
DFT Investigation of the Molecular Properties of the Dimethylglyoximato Complexes [M(Hdmg)2] (M = Ni, Pd, Pt)



Abstract
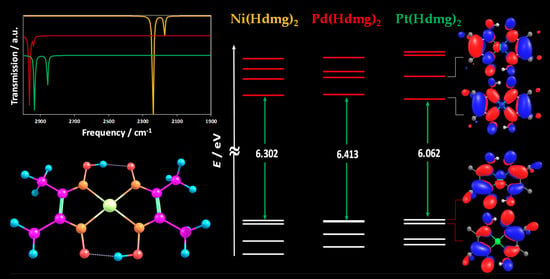
:
1. Introduction
2. Experimental Section
3. Results and Discussion
3.1. DFT-Optimised Molecular Geometries
3.2. Infrared (IR) Spectroscopy
3.3. Energies and Compositions of the Frontier Orbitals
3.4. UV-vis Absorption Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, S.Y.; Reddy, K.D.; Manna, A.K. Structural, Electronic, and Spectral Properties of Metal Dimethylglyoximato [M(DMG)2; M = Ni2+, Cu2+] Complexes: A Comparative Theoretical Study. J. Phys. Chem. A 2019, 123, 9166–9174. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.G.; Mandal, N.; Datta, A.; Datta, D. On bonding in bis(dimethylglyoximato)nickel(II). Comput. Theor. Chem. 2017, 1114, 118–124. [Google Scholar] [CrossRef]
- Liu, K.; Orimoto, Y.; Aoki, Y. Theoretical investigation of the pressure-induced insulator-to-metal-to-insulator transitions in one-dimensional bis(dimethylglyoximato) platinum(II), Pt(dmg)2. Polyhedron 2015, 87, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Bruce-Smith, I.F.; Zakharov, B.A.; Stare, J.; Boldyreva, E.V.; Pulham, C.R. Structural Properties of Nickel Dimethylglyoxime at High Pressure: Single-Crystal X-ray Diffraction and DFT Studies. J. Phys. Chem. C 2014, 118, 24705–24713. [Google Scholar] [CrossRef]
- Kovacs, A. Theoretical study of the strong intramolecular hydrogen bond and metal–ligand interactions in group 10 (Ni, Pd, Pt) bis(dimethylglyoximato) complexes. J. Organomet. Chem. 2007, 692, 5383–5389. [Google Scholar] [CrossRef]
- Di Bella, S.; Casarin, M.; Fragala, I.; Granozzi, G.; Marks, T.J. Electronic Structure of Tetracoordinate Transition-Metal Complexes. 2. Comparative Theoretical ab Initio/Hartree-Fock-Slater and UV-Photoelectron Spectroscopic Studies of Building Blocks for Low-Dimensional Conductors: Glyoximate Complexes of Palladium(II) and Platinum(II). Inorg. Chem. 1988, 27, 3993–4002. [Google Scholar]
- Yoshida, K. Epitaxial Growth of Bis(dimethylglyoximato)platinum(II) Accompanied by Hole Formation. Cryst. Growth Des. 2020, 20, 7271–7275. [Google Scholar] [CrossRef]
- Takeda, K.; Sasaki, T.; Hayashi, J.; Kagami, S.; Shirotani, I.; Yakushi, K. X-ray and optical studies of one-dimensional bis(dimethylglyoximato)Pd(II), Pd(dmg)2 at high pressures. J. Phys. Conf. Ser. 2010, 215, 012065. [Google Scholar] [CrossRef]
- Thomas, T.W.; Underhill, A.E. Metal–metal interactions in transition-metal complexes containing infinite chains of metal atoms. Chem. Soc. Rev. 1972, 1, 99–120. [Google Scholar] [CrossRef]
- Tauzher, G.; Dreos, R.; Felluga, A.; Nardin, G.; Randaccio, L.; Stener, M. Intramolecular and intermolecular O‒H‒O hydrogen bond in some nickel(II) complexes with tridentate amino‒oxime ligands. Inorg. Chim. Acta 2003, 355, 361–367. [Google Scholar] [CrossRef]
- Li, D.-X.; Xu, D.-J.; Xu, Y.-Z. Redetermination of bis(dimethylglyoximato-[κ]2-N,N’)nickel(II). Acta Crystallogr. Sect. E Struct. Rep. Online 2003, 59, m1094–m1095. [Google Scholar] [CrossRef] [Green Version]
- Konno, M.; Okamoto, T.; Shirotani, I. Structure changes and proton transfer between O...O in bis(dimethylglyoximato)platinum(II) at low temperature (150 K) and at high pressures (2.39 and 3.14 GPa). Acta Crystallogr. Sect. B Struct. Sci. 1989, 45, 142–147. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.S.; Salinas, B.E.V.; Schlemper, E.O. Three-dimensional determinations of the crystal structures of bis(dimethylglyoximato)palladium(II) and bis(dimethylglyoximato)platinum(II). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1979, 35, 628–633. [Google Scholar] [CrossRef]
- Murmann, R.K.; Schlemper, E.O. On the crystal structure of bis(glyoximato)nickel(II). Acta Crystallogr. 1967, 23, 667–669. [Google Scholar] [CrossRef]
- Martin, J.D.; Hogan, P.; Abboud, K.A.; Dahmen, K.-H. Variations on Nickel Complexes of the vic-Dioximes: An Understanding of Factors Affecting Volatility toward Improved Precursors for Metal-Organic Chemical Vapor Deposition of Nickel. Chem. Mater. 1998, 10, 2525–2532. [Google Scholar] [CrossRef]
- Caton, J.E.; Banks, C.V. Studies of the spectra of copper dimethyl-glyoxime, nickel dimethylglyoxime and nickel ethylmethylglyoxime in various solvents. Talanta 1966, 13, 967–977. [Google Scholar] [CrossRef]
- Burger, K.; Ruff, I.; Ruff, F. Some theoretical and practical problems in the use of organic reagents in chemical analysis—IV: Infra-red and ultra-violet spectrophotometric study of the dimethylglyoxime complexes of transition metals. J. Inorg. Nucl. Chem. 1965, 27, 179–190. [Google Scholar] [CrossRef]
- Frasson, E.; Panattoni, C. X-ray studies on the metal complexes with the glyoximes. IV. Structure of Ni-methyl-ethyl-glyoxime. Acta Crystallogr. 1960, 13, 893–898. [Google Scholar] [CrossRef]
- Williams, D.E.; Wohlauer, G.; Rundle, R.E. Crystal Structures of Nickel And Palladium Dimethylglyoximes. J. Am. Chem. Soc. 1959, 81, 755–756. [Google Scholar] [CrossRef]
- Frasson, E.; Panattoni, C.; Zannetti, R. X-ray studies on the metal complexes with the glyoximes. II. Structure of the Pt-dimethylglyoxime. Acta Crystallogr. 1959, 12, 1027–1031. [Google Scholar] [CrossRef]
- Blinc, R.; Hadži, D. Infrared spectra and hydrogen bonding in the nickel–dimethylglyoxime and related complexes. J. Chem. Soc. 1958, 4536–4540. [Google Scholar] [CrossRef]
- Huila, M.F.G.; Lukin, N.; Parussulo, A.L.A.; Oliveira, P.V.; Kyohara, P.K.; Araki, K.; Toma, H.E. Unraveling the Mysterious Role of Palladium in Feigl bis(dimethylglyoximate)nickel(II) Spot Tests by Means of Confocal Raman Microscopy. Anal. Chem. 2012, 84, 3067–3069. [Google Scholar] [CrossRef]
- Sharpe, A.G.; Wakefield, D.B. The basis of the selectivity of dimethylglyoxime as a reagent in gravimetric analysis. J. Chem. Soc. 1957, 281–285. [Google Scholar] [CrossRef]
- Godycki, L.E.; Rundle, R.E. The structure of nickel dimethylglyoxime. Acta Crystallogr. 1953, 6, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Gerasimchuk, N. Editorial (Thematic Issue: Recent Advances in Chemistry and Applications of Oximes and their Metal Complexes: Part I). Curr. Inorg. Chem. 2015, 5, 3–4. [Google Scholar] [CrossRef]
- Takeda, K.; Shirotani, I.; Yakushi, K. Pressure-Induced Insulator-to-Metal-to-Insulator Transitions in One-Dimensional Bis(dimethylglyoximato)platinum(II), Pt(dmg)2. Chem. Mater. 2000, 12, 912–916. [Google Scholar] [CrossRef]
- Shirotani, I.; Suzuki, K.; Yagi, T. Pressure-sensitive Optical Properties of Bis(1,2-dionedioximato)-Palladium(II) Complexes. Proc. Jpn. Acad. Ser. B 1992, 68, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Hayashi, J.; Shirotani, I.; Fukuda, H.; Yakushi, K. Structural, Optical, and Electrical Properties of One-Dimensional Bis(Dimethylglyoximato)nickel(II), Ni(dmg)2 at High Pressure. Mol. Cryst. Liq. Cryst. 2006, 460, 131–144. [Google Scholar] [CrossRef]
- Shirotani, I.; Suzuki, K.; Suzuki, T.; Yagi, T.; Tanaka, M. Absorption Spectra and Electrical Resistivities of Bis(1,2-dione dioximato)nickel(II) Complexes at High Pressures. Bull. Chem. Soc. Jpn. 1992, 65, 1078–1083. [Google Scholar] [CrossRef]
- Shirotani, I.; Inagaki, Y.; Utsumi, W.; Yagi, T. Pressure-sensitive absorption spectra of thin films of bis(diphenylglyoximato)platinum(II), Pt(dpg)2: Potential application as an indicator of pressure. J. Mater. Chem. 1991, 1, 1041–1043. [Google Scholar] [CrossRef]
- Ferraro, J.R. Some possible new internal pressure calibrants. Inorg. Nucl. Chem. Lett. 1970, 6, 823–825. [Google Scholar] [CrossRef]
- Kamnoet, P.; Aeungmaitrepirom, W.; Menger, R.F.; Henry, C.S. Highly selective simultaneous determination of Cu(II), Co(II), Ni(II), Hg(II), and Mn(II) in water samples using microfluidic paper-based analytical de-vices. Analyst 2021, 146, 2229–2239. [Google Scholar] [CrossRef]
- Mirhashemi, A.; Ghorbani, Y.; Sadighi, S. Synthesis and evaluation of Fe3O4–Al2O3/SDS–DMG adsorbent for extraction and preconcentration of Pd(II) from real samples. J. Iran. Chem. Soc. 2020, 17, 2073–2081. [Google Scholar] [CrossRef]
- Olorundare, F.O.G.; Nkosi, D.; Arotiba, O.A. Voltammetric Determination of Nitrophenols at a Nickel Dimethylglyoxime Complex—Gold Nanoparticle Modified Glassy Carbon Electrode. Int. J. Electrochem. Sci. 2016, 11, 7318–7332. [Google Scholar] [CrossRef]
- Andris, B.; Prazsky, M.; Sebesta, F. Rapid separation and determination of 107Pd in radioactive waste produced during NPP A-1 decommissioning. J. Radioanal. Nucl. Chem. 2015, 304, 123–126. [Google Scholar] [CrossRef]
- Ma, J.; Meng, W.; Zhang, L.; Li, F.; Li, T. Effective oil–water mixture separation and photocatalytic dye de-contamination through nickel-dimethylglyoxime microtubes coated superhydrophobic and superoleophilic films. RSC Adv. 2021, 11, 5035–5043. [Google Scholar] [CrossRef]
- Qi, P.; Gu, Y.; Sun, H.; Lian, Y.; Yuan, X.; Hu, J.; Deng, Z.; Yao, H.-C.; Guo, J.; Peng, Y. Active Nickel Derived from Coordination Complex with Weak Inter/Intra-molecular Interactions for Efficient Hydrogen Evolution via a Tandem Mechanism. J. Catal. 2020, 389, 29–37. [Google Scholar] [CrossRef]
- Sarkar, B.; Kwek, W.; Verma, D.; Kim, J. Effective vacuum residue upgrading using sacrificial nickel(II) dimethylglyoxime complex in supercritical methanol. Appl. Catal A Gen. 2017, 545, 148–158. [Google Scholar] [CrossRef]
- Titova, Y.Y.; Belykh, L.B.; Shmidt, F.K. Formation and properties of Ziegler systems based on nickel bis(dimethylglyoximate) in catalysis of hydrogenation reactions. Russ. J. Gen. Chem. 2014, 84, 2413–2420. [Google Scholar] [CrossRef]
- Feng, J.-J.; Zhou, D.-L.; Xi, H.-X.; Chen, J.-R.; Wang, A.-J. Facile synthesis of porous worm-like Pd nanotubes with high electrocatalytic activity and stability towards ethylene glycol oxidation. Nanoscale 2013, 5, 6754–6757. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Li, Q.; Zhang, J.; Wu, J.; Zhao, T.; Liu, Z.; Zhou, L.; He, H.; Li, B.; Zhang, G. Engineering Surface Atomic Architecture of NiTe Nanocrystals Toward Efficient Electrochemical N2 Fixation. Adv. Funct. Mater. 2020, 30, 2004208. [Google Scholar] [CrossRef]
- Wang, T.; Li, F.; Huang, H.; Yin, S.; Chen, P.; Jin, P.; Chen, Y. Porous Pd‒PdO Nanotubes for Methanol Electrooxidation. Adv. Funct. Mater. 2020, 30, 2000534. [Google Scholar] [CrossRef]
- Dong, X.; Li, J.; Wei, D.; Li, R.; Qu, K.; Wang, L.; Xu, S.; Kang, W.; Li, H. Pd(II)/Ni(II)-dimethylglyoxime derived Pd‒Ni‒P@N-doped carbon hybrid nanocatalysts for oxygen reduction reaction. Appl. Surf. Sci. 2019, 479, 273–279. [Google Scholar] [CrossRef]
- Kordatos, K.; Vlasopoulos, A.; Strikos, S.; Ntziouni, A.; Gavela, S.; Trasobares, S.; Kasselouri-Rigopoulou, V. Synthesis of carbon nanotubes by pyrolysis of solid Ni(dmg)2. Electrochim. Acta 2009, 54, 2466–2472. [Google Scholar] [CrossRef]
- Dakhel, A.; Ali-Mohamed, A.Y. Characterisation and ac-electrical investigation of sublimated bis(dimethylglyoximato)palladium(II) thin films. J. Organomet. Chem. 2006, 691, 3760–3764. [Google Scholar] [CrossRef]
- Dakhel, A.A.; Ahmed, Y.A.-M.; Henari, F. Structural and optical studies of evaporated bis(dimethylglyoximato)nickel(II) thin films. Opt. Mater. 2006, 28, 925–929. [Google Scholar] [CrossRef]
- Suzuki, I.; Honjo, T.; Terada, K. Vacuum Sublimation Behavior of Nickel(II), Palladium(II), and Platinum(II) Chelates with Dimethylglyoxime. Bull. Chem. Soc. Jpn. 1990, 63, 3686–3688. [Google Scholar] [CrossRef] [Green Version]
- Iwai, S.; Kamata, T.; Murata, S.; Yamamoto, K.; Ohta, T. Wavepacket motion during thermalization of self-trapped exciton driven by an intramolecular vibration in one-dimensional platinum dimethylglyoxime complex. J. Lumin. 2000, 87–89, 629–632. [Google Scholar] [CrossRef]
- Kamata, T.; Fukaya, T.; Matsuda, H.; Mizukami, F. Reversible control of the nonlinear optical activity of one-dimensional metal complexes. Appl. Phys. Lett. 1994, 65, 1343–1345. [Google Scholar] [CrossRef]
- Kamata, T.; Curran, S.; Roth, S.; Fukaya, T.; Matsuda, H.; Mizukami, F. Third-order nonlinear optical prop-erties of evaporated thin films of platinum-alkyldionedioxime complexes: Effects of metal—Metal distance. Synth. Met. 1996, 83, 267–271. [Google Scholar] [CrossRef]
- Kamata, T.; Fukaya, T.; Kodzasa, T.; Matsuda, H.; Mizukami, F. Third-order nonlinear optical properties of bis(dimethylglyoximato)metal(II). Synth. Met. 1995, 71, 1725–1726. [Google Scholar] [CrossRef]
- Yang, B.; Li, J.; Zhang, L.; Xu, G. A molecularly imprinted electrochemiluminescence sensor based on the mimetic enzyme catalytic effect for ultra-trace Ni2+ determination. Analyst 2016, 141, 5822–5828. [Google Scholar] [CrossRef] [PubMed]
- Benoit, S.L.; Maier, R.J. The nickel-chelator dimethylglyoxime inhibits human amyloid beta peptide in vitro aggregation. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, L.; Batchelor, L.K.; Dyson, P.J.; Zacchini, S.; Schoch, S.; Pampaloni, G.; Marchetti, F. α-Diimine homologues of cisplatin: Synthesis, speciation in DMSO/water and cytotoxicity. New J. Chem. 2018, 42, 17453–17463. [Google Scholar] [CrossRef]
- Kluge, T.; Bette, M.; Rüffer, T.; Bruhn, C.; Wagner, C.; Ströhl, D.; Schmidt, J.; Steinborn, D. Activation of Acetyl Ligands through Hydrogen Bonds: A New Way to Platinum(II) Complexes Bearing Protonated Imino-acetyl Ligands. Organometallics 2013, 32, 7090–7106. [Google Scholar] [CrossRef]
- Schwieger, S.; Heinemann, F.W.; Wagner, C.; Kluge, R.; Damm, C.; Israel, G.; Steinborn, D. A Bis(acetyl)-Bridged Platinum(II) Coordination Polymer as a Building Block for Diacetylplatinum(II) Complexes and Platina-β-diketones. Organometallics 2009, 28, 2485–2493. [Google Scholar] [CrossRef]
- Nagakura, S.; Ohashi, Y.; Hanazaki, I. Spectroscopic study of the interaction between the central metal ions in the crystals of bis(dimethylglyoximato)nickel(II) and related complexes. Inorg. Chem. 1970, 9, 2551–2556. [Google Scholar] [CrossRef]
- Kurz, H.; Schötz, K.; Papadopoulos, I.; Heinemann, F.W.; Maid, H.; Guldi, D.M.; Hörner, G.; Weber, B. A Flu-orescence-Detected Coordination-Induced Spin State Switch. J. Am. Chem. Soc. 2021, 143, 3466–3480. [Google Scholar] [CrossRef]
- Berkefeld, A.; Fröhlich, M.; Kordan, M.; Hörner, G.; Schubert, H. Selective metalation of phenol-type proligands for preparative organometallic chemistry. Chem. Commun. 2020, 56, 3987–3990. [Google Scholar] [CrossRef]
- Heil, A.; Marian, C.M. Structure—Emission Property Relationships in Cyclometalated Pt(II) β-Diketonate Complexes. Inorg. Chem. 2019, 58, 6123–6136. [Google Scholar] [CrossRef]
- Garbe, S.; Krause, M.; Klimpel, A.; Neundorf, I.; Lippmann, P.; Ott, I.; Brünkink, D.; Strassert, C.A.; Doltsinis, N.L.; Klein, A. Cyclometalated Pt Complexes of CNC Pincer Ligands: Luminescence and Cytotoxic Properties. Organometallics 2020, 39, 746–756. [Google Scholar] [CrossRef]
- Föller, J.; Friese, D.H.; Riese, S.; Kaminski, J.M.; Metz, S.; Schmidt, D.; Wuerthner, F.; Lambert, C.; Marian, C.M. On the photophysical properties of IrIII, PtII, and PdII (phenylpyrazole) (phenyldipyrrin) complexes. Phys. Chem. Chem. Phys. 2020, 22, 3217–3233. [Google Scholar] [CrossRef]
- Mews, N.M.; Reimann, M.; Hörner, G.; Kaupp, M.; Schubert, H.; Berkefeld, A. A four-parameter system for rationalising the electronic properties of transition metal–radical ligand complexes. Dalton Trans. 2020, 49, 9735–9742. [Google Scholar] [CrossRef] [PubMed]
- Haseloer, A.; Denkler, L.M.; Jordan, R.; Reimer, M.; Olthof, S.; Schmidt, I.; Meerholz, K.; Hörner, G.; Klein, A. Ni, Pd, and Pt complexes of a tetradentate dianionic thiosemicarbazone-based O^N^N^S ligand. Dalton Trans. 2021, 50, 4311–4322. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, T.; Buss, S.; Petrovskii, S.K.; Grachova, E.V.; Krause, M.; Klein, A.; Strassert, C.A.; Koshevoy, I.O.; Hirva, P. Photophysics and Excited State Dynamics of Cyclometalated [M(C^N^N)(CN)] (M = Ni, Pd, Pt) Complexes: A Theoretical and Experimental Study. Inorg. Chem. 2021. [Google Scholar] [CrossRef]
- Krause, M.; von der Stück, R.; Brünink, D.; Buss, S.; Doltsinis, N.L.; Strassert, C.A.; Klein, A. Platinum and palladium complexes of tridentate −C^N^N (phen-ide)-pyridine-thiazol ligands—A case study involving spectroelectrochemistry, photoluminescence spectroscopy and TD-DFT calculations. Inorg. Chim. Acta 2021, 518, 120093. [Google Scholar] [CrossRef]
- Poirier, S.; Lynn, H.; Reber, C. Variation of M·H–C Interactions in Square-Planar Complexes of Nickel(II), Palladium(II), and Platinum(II) Probed by Luminescence Spectroscopy and X-ray Diffraction at Variable Pressure. Inorg. Chem. 2018, 57, 7713–7723. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Barone, V.; Cossi, M.; Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Roy, L.E.; Hay, P.J.; Martin, R.L. Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Rohlfing, C.M.; Hay, P.J.; Martin, R.L. An effective core potential investigation of Ni, Pd, and Pt and their monohydrides. J. Chem. Phys. 1986, 85, 1447–1455. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Potocny, A.M.; Pistner, A.J.; Yap, G.P.A.; Rosenthal, J. Electrochemical, Spectroscopic, and 1O2 Sensitization Characteristics of Synthetically Accessible Linear Tetrapyrrole Complexes of Palladium and Platinum. Inorg. Chem. 2017, 56, 12703–12711. [Google Scholar] [CrossRef]
- Kar, P.; Yoshida, M.; Kobayashi, A.; Routaboul, L.; Braunstein, P.; Kato, M. Colour tuning by the stepwise synthesis of mononuclear and homo- and hetero-dinuclear platinum(II) complexes using a zwitterionic quinonoid ligand. Dalton Trans. 2016, 45, 14080–14088. [Google Scholar] [CrossRef]
- Phadnis, P.P.; Jain, V.K.; Schurr, T.; Klein, A.; Lissner, F.; Schleid, T.; Kaim, W. Synthesis, spectroscopy, structure and photophysical properties of dinaphthylmethylarsine complexes of palladium(II) and platinum(II). Inorg. Chim. Acta 2005, 358, 2609–2617. [Google Scholar] [CrossRef]
- Dey, D.; Kumbhare, L.B.; Jain, V.K.; Klein, A.; Schurr, T.; Kaim, W.; Belaj, F. Structural Varieties in 1-Dimethylaminopropyl-2-chalcogenolate and 2-Dimethylaminopropyl-1-chalcogenolate (S, Se, Te) Complexes of Palladium(II) and Platinum(II): Synthesis, Spectroscopy and Structures. Eur. J. Inorg. Chem. 2004, 22, 4510–4520. [Google Scholar] [CrossRef]
- Phadnis, P.P.; Jain, V.K.; Klein, A.; Weber, M.; Kaim, W. Configurational selectivity in benzyldimethylarsine complexes of palladium(II) and platinum(II): Synthesis, spectroscopy and structures. Inorg. Chim. Acta 2003, 346, 119–128. [Google Scholar] [CrossRef]
- Huheey, J.E.; Huheey, C.L. Anomalous properties of elements that follow “long periods” of elements. J. Chem. Educ. 1972, 49, 227–230. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Rundle, R.E.; Parasol, M. O–H Stretching Frequencies in Very Short and Possibly Symmetrical Hydrogen Bonds. J. Chem. Phys. 1952, 20, 1487–1488. [Google Scholar] [CrossRef]
- Caton, J.E.; Banks, C.V. Hydrogen bonding in some copper(II) and nickel(II) vic-dioximes. Inorg. Chem. 1967, 6, 1670–1675. [Google Scholar] [CrossRef]
- Orel, B.; Penko, M.; Hadzi, D. Infrared and resonance enhanced Raman spectra of some metal vic-dioximes with short hydrogen bonds. Spectrochim. Acta Part A Mol. Spectrosc. 1980, 36, 859–864. [Google Scholar] [CrossRef]
- Szabó, A.; Kovács, A. Vibrational analysis of the bis(dimethylglyoximato)nickel(II) complex. J. Mol. Struct. 2003, 651–653, 547–553. [Google Scholar] [CrossRef]
- Várhelyi, C.; Kovács, A.; Gömöry, A.; Várhelyi, C.; Pokol, G.; Farkas, G.; Sohár, P. Comparative spectral and thermal studies of [Pt(DioxH)2] chelates. J. Coord. Chem. 2009, 62, 2429–2437. [Google Scholar] [CrossRef]
- Bigotto, A.; Galasso, V.; De Alti, G. Infrared spectra of transition metal glyoximates. Spectrochim. Acta Part A Mol. Spectrosc. 1971, 27, 1659–1670. [Google Scholar] [CrossRef]
- Kohler, U.; Hausen, H.-D.; Weidlein, J. Bis(dimethylmetall(III)glyoximato)metallate(II) (metall(III) = Al, Ga, In, metall(II) = Ni, Pd, Pt, Cu). J. Organomet. Chem. 1984, 272, 337–350. [Google Scholar] [CrossRef]
- Yoshida, T. An X-Ray Photoelectron Spectroscopic Study of Dioxime Metal Complexes. Bull. Chem. Soc. Jpn. 1978, 51, 3257–3260. [Google Scholar] [CrossRef] [Green Version]
- Basu, G.; Cook, G.M.; Belford, R.L. The Green Band of Crystalline Nickel Dimethylglyoxime. I. Mixed Crystals with Palladium, Platinum, and Copper and the Questions of Nonlocalized or d-p Transitions. Inorg. Chem. 1964, 3, 1361–1368. [Google Scholar] [CrossRef]
- Nishida, Y.; Kozuka, M.; Nakamoto, K. Resonance raman spectra of [Ni(dmg)2], [Pd(dmg)2] and [Pt(dmg)2] in the solid state (dmgH = dimethylglyoxime). Inorg. Chim. Acta 1979, 34, L273–L275. [Google Scholar] [CrossRef]
- Kaim, W.; Kohlmann, S.; Ernst, S.; Olbrich-Deussner, B.; Bessenbacher, C.; Schulz, A. What determines the solvatochromism of metal-to-ligand charge transfer transitions? A demonstration involving 17 tungsten carbonyl complexes. J. Organomet. Chem. 1987, 321, 215–226. [Google Scholar] [CrossRef]
- Manuta, D.M.; Lees, A.J. Solvatochromism of the Metal to Ligand Charge-Transfer Transitions of Zerovalent Tungsten Carbonyl Complexes. Inorg. Chem. 1986, 25, 3212–3218. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| O‒H | O‒H | C=N | N‒O | N‒O | Reference |
|---|---|---|---|---|---|
| Stretching | Bending | Stretching | Stretching | Stretching | |
| 2174 (sym) 2240 (asym) | 1810 | 1647 and 1677 | 1222 and 1297 | 1121 | Our Calc. |
| 2429 (sym) 2540 (asym) | 1600–1823 | 1637 | 1336 (sym) 1365 (asym) | 1261 (sym) 1261 (asym) | Calc. [1] b |
| 2585 | 1782 | 1529 and 1590 | 1249 | 1120 | Calc. [85] c |
| 2350 | 1780 | 1560 | 1235 | 1100 | Exp. [21] |
| 2350 | - | 1576 | 1241 | 1103 | Exp. [17] |
| - | 1790 | 1550 and 1572 | 1240 | 1101 | Exp. [87] |
| O‒H | O‒H | C=N | N‒O | N‒O | Reference |
|---|---|---|---|---|---|
| Stretching | Bending | Stretching | Stretching | Stretching | |
| 2939 (sym) 2964 (asym) | 1786 | 1611 and 1621 | 1217 and 1313 | 1114 | Our results |
| 2340 | 1710 | 1550 and 1500 | 1250 | 1090 | Exp. [21] |
| 2340 | - | 1552 | 1259 | 1091 | Exp. [17] |
| O‒H | O‒H | C=N | N‒O | N‒O | Reference |
|---|---|---|---|---|---|
| Stretching | Bending | Stretching | Stretching | Stretching | |
| 2857 (sym) 2934 (asym) | 1788 | 1619 | 1217 and 1320 | 1119 | Our results |
| 2350 | - | 1550 | 1262 | 1089 | Exp. [17] |
| [M(Hdmg)2] | [Ni(Hdmg)2] | [Pd(Hdmg)2] | [Pt(Hdmg)2] |
|---|---|---|---|
| λ3 (ε) b | 170 (4.3) | 171 (4.1) | 176 (6.5) |
| λ2 (ε) b | 211 (4.1) | 231 (1.9) | 250 (2.5) |
| λ1 (ε) b | 310 (6.2) | 312 (4.0) | 350 (2.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, M.; Klein, A. DFT Investigation of the Molecular Properties of the Dimethylglyoximato Complexes [M(Hdmg)2] (M = Ni, Pd, Pt). Inorganics 2021, 9, 47. https://doi.org/10.3390/inorganics9060047
Niazi M, Klein A. DFT Investigation of the Molecular Properties of the Dimethylglyoximato Complexes [M(Hdmg)2] (M = Ni, Pd, Pt). Inorganics. 2021; 9(6):47. https://doi.org/10.3390/inorganics9060047
Chicago/Turabian StyleNiazi, Maryam, and Axel Klein. 2021. "DFT Investigation of the Molecular Properties of the Dimethylglyoximato Complexes [M(Hdmg)2] (M = Ni, Pd, Pt)" Inorganics 9, no. 6: 47. https://doi.org/10.3390/inorganics9060047
APA StyleNiazi, M., & Klein, A. (2021). DFT Investigation of the Molecular Properties of the Dimethylglyoximato Complexes [M(Hdmg)2] (M = Ni, Pd, Pt). Inorganics, 9(6), 47. https://doi.org/10.3390/inorganics9060047