S. Adiabaticity of the Proton-Coupled Electron-Transfer Step in the Reduction of Superoxide Effected by Nickel-Containing Superoxide Dismutase Metallopeptide-Based Mimics. J. Phys. Chem. B 2015, 119, 5453–5461. [Google Scholar] [CrossRef]
- Pelmenschikov, V.; Siegbahn, P.E.M. Nickel Superoxide Dismutase Reaction Mechanism Studied by Hybrid Density Functional Methods. J. Am. Chem. Soc. 2006, 128, 7466–7475. [Google Scholar] [CrossRef]
- Krämer, T.; Kampa, M.; Lubitz, W.; van Gastel, M.; Neese, F. Theoretical Spectroscopy of the NiII Intermediate States in the Catalytic Cycle and the Activation of [NiFe] Hydrogenases. ChemBioChem 2013, 14, 1898–1905. [Google Scholar] [CrossRef]
- Shearer, J. Use of a Metallopeptide-Based Mimic Provides Evidence for a Proton-Coupled Electron-Transfer Mechanism for Superoxide Reduction By Nickel-Containing Superoxide Dismutase. Angew. Chem. Int. Ed. 2013, 52, 2569–2572. [Google Scholar] [CrossRef] [PubMed]
- Barondeau, D.P.; Kassmann, C.J.; Bruns, C.K.; Tainer, J.A.; Getzoff, E.D. Nickel Superoxide Dismutase Structure and Mechanism. Biochemistry 2004, 43, 8038–8047. [Google Scholar] [CrossRef] [PubMed]
- Wuerges, J.; Lee, J.-W.; Yim, Y.-I.; Yim, H.-S.; Kang, S.-O.; Carugo, K.D. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc. Natl. Acad. Sci. USA 2004, 101, 8569–8574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.C.; Guce, A.I.; Johnson, O.E.; Brunold, T.C.; Cabelli, D.E.; Garman, S.C.; Maroney, M.J. Nickel Superoxide Dismutase: Structural and Functional Roles of His1 and Its H-Bonding Network. Biochemistry 2015, 54, 1016–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, J.; Long, L.M. A Nickel Superoxide Dismutase Maquette That Reproduces the Spectroscopic and Functional Properties of the Metalloenzyme. Inorg. Chem. 2006, 45, 2358–2360. [Google Scholar] [CrossRef]
- Neupane, K.P.; Shearer, J. The Influence of Amine/Amide versus Bisamide Coordination in Nickel Superoxide Dismutase. Inorg. Chem. 2006, 45, 10552–10566. [Google Scholar] [CrossRef] [PubMed]
- Neupane, K.P.; Gearty, K.; Francis, A.; Shearer, J. Probing Variable Axial Ligation in Nickel Superoxide Dismutase Utilizing Metallopeptide-Based Models: Insight into the Superoxide Disproportionation Mechanism. J. Am. Chem. Soc. 2007, 129, 14605–14618. [Google Scholar] [CrossRef]
- Shearer, J.; Neupane, K.P.; Callan, P.E. Metallopeptide Based Mimics with Substituted Histidines Approximate a Key Hydrogen Bonding Network in the Metalloenzyme Nickel Superoxide Dismutase. Inorg. Chem. 2009, 48, 10560–10571. [Google Scholar] [CrossRef] [Green Version]
- Tietze, D.; Breitzke, H.; Imhof, D.; Koeth, E.; Weston, J.; Buntkowsky, G. New insight into the mode of action of nickel superoxide dismutase by investigating metallopeptide substrate models. Chem. - Eur. J. 2009, 15, 517–523. [Google Scholar] [CrossRef]
- Tietze, D.; Voigt, S.; Mollenhauer, D.; Tischler, M.; Imhof, D.; Gutmann, T.; Gonzalez, L.; Ohlenschlaeger, O.; Breitzke, H.; Goerlach, M.; et al. Revealing the Position of the Substrate in Nickel Superoxide Dismutase: A Model Study. Angew. Chem. Int. Ed. 2011, 50, 2946–2950. [Google Scholar] [CrossRef]
- Tietze, D.; Sartorius, J.; Koley Seth, B.; Herr, K.; Heimer, P.; Imhof, D.; Mollenhauer, D.; Buntkowsky, G. New insights into the mechanism of nickel superoxide degradation from studies of model peptides. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tietze, D.; Koley Seth, B.; Brauser, M.; Tietze, A.A.; Buntkowsky, G. NiII Complex Formation and Protonation States at the Active Site of a Nickel Superoxide Dismutase-Derived Metallopeptide: Implications for the Mechanism of Superoxide Degradation. Chem. - Eur. J. 2018, 24, 15879–15888. [Google Scholar] [CrossRef] [PubMed]
- Shearer, J.; Callan, P.E.; Amie, J. Use of Metallopeptide Based Mimics Demonstrates That the Metalloprotein Nitrile Hydratase Requires Two Oxidized Cysteinates for Catalytic Activity. Inorg. Chem. 2010, 49, 9064–9077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.; Flores, M.; Roy, S.; Schmitt, J.C.; Hamilton, G.A.; Hartnett, H.E.; Shearer, J.M.; Jones, A.K. Sequential oxidations of thiolates and the cobalt metallocenter in a synthetic metallopeptide: Implications for the biosynthesis of nitrile hydratase. Inorg. Chem. 2013, 52, 5236–5245. [Google Scholar] [CrossRef] [PubMed]
- Martinage, O.; Le Clainche, L.; Czarny, B.; Dugave, C. Synthesis and biological evaluation of a new triazole-oxotechnetium complex. Org. Biomol. Chem. 2012, 10, 6484–6490. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. A colorimetric method for determining low concentrations of mercaptans. Arch Biochem Biophys 1958, 74, 443–450. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Weigend, F.A. Reinhart, Balanced basis sets of split valence, triple zeta valance and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Hofener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Chem. Phys. Phys. Chem. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.E.; Keah, H.H.; Gomme, P.T.; Stanton, P.G.; Hearn, M.T. Synthesis of peptide amides using Fmoc-based solid-phase procedures on 4-methylbenzhydrylamine resins. Int. J. Pept. Protein Res. 1995, 46, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Colpas, G.J.; Maroney, M.J.; Bagyinka, C.; Kumar, M.; Willis, W.S.; Suib, S.L.; Mascharak, P.K.; Baidya, N. X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases. Inorg. Chem. 1991, 30, 920–928. [Google Scholar] [CrossRef]
- Denny, J.A.; Darensbourg, M.Y. Metallodithiolates as Ligands in Coordination, Bioinorganic, and Organometallic Chemistry. Chem. Rev. 2015, 115, 5248–5273. [Google Scholar] [CrossRef]
- Jenkins, R.M.; Singleton, M.L.; Leamer, L.A.; Reibenspies, J.H.; Darensbourg, M.Y. Orientation and Stereodynamic Paths of Planar Monodentate Ligands in Square Planar Nickel N2S Complexes. Inorg. Chem. 2010, 49, 5503–5514. [Google Scholar] [CrossRef]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Kirchner, B.; Wennmohs, F.; Ye, S.; Neese, F. Theoretical bioinorganic chemistry: The electronic structure makes a difference. Curr. Opin. Chem. Biol. 2007, 11, 134–141. [Google Scholar] [CrossRef]
- Jenkins, R.M.; Singleton, M.L.; Almaraz, E.; Relbenspies, J.H.; Darensbourg, M.Y. Imidazole-Containing (N3S)-Ni-II Complexes Relating to Nickel Containing Biomolecules. Inorg. Chem. 2009, 48, 7280–7293. [Google Scholar] [CrossRef]
- Grapperhaus, C.A.; Darensbourg, M.Y. Oxygen Capture by Sulfur in Nickel Thiolates. Acc. Chem. Res. 1998, 31, 451–459. [Google Scholar] [CrossRef]
- Green, K.N.; Brothers, S.M.; Jenkins, R.M.; Carson, C.E.; Grapperhaus, C.A.; Darensbourg, M.Y. An Experimental and Computational Study of Sulfur-Modified Nucleophilicity in a Dianionic NiN2S2 Complex. Inorg. Chem. 2007, 46, 7536–7544. [Google Scholar] [CrossRef] [PubMed]
- Shearer, J.; Dehestani, A.; Abanda, F. Probing Variable Amine/Amide Ligation in NiIIN2S2 Complexes Using Sulfur K-Edge and Nickel L-Edge X-ray Absorption Spectroscopies: Implications for the Active Site of Nickel Superoxide Dismutase. Inorg. Chem. 2008, 47, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Mullins, C.S.; Grapperhaus, C.A.; Kozlowski, P.M. Density functional theory investigations of NiN2S2 reactivity as a function of nitrogen donor type and N–H···S hydrogen bonding inspired by nickel-containing superoxide dismutase. J. Biol. Inorg. Chem. 2006, 11, 617–625. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LUMO | HOMO | HOMO-1 | HOMO-2 | HOMO-3 | |
|---|---|---|---|---|---|
| Ni doubly-protonated unprotonated | 61.4 51.7 | 48.9 29.9 | 76.8 16.2 | 29.9 76.3 | 80.5 61.7 |
| S1 doubly-protonated unprotonated | 6.0 11.5 | 1.6 9.9 | 2.4 55.7 | 7.4 5.6 | 0.0 0.3 |
| S2 doubly-protonated unprotonated | 3.9 11.6 | 0.2 46.1 | 1.1 10.1 | 0.1 6.5 | 6.4 4.9 |
| N doubly-protonated unprotonated | 3.9 2.1 | 28.5 0.2 | 2.7 10.0 | 3.3 11.0 | 0.0 5.3 |
| O doubly-protonated unprotonated | 7.4 3.6 | 0.2 0.6 | 6.7 3.0 | 41.0 0.9 | 3.7 4.8 |
| E doubly-protonated unprotonated | 5.36 5.12 | 0.29 0.73 | 0.00 0.42 | −0.07 0.00 | −0.49 −0.22 |
| LUMO+1 | LUMO | HOMO | HOMO-1 | HOMO-2 | HOMO-3 | HOMO-4 | HOMO-5 | |
|---|---|---|---|---|---|---|---|---|
| Ni1 | 0.3 | 21.1 | 28.2 | 4.8 | 11.8 | 20.3 | 7.5 | 18.5 |
| Ni2 | 50.6 | 0.4 | 4.3 | 23.3 | 11.7 | 1.7 | 24.6 | 38.4 |
| S3 | 0.4 | 16.4 | 10.4 | 1.5 | 1.2 | 10.2 | 0.1 | 0.4 |
| S4 | 0.4 | 17.1 | 5.2 | 0.2 | 0.6 | 88 | 0.8 | 7.3 |
| S5 | 6.1 | 2.0 | 4.2 | 2.0 | 9.5 | 0.6 | 1.2 | 1.1 |
| S6 | 8.7 | 2.4 | 9.5 | 1.4 | 8.9 | 0.4 | 6.0 | 1.4 |
| S7 | 0.1 | 5.1 | 21.9 | 4.6 | 15.6 | 30.0 | 6.1 | 5.1 |
| S8 | 10.1 | 0.0 | 3.2 | 49.2 | 15.2 | 0.4 | 2.3 | 4.3 |
| N9 | 0.2 | 1.6 | 0.4 | 0.2 | 1.7 | 6.1 | 5.7 | 5.5 |
| N10 | 3.0 | 0.0 | 0.2 | 0.9 | 5.0 | 0.8 | 14.9 | 3.8 |
| E | 3.76 | 3.62 | 0.00 | −0.30 | −0.54 | −0.69 | −0.72 | −1.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keegan, B.C.; Ocampo, D.; Shearer, J. pH Dependent Reversible Formation of a Binuclear Ni2 Metal-Center within a Peptide Scaffold. Inorganics 2019, 7, 90. https://doi.org/10.3390/inorganics7070090
Keegan BC, Ocampo D, Shearer J. pH Dependent Reversible Formation of a Binuclear Ni2 Metal-Center within a Peptide Scaffold. Inorganics. 2019; 7(7):90. https://doi.org/10.3390/inorganics7070090
Chicago/Turabian StyleKeegan, Brenna C., Daniel Ocampo, and Jason Shearer. 2019. "pH Dependent Reversible Formation of a Binuclear Ni2 Metal-Center within a Peptide Scaffold" Inorganics 7, no. 7: 90. https://doi.org/10.3390/inorganics7070090
APA StyleKeegan, B. C., Ocampo, D., & Shearer, J. (2019). pH Dependent Reversible Formation of a Binuclear Ni2 Metal-Center within a Peptide Scaffold. Inorganics, 7(7), 90. https://doi.org/10.3390/inorganics7070090