“Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition



Abstract
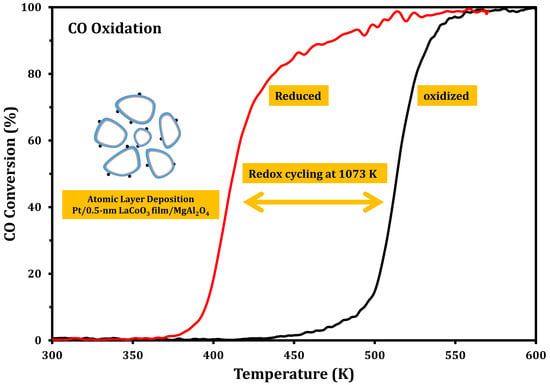
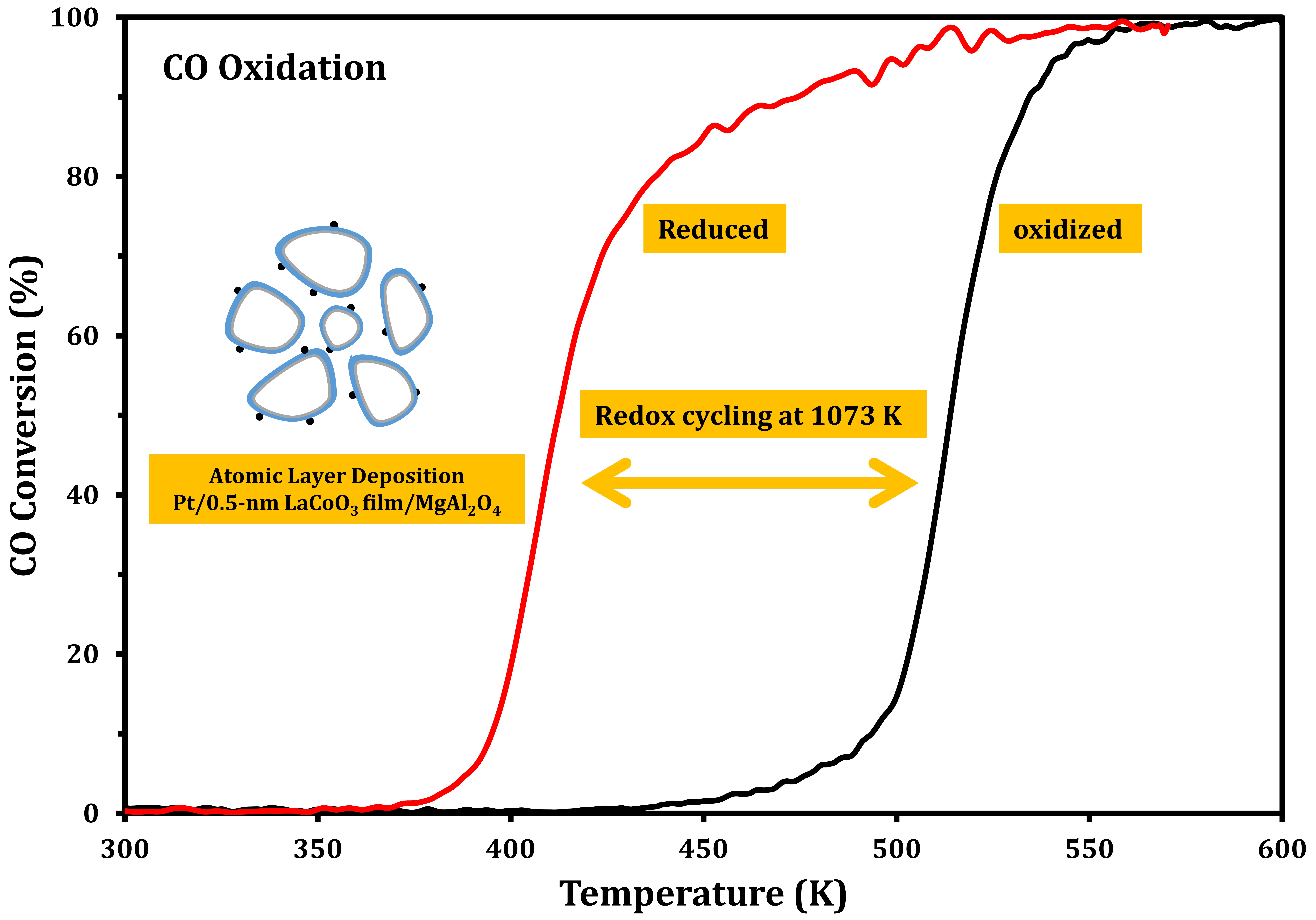
:
1. Introduction
2. Results
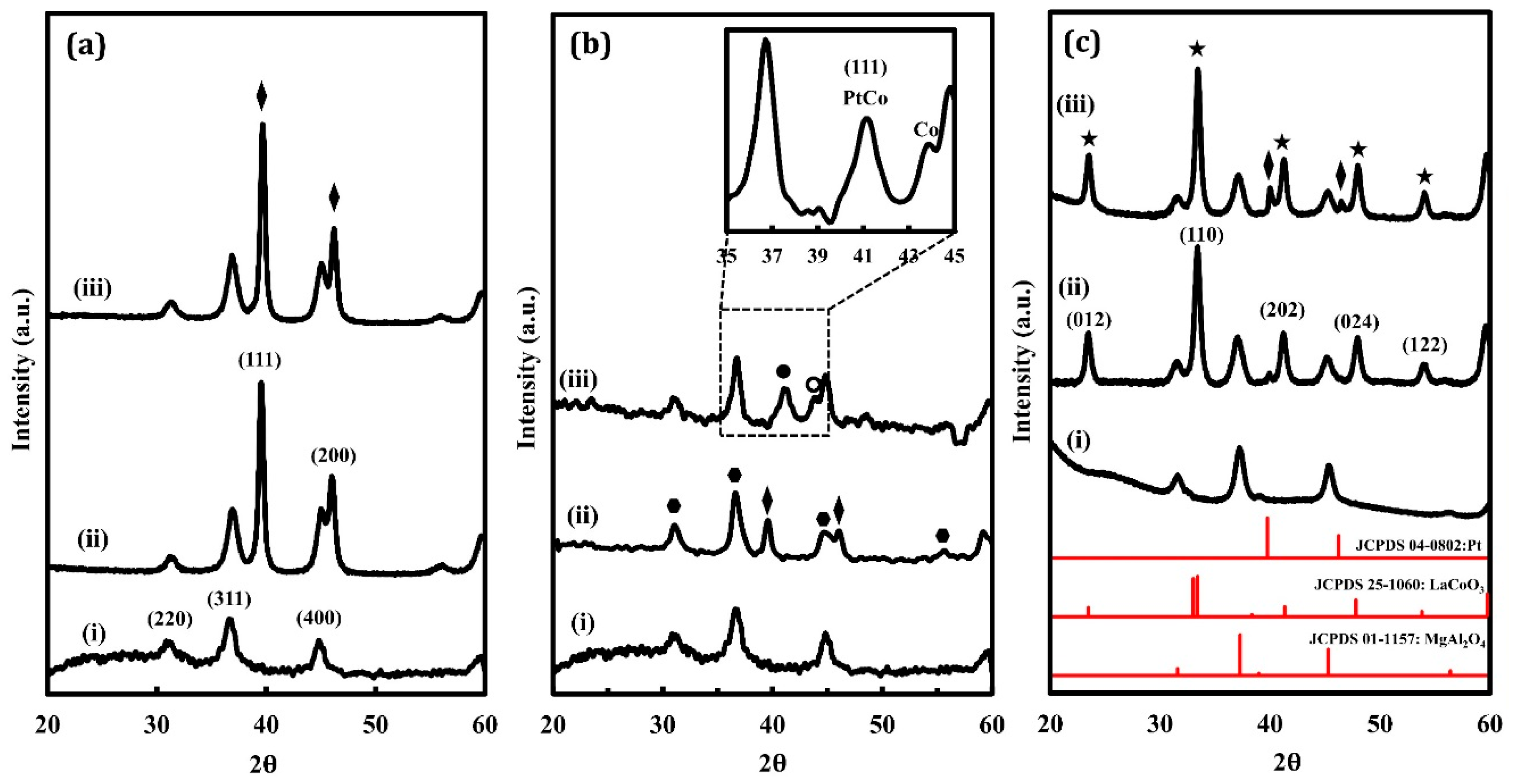
2.1. Catalyst Characterization
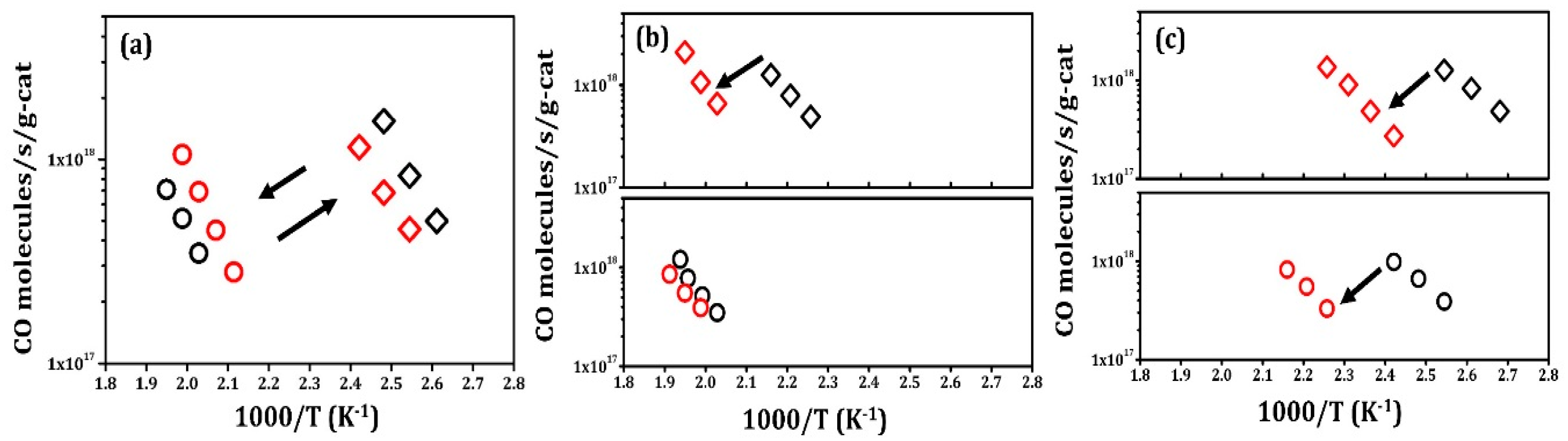
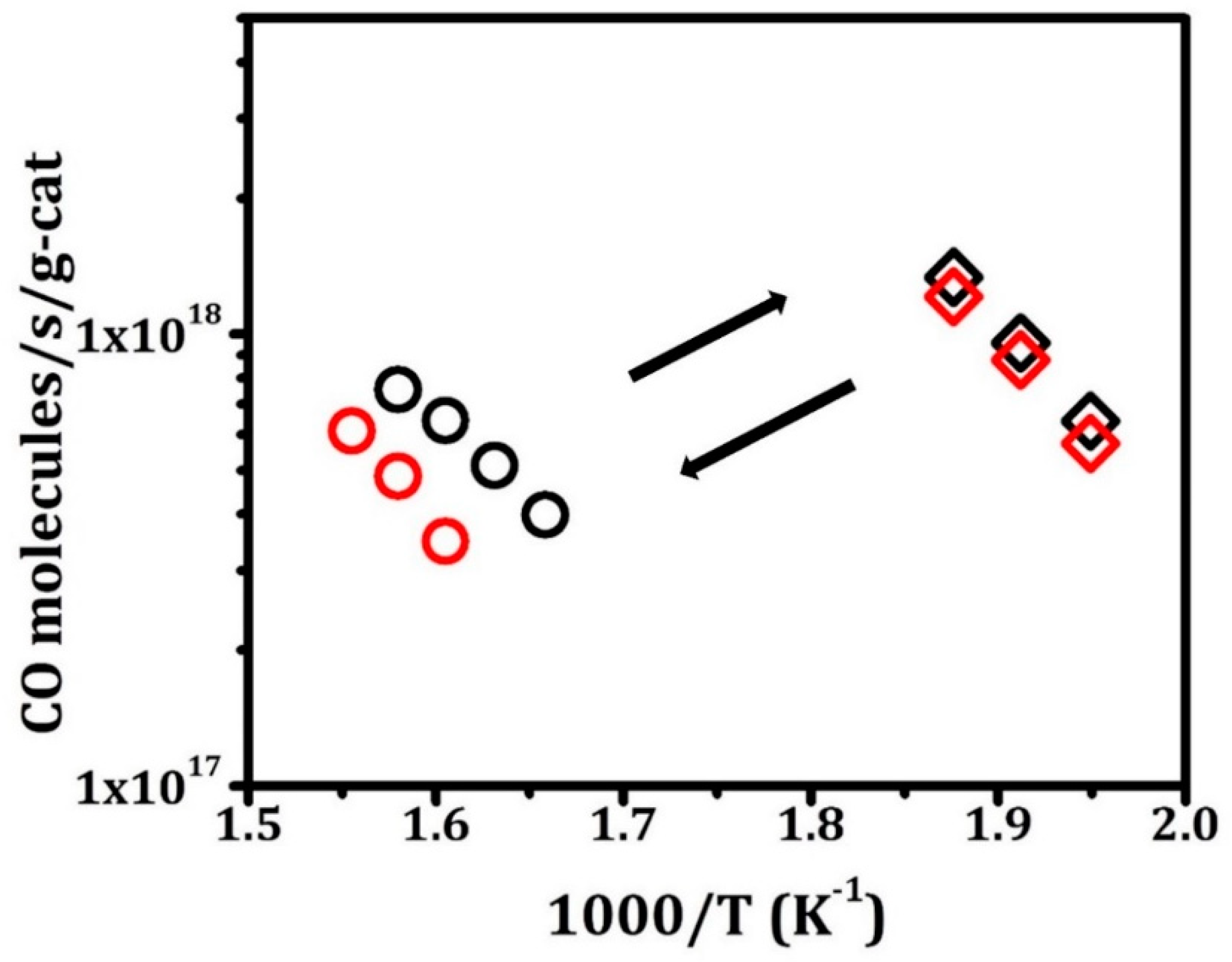
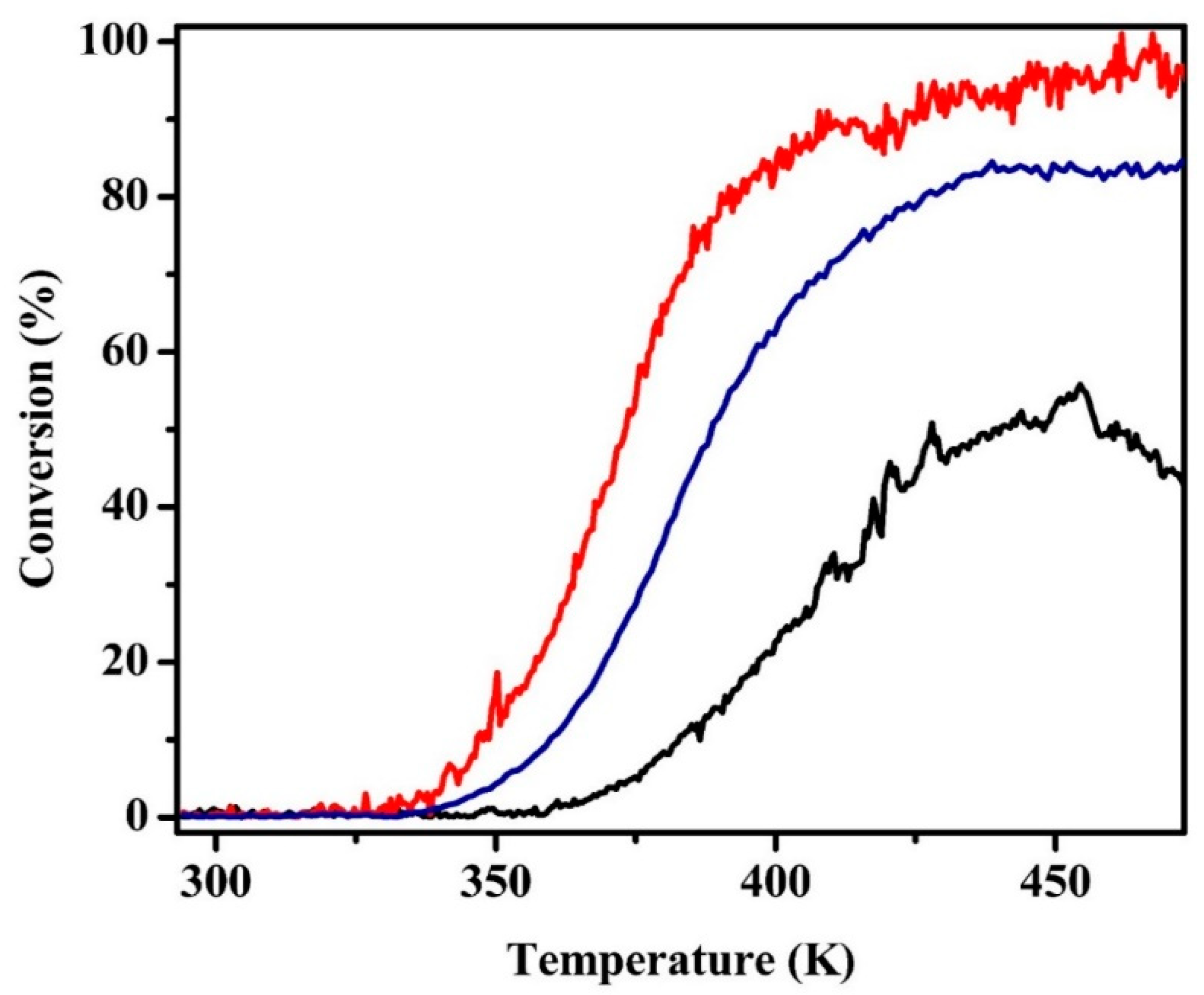
2.2. CO Oxidation Rates
2.3. WGS and Toluene Hydrogenation
3. Discussion
4. Experimental Methods
4.1. Sample Preparation
4.2. Characterization Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Farrauto, R.J.; Deeba, M.; Alerasool, S. Gasoline automobile catalysis and its historical journey to cleaner air. Nat. Catal. 2019, 2, 603–613. [Google Scholar] [CrossRef]
- Nishihata, Y.; Mizuki, J.; Akao, T.; Tanaka, H.; Uenishi, M.; Kimura, M.; Okamoto, T.; Hamada, N. Self-regeneration of a Pd-perovskite catalyst for automotive emissions control. Nature 2002, 418, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Katz, M.B.; Dai, S.; Zhang, K.; Du, X.; Graham, G.W.; Pan, X.Q. New Atomic-Scale Insight into Self-Regeneration of Pt-CaTiO3 Catalysts: Incipient Redox-Induced Structures Revealed by a Small-Angle Tilting STEM Technique. J. Phys. Chem. C 2017, 121, 17348–17353. [Google Scholar] [CrossRef]
- Dai, S.; Zhang, S.; Katz, M.B.; Graham, G.W.; Pan, X.Q. In Situ Observation of Rh-CaTiO3 Catalysts during Reduction and Oxidation Treatments by Transmission Electron Microscopy. ACS Catal. 2017, 7, 1579–1582. [Google Scholar] [CrossRef]
- Neagu, D.; Oh, T.-S.; Miller, D.N.; Ménard, H.; Bukhari, S.M.; Gamble, S.R.; Gorte, R.J.; Vohs, J.M.; Irvine, J.T. Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution. Nat. Commun. 2015, 6, 8120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onn, T.M.; Monai, M.; Dai, S.; Fonda, E.; Montini, T.; Pan, X.Q.; Graham, G.W.; Fornasiero, P.; Gorte, R.J. Smart Pd Catalyst with Improved Thermal Stability Supported on High-Surface-Area LaFeO3 Prepared by Atomic Layer Deposition. J. Am. Chem. Soc. 2018, 140, 4841–4848. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Jang, J.B.; Zhang, L.; Stach, E.A.; Gorte, R.J. Improved Coking Resistance of “Intelligent” Ni Catalysts Prepared by Atomic Layer Deposition. ACS Catal. 2018, 8, 7679–7687. [Google Scholar] [CrossRef]
- Lin, C.; Foucher, A.C.; Ji, Y.; Curran, C.D.; Stach, E.A.; McIntosh, S.; Gorte, R.J. “Intelligent” Pt Catalysts Studied on High-Surface-Area CaTiO3 Films. ACS Catal. 2019, 9, 7318–7327. [Google Scholar] [CrossRef]
- Tanaka, H.; Taniguchi, M.; Uenishi, M.; Kajita, N.; Tan, I.; Nishihata, Y.; Mizuki, J.I.; Narita, K.; Kimura, M.; Kaneko, K. Self-regenerating Rh- and Pt-based perovskite catalysts for automotive-emissions control. Angew. Chem. Int. Ed. 2006, 45, 5998–6002. [Google Scholar] [CrossRef]
- Bidrawn, F.; Kungas, R.; Vohs, J.M.; Gorte, R.J. Modeling Impedance Response of SOFC Cathodes Prepared by Infiltration. J. Electrochem. Soc. 2011, 158, B514–B525. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.W.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W.J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Bahlawane, N. Kinetics of methane combustion over CVD-made cobalt oxide catalysts. Appl. Catal. B-Environ. 2006, 67, 168–176. [Google Scholar] [CrossRef]
- Mergler, Y.J.; van Aalst, A.; van Delft, J.; Nieuwenhuys, B.E. Promoted Pt catalysts for automotive pollution control: Characterization of Pt/SiO2, Pt/CoOx/SiO2, and Pt/MnOx/SiO2 catalysts. J. Catal. 1996, 161, 310–318. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Espro, C.; Fazio, B.; Palella, A.; Spadaro, L. A New Class of MnCeOx Materials for the Catalytic Gas Exhausts Emission Control: A Study of the CO Model Compound Oxidation. Top. Catal. 2019, 62, 259–265. [Google Scholar] [CrossRef]
- Arena, F.; Chio, R.D.; Espro, C.; Fazio, B.; Palella, A.; Spadaro, L. A definitive assessment of the CO oxidation pattern of a nanocomposite MnCeOx catalyst. React. Chem. Eng. 2018, 3, 293–300. [Google Scholar] [CrossRef]
- Mao, X.; Foucher, A.; Stach, E.A.; Gorte, R.J. A Study of Support Effects for CH4 and CO Oxidation over Pd Catalysts on ALD-Modified Al2O3. Catal. Lett. 2019, 149, 905–915. [Google Scholar] [CrossRef]
- Steiger, P.; Alxneit, I.; Ferri, D. Nickel incorporation in perovskite-type metal oxides-Implications on reducibility. Acta Mater. 2019, 164, 568–576. [Google Scholar] [CrossRef]
- Mergler, Y.; Hoebink, J.; Nieuwenhuys, B. CO Oxidation over a Pt/CoOx/SiO2 Catalyst: A Study Using Temporal Analysis of Products. J. Catal. 1997, 167, 305–313. [Google Scholar] [CrossRef]
- Cargnello, M.; Doan-Nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of Metal Nanocrystal Size Reveals Metal-Support Interface Role for Ceria Catalysts. Science 2013, 341, 771–773. [Google Scholar] [CrossRef] [Green Version]
- Gorte, R.J. Ceria in Catalysis: From Automotive Applications to the Water Gas Shift Reaction. AIChE J. 2010, 56, 1126–1135. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Goodman, D.W. High-Pressure Catalytic Reactions over Single-Crystal Metal-Surfaces. Surf. Sci. Rep. 1991, 14, 1–107. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Willigan, R.R.; Dardas, Z.; Vanderspurt, T.H. Water gas shift activity of noble metals supported on ceria-zirconia oxides. AIChE J. 2006, 52, 1888–1894. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B-Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Onn, T.M.; Kungas, R.; Fornasiero, P.; Huang, K.; Gorte, R.J. Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation. Inorganics 2018, 6, 34. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BET S.A. (±2 m2/g) | Metal Oxide Loading (±1.5 wt %) | Pt Loading (±0.2 wt %) | Film Thickness (nm) | |
|---|---|---|---|---|
| Pt/LCO/MAO | 58 | 30 | 2.9 | 0.5 |
| Pt/La/MAO | 83 | 20 | 2.7 | 0.3 |
| Pt/Co/MAO | 81 | 20 | 2.8 | 0.3 |
| Pt/MAO | 118 | - | 3.1 | - |
| 1073 K Oxidized | 1073 K Reduced | |||
|---|---|---|---|---|
| Measured | Estimated | Measured | Estimated | |
| Pt/LCO/MAO | - | 2 | - | 40 |
| Pt/MAO | 0.1 | 0.1 | 0.1 | 0.4 |
| Pt/Co/MAO | 7 | 6.5 | 13 | 13 |
| Pt/La/MAO | 1.3 | 1.4 | 1.4 | 2.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, X.; Foucher, A.C.; Stach, E.A.; Gorte, R.J. “Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition. Inorganics 2019, 7, 113. https://doi.org/10.3390/inorganics7090113
Mao X, Foucher AC, Stach EA, Gorte RJ. “Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition. Inorganics. 2019; 7(9):113. https://doi.org/10.3390/inorganics7090113
Chicago/Turabian StyleMao, Xinyu, Alexandre C. Foucher, Eric A. Stach, and Raymond J. Gorte. 2019. "“Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition" Inorganics 7, no. 9: 113. https://doi.org/10.3390/inorganics7090113
APA StyleMao, X., Foucher, A. C., Stach, E. A., & Gorte, R. J. (2019). “Intelligent” Pt Catalysts Based on Thin LaCoO3 Films Prepared by Atomic Layer Deposition. Inorganics, 7(9), 113. https://doi.org/10.3390/inorganics7090113