Peroxidized Linoleic Acid, 13-HPODE, Alters Gene Expression Profile in Intestinal Epithelial Cells

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Lipid Peroxide
2.3. Treatment of Cells with 13-HPODE/LA
2.4. Total RNA Extraction and Quantification
2.5. RNA-seq Library Preparation and Sequencing
2.6. Sequence Data Processing
2.7. Enrichment Analyses
2.8. Validation via qRT-PCR
3. Results
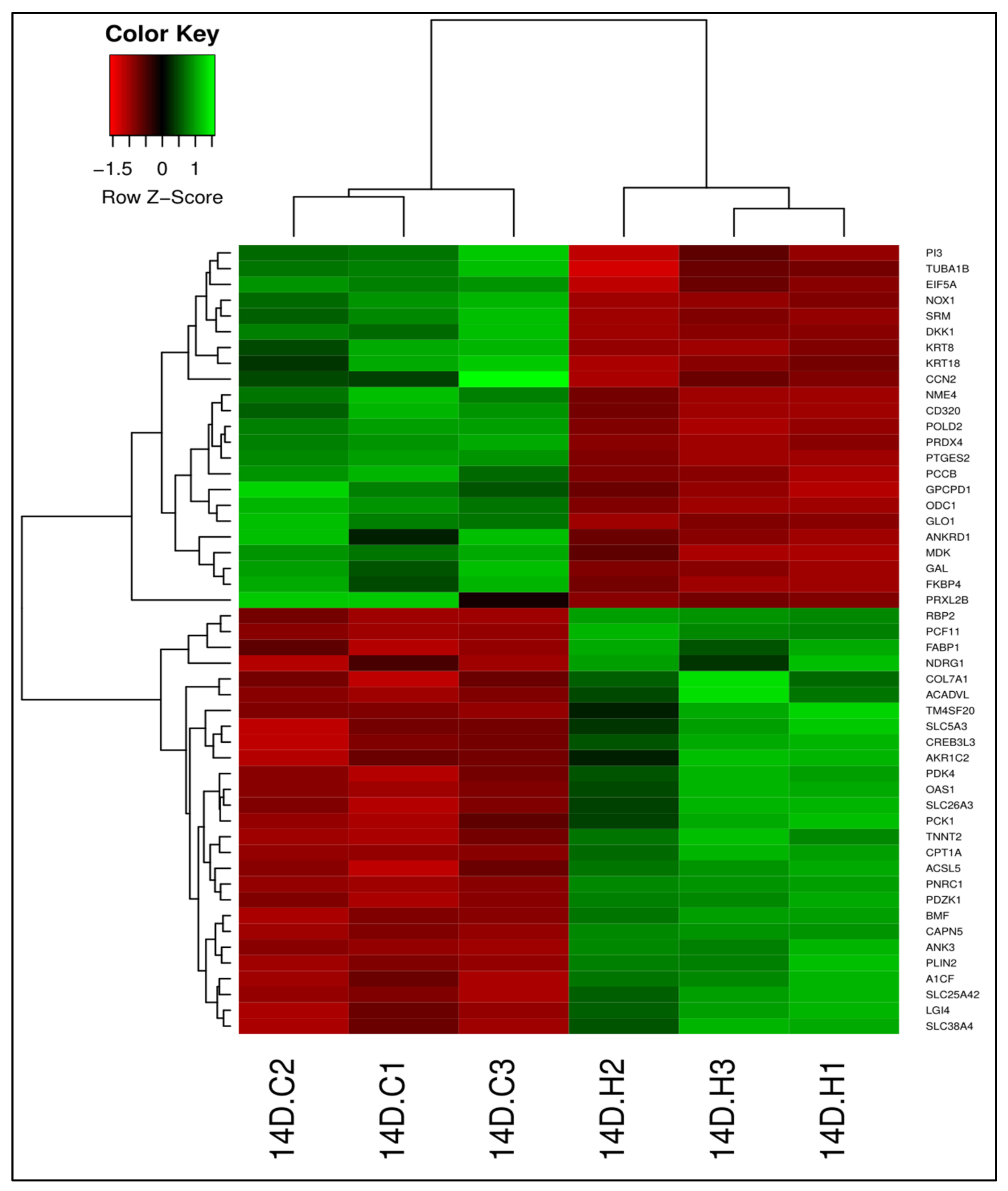
3.1. 13-HPODE-Treated Caco-2 Cells
3.1.1. Differential Gene Expression
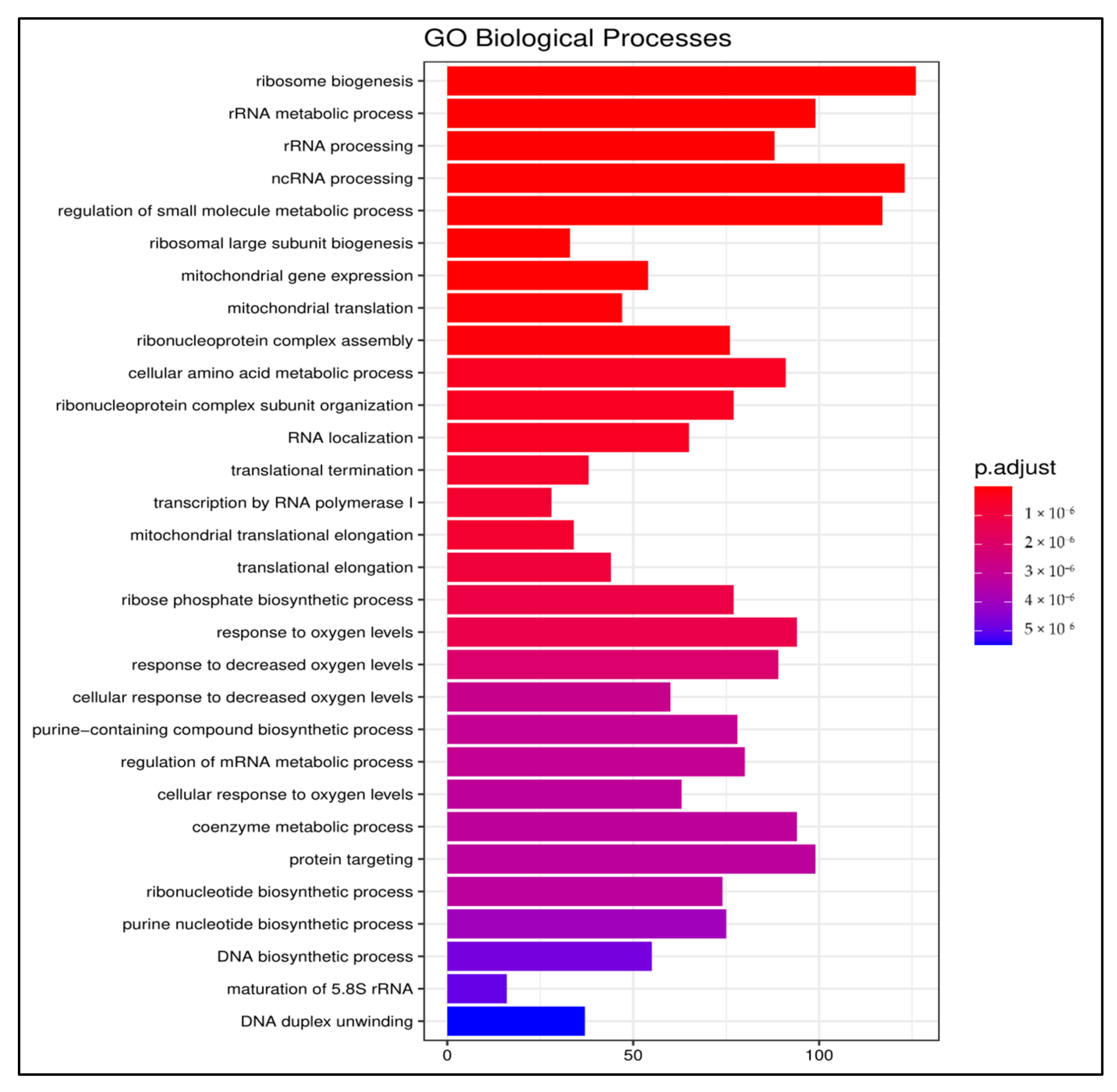
3.1.2. Gene Ontology
3.1.3. Pathway Enrichment Analysis
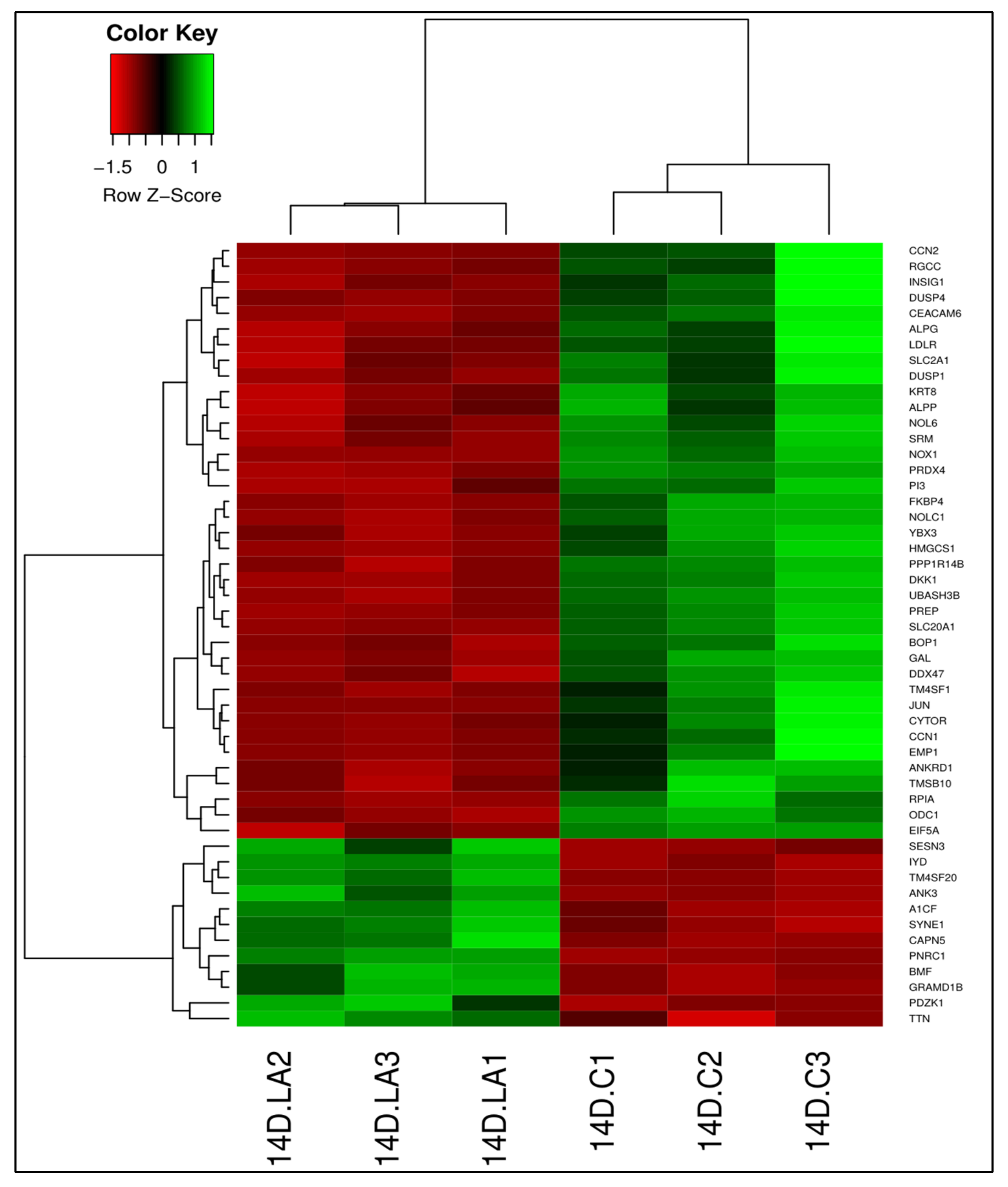
3.2. LA-Treated Caco-2 Cells
3.2.1. Differential Gene Expression
3.2.2. Gene Ontology
3.2.3. Pathway Enrichment Analysis
3.3. Differential Gene Expression between 13-HPODE-Treated and LA-Treated Cells
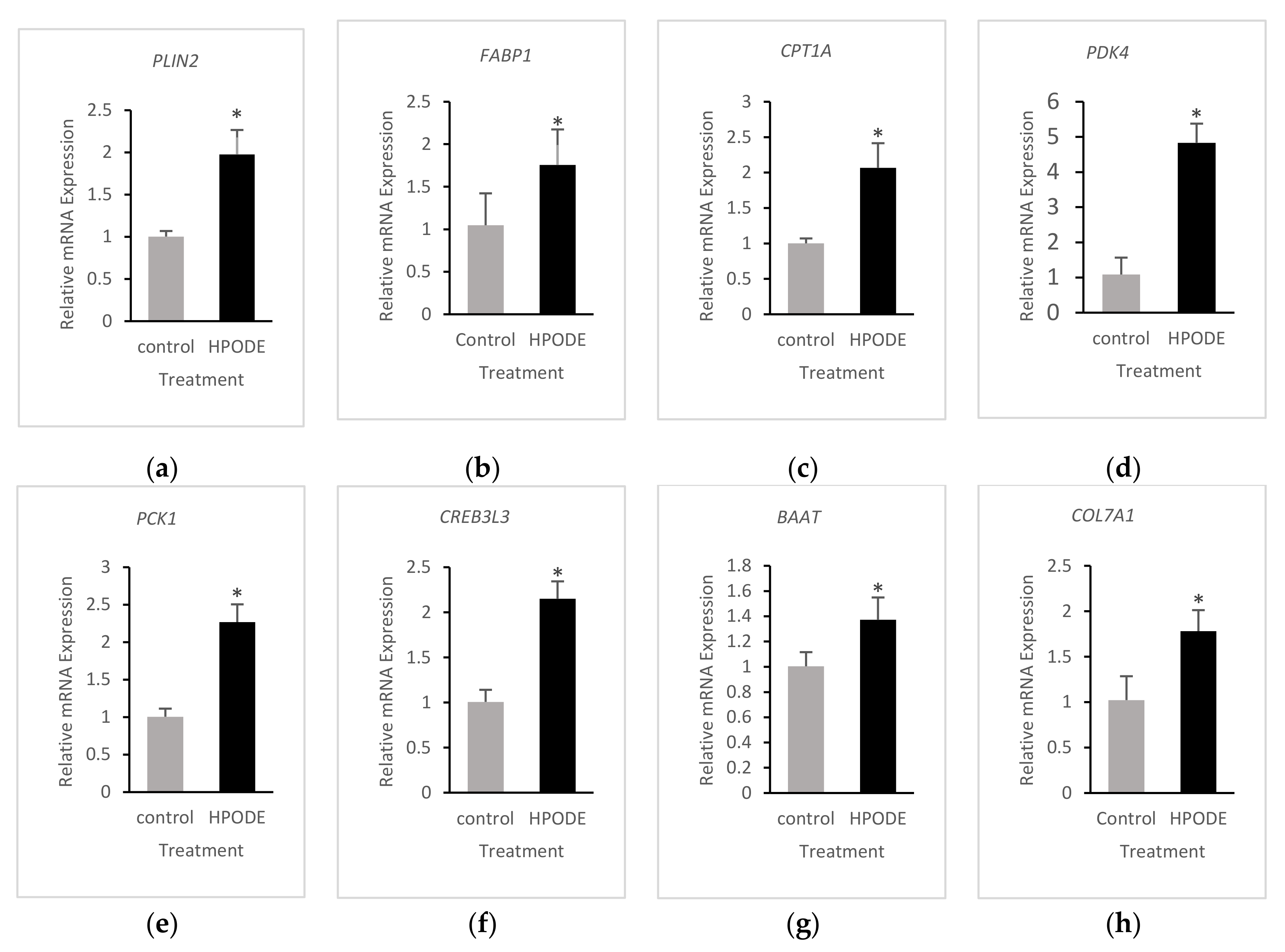
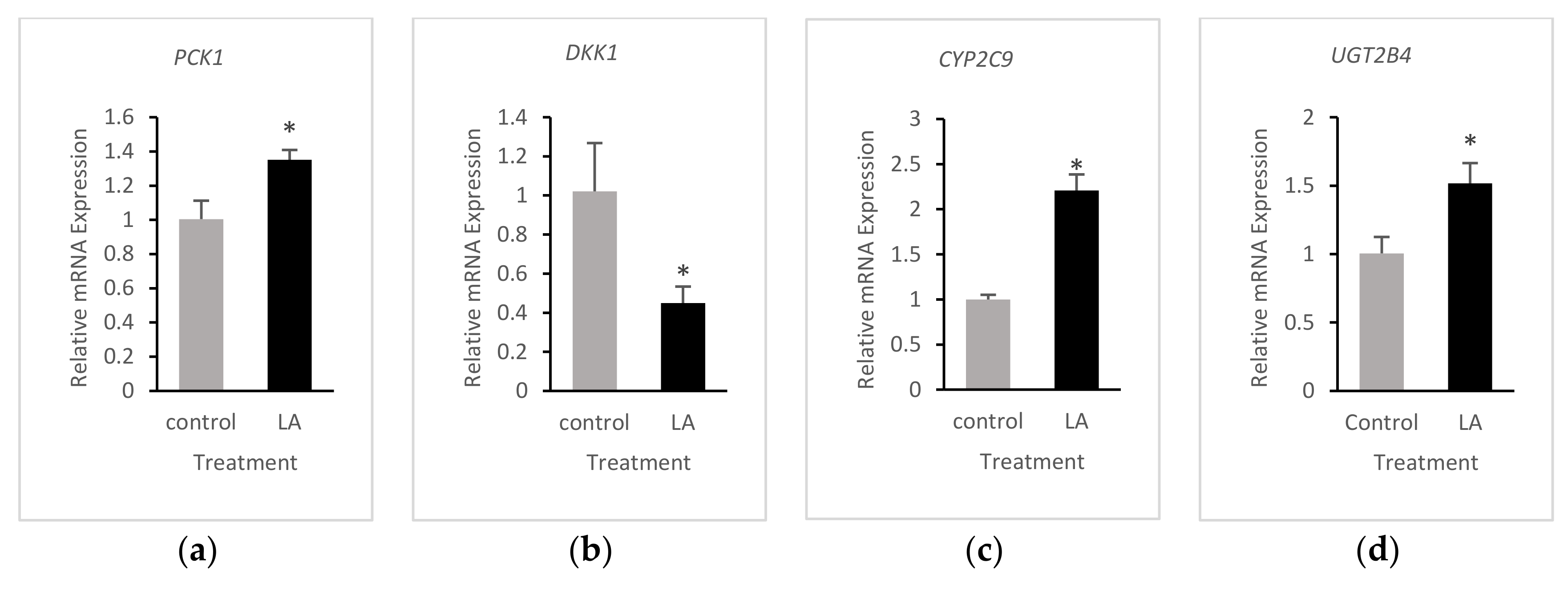
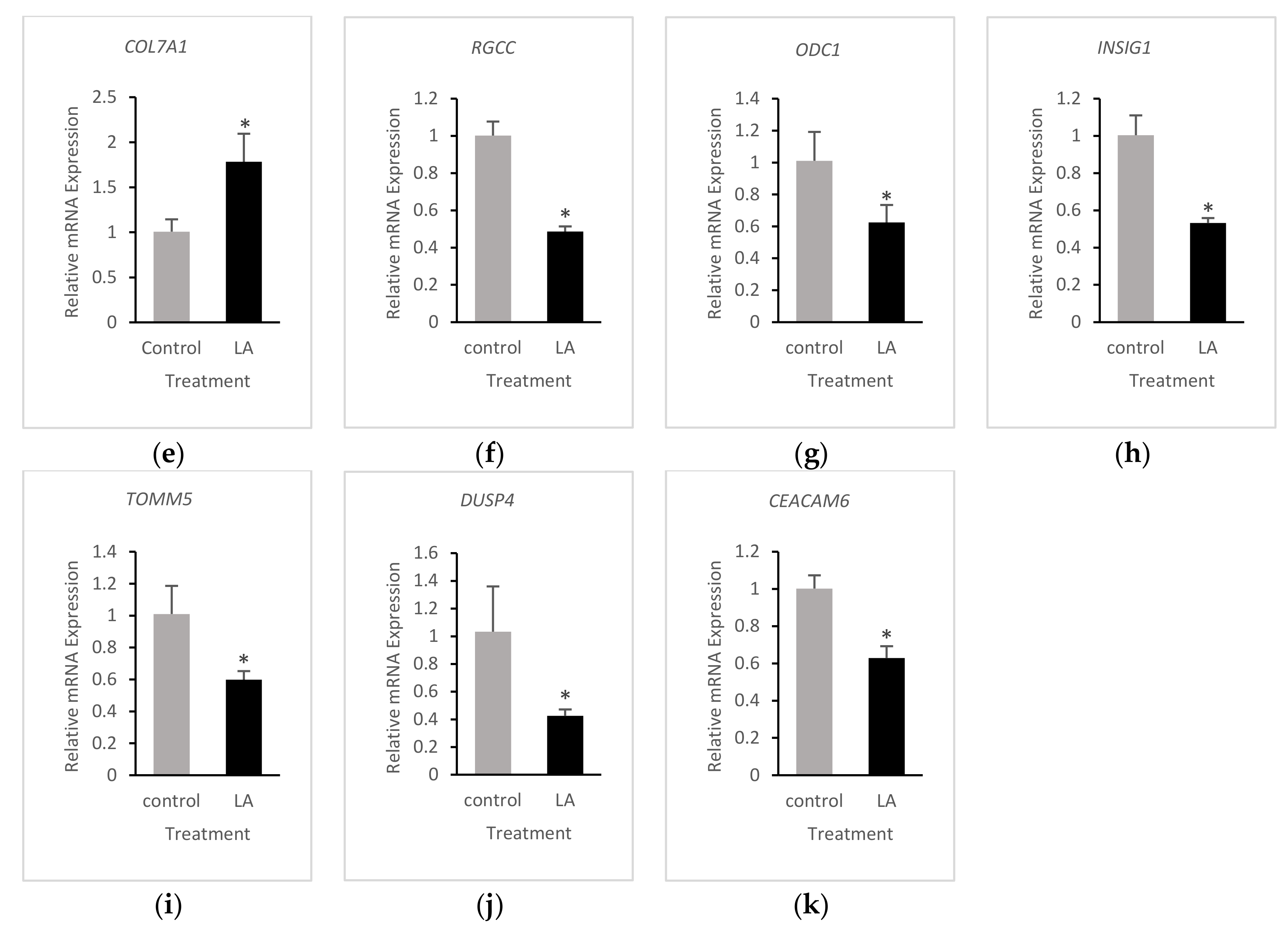
3.4. Validation of RNA-Seq Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, X.; Hussain, M.M. Gut triglyceride production. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2012, 1821, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lise Halvorsen, B.; Blomhoff, R. Determination of lipid oxidation products in vegetable oils and marine omega-3 supplements. Food Nutr. Res. 2011, 55, 5792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penumetcha, M.; Khan, N.; Parthasarathy, S. Dietary oxidized fatty acids: An atherogenic risk? J. Lipid Res. 2000, 41, 1473–1480. [Google Scholar] [CrossRef]
- Staprans, I.; Rapp, J.H.; Pan, X.M.; Feingold, K.R. The effect of oxidized lipids in the diet on serum lipoprotein peroxides in control and diabetic rats. J. Clin. Investig. 1993, 92, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Sottero, B.; Rossin, D.; Poli, G.; Biasi, F. Lipid Oxidation Products in the Pathogenesis of Inflammation-related Gut Diseases. Curr. Med. Chem. 2018, 25, 1311–1326. [Google Scholar] [CrossRef]
- Staprans, I.; Pan, X.M.; Miller, M.; Rapp, J.H. Effect of dietary lipid peroxides on metabolism of serum chylomicrons in rats. Am. J. Physiol. Liver Physiol. 1993, 264, G561–G568. [Google Scholar] [CrossRef]
- Wijeratne, S.S.K.; Cuppett, S.L. Lipid Hydroperoxide Induced Oxidative Stress Damage and Antioxidant Enzyme Response in Caco-2 Human Colon Cells. J. Agric. Food Chem. 2006, 54, 4476–4481. [Google Scholar] [CrossRef]
- Bhaskar, N.; Narasimhulu, C.A.; Keewan, E.; Rohr, M.; Parthasarathy, S. Proinflammatory Properties of Peroxidized Fat May Contribute to the Etiology of Crohn’s Disease. J. Med. Food. 2019, 22, 162–169. [Google Scholar] [CrossRef]
- Rohr, M.; Narasimhulu, C.A.; Keewan, E.; Hamid, S.; Parthasarathy, S. The dietary peroxidized lipid, 13-HPODE, promotes intestinal inflammation by mediating granzyme B secretion from natural killer cells. Food Funct. 2020, 11, 9526–9534. [Google Scholar] [CrossRef]
- Bernotti, S.; Seidman, E.; Sinnett, D.; Brunet, S.; Dionne, S.; Delvin, E.; Levy, E. Inflammatory reaction without endogenous antioxidant response in Caco-2 cells exposed to iron/ascorbate-mediated lipid peroxidation. Am. J. Physiol. Liver Physiol. 2003, 285, G898–G906. [Google Scholar] [CrossRef] [Green Version]
- Staprans, I.; Pan, X.-M.; Rapp, J.H.; Feingold, K.R. The role of dietary oxidized cholesterol and oxidized fatty acids in the development of atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Staprãns, I.; Rapp, J.H.; Pan, X.M.; Kim, K.Y.; Feingold, K.R. Oxidized lipids in the diet are a source of oxidized lipid in chylomicrons of human serum. Arter. Thromb. A J. Vasc. Biol. 1994, 14, 1900–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staprans, I.; Rapp, J.H.; Pan, X.M.; Feingold, K.R. Oxidized lipids in the diet are incorporated by the liver into very low density lipoprotein in rats. J. Lipid Res. 1996, 37, 420–430. [Google Scholar] [CrossRef]
- Khan-Merchant, N.; Penumetcha, M.; Meilhac, O.; Parthasarathy, S. Oxidized Fatty Acids Promote Atherosclerosis Only in the Presence of Dietary Cholesterol in Low-Density Lipoprotein Receptor Knockout Mice. J. Nutr. 2002, 132, 3256–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penumetcha, M.; Khan-Merchant, N.; Parthasarathy, S. Enhanced solubilization and intestinal absorption of cholesterol by oxidized linoleic acid. J. Lipid Res. 2002, 43, 895–903. [Google Scholar] [CrossRef]
- Stierum, R.; Gaspari, M.; Dommels, Y.; Ouatas, T.; Pluk, H.; Jespersen, S.; Vogels, J.; Verhoeckx, K.; Groten, J.; Van Ommen, B. Proteome analysis reveals novel proteins associated with proliferation and differentiation of the colorectal cancer cell line Caco-2. Biochim. Biophys. Acta Proteins Proteom. 2003, 1650, 73–91. [Google Scholar] [CrossRef]
- Hussain, M.M. Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 2014, 25, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauli, A.M.; Whittimore, J.D. Using Caco-2 Cells to Study Lipid Transport by the Intestine. J. Vis. Exp. 2015, e53086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepinskas, G.; Kvietys, P.R.; Aw, T.Y. ω3-Lipid peroxides injure CaCo-2 cells: Relationship to the development of reduced glutathione antioxidant systems. Gastroenterology 1994, 107, 80–86. [Google Scholar] [CrossRef]
- Wang, T.-G.; Gotoh, Y.; Jennings, M.H.; Rhoads, C.A.; Aw, T.Y. Lipid hydroperoxide-induced apoptosis in human colonic CaCo-2 cells is associated with an early loss of cellular redox balance. FASEB J. 2000, 14, 1567–1576. [Google Scholar] [CrossRef]
- Gotoh, Y.; Noda, T.; Iwakiri, R.; Fujimoto, K.; Rhoads, C.A.; Aw, T.Y. Lipid peroxide-induced redox imbalance differentially mediates CaCo-2 cell proliferation and growth arrest. Cell Prolif. 2002, 35, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. 2020, 11, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMeo, M.T.; Mutlu, E.A.; Keshavarzian, A.; Tobin, M.C. Intestinal Permeation and Gastrointestinal Disease. J. Clin. Gastroenterol. 2002, 34, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Duerr, M.A.; Hermanson, D.J.; Hedde, P.N.; Wong, J.S.; Vallim, T.Q.D.A.; Cravatt, B.F.; Gratton, E.; Ford, D.A.; et al. Intestinal Phospholipid Remodeling Is Required for Dietary-Lipid Uptake and Survival on a High-Fat Diet. Cell Metab. 2016, 23, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Keewan, E.; Narasimhulu, C.A.; Rohr, M.; Hamid, S.; Parthasarathy, S. Are Fried Foods Unhealthy? The Dietary Peroxidized Fatty Acid, 13-HPODE, Induces Intestinal Inflammation In Vitro and In Vivo. Antioxidants 2020, 9, 926. [Google Scholar] [CrossRef]
- Whelan, J.; Fritsche, K. Linoleic Acid. Adv. Nutr. 2013, 4, 311–312. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; O’Keefe, J.H. Omega-6 vegetable oils as a driver of coronary heart disease: The oxidized linoleic acid hypothesis. Open Heart 2018, 5, e000898. [Google Scholar] [CrossRef] [Green Version]
- Salerno, J.W.; Smith, D.E. The use of sesame oil and other vegetable oils in the inhibition of human colon cancer growth in vitro. Anticancer Res. 1991, 11, 209–215. [Google Scholar]
- Rong, R.; Ramachandran, S.; Penumetcha, M.; Khan, N.; Parthasarathy, S. Dietary oxidized fatty acids may enhance intestinal apolipoprotein A-I production. J. Lipid Res. 2002, 43, 557–564. [Google Scholar] [CrossRef]
- Auerbach, B.J.; Kiely, J.S.; Cornicelli, J.A. A spectrophotometric microtiter-based assay for the detection of hydroperoxy derivatives of linoleic acid. Anal. Biochem. 1992, 201, 375–380. [Google Scholar] [CrossRef]
- Andrews, S. FastQC—A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 23 January 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods. 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363. [Google Scholar] [CrossRef]
- Pazienza, V.; Vinciguerra, M.; Mazzoccoli, G. PPARs Signaling and Cancer in the Gastrointestinal System. PPAR Res. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zińczuk, J.; Maciejczyk, M.; Zaręba, K.; Romaniuk, W.; Markowski, A.; Kędra, B.; Zalewska, A.; Pryczynicz, A.; Matowicka-Karna, J.; Guzińska-Ustymowicz, K. Antioxidant Barrier, Redox Status, and Oxidative Damage to Biomolecules in Patients with Colorectal Cancer. Can Malondialdehyde and Catalase Be Markers of Colorectal Cancer Advancement? Biomolecules 2019, 9, 637. [Google Scholar]
- Espinola-Klein, C.; Rupprecht, H.J.; Bickel, C.; Schnabel, R.; Genth-Zotz, S.; Torzewski, M.; Lackner, K.; Münzel, T.; Blankenberg, S. Glutathione Peroxidase-1 Activity, Atherosclerotic Burden, and Cardiovascular Prognosis. Am. J. Cardiol. 2007, 99, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Matsuda, M.; Fukuhara, A.; Komuro, R.; Shimomura, I. Dysregulated glutathione metabolism links to impaired insulin action in adipocytes. Am. J. Physiol. Metab. 2009, 296, E1326–E1334. [Google Scholar] [CrossRef] [Green Version]
- Wojtal, K.A.; Eloranta, J.J.; Hruz, P.; Gutmann, H.; Drewe, J.; Staumann, A.; Beglinger, C.; Fried, M.; Kullak-Ublick, G.A.; Vavricka, S.R. Changes in mRNA Expression Levels of Solute Carrier Transporters in Inflammatory Bowel Disease Patients. Drug Metab. Dispos. 2009, 37, 1871–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumann, T.; König, J.; Henke, C.; Willmes, D.M.; Bornstein, S.R.; Jordan, J.; Fromm, M.F.; Birkenfeld, A.L. Solute Carrier Transporters as Potential Targets for the Treatment of Metabolic Disease. Michel MC, editor. Pharmacol. Rev. 2020, 72, 343–379. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxy radicals, lipid peroxidation and DNA damage. Toxicology 2002, 181–182, 219–222. [Google Scholar] [CrossRef]
- Eilertsen, K.J.; Kazmierski, S.T.; Keller, T.C.S. Cellular titin localization in stress fibers and interaction with myosin II filaments in vitro. J. Cell Biol. 1994, 126, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.N.; Huong, H.T.; Song, C.H.; Lee, J.H.; Han, H.J. Linoleic acid stimulates gluconeogenesis via Ca2+/PLC, cPLA2, and PPAR pathways through GPR40 in primary cultured chicken hepatocytes. Am. J. Physiol. Physiol. 2008, 295, C1518–C1527. [Google Scholar] [CrossRef] [Green Version]
- de Vogel-van den Bosch, H.M.; Bünger, M.; de Groot, P.J.; Bosch-Vermeulen, H.; Hooiveld, G.J.E.J.; Müller, M. PPARalpha-mediated effects of dietary lipids on intestinal barrier gene expression. BMC Genom. 2008, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Bünger, M.; van den Bosch, H.M.; van der Meijde, J.; Kersten, S.; Hooiveld, G.J.E.J.; Müller, M. Genome-wide analysis of PPARα activation in murine small intestine. Physiol. Genom. 2007, 30, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Westerweel, P.E.; Luyten, R.K.M.A.C.; Koomans, H.A.; Derksen, R.H.W.M.; Verhaar, M.C. Premature atherosclerotic cardiovascular disease in systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Reinaud, O.; Delaforge, M.; Boucher, J.L.; Rocchiccioli, F.; Mansuy, D. Oxidative metabolism of linoleic acid by human leukocytes. Biochem. Biophys. Res. Commun. 1989, 161, 883–891. [Google Scholar] [CrossRef]
- Ramana, K.V.; Srivastava, S.; Singhal, S.S. Lipid Peroxidation Products in Human Health and Disease. Oxid. Med. Cell. Longev. 2013, 2013, 583438. [Google Scholar] [CrossRef]
- Cima, I.; Corazza, N.; Dick, B.; Fuhrer, A.; Herren, S.; Jakob, S.; Ayuni, E.; Mueller, C.; Brunner, T. Intestinal Epithelial Cells Synthesize Glucocorticoids and Regulate T Cell Activation. J. Exp. Med. 2004, 200, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidler, D.; Renzulli, P.; Schnoz, C.; Berger, B.; Schneider-Jakob, S.; Flück, C.; Inderbitzin, D.; Corazza, N.; Candinas, D.; Brunner, T. Colon cancer cells produce immunoregulatory glucocorticoids. Oncogene. 2011, 30, 2411–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, M.A.; Ye, D.; Kennedy, J.C.; Al-Sadi, R.; Shepela, C.; Ma, T.Y. Mechanism of glucocorticoid regulation of the intestinal tight junction barrier. Am. J. Physiol. Liver Physiol. 2007, 292, G590–G598. [Google Scholar] [CrossRef] [Green Version]
- Wilckens, T.; De Rijk, R. Glucocorticoids and immune function: Unknown dimensions and new frontiers. Immunol. Today 1997, 18, 418–424. [Google Scholar] [CrossRef]
- Bruder, E.; Raff, H.; Goodfriend, T. An Oxidized Derivative of Linoleic Acid Stimulates Dehydroepiandrosterone Production by Human Adrenal Cells. Horm. Metab. Res. 2006, 38, 803–806. [Google Scholar] [CrossRef]
- Bruder, E.D.; Ball, D.L.; Goodfriend, T.L.; Raff, H. An oxidized metabolite of linoleic acid stimulates corticosterone production by rat adrenal cells. Am. J. Physiol. Integr. Comp. Physiol. 2003, 284, R1631–R1635. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chem. Biol. Interact. 2015, 234, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Vangaveti, V.N.; Jansen, H.; Kennedy, R.L.; Malabu, U.H. Hydroxyoctadecadienoic acids: Oxidised derivatives of linoleic acid and their role in inflammation associated with metabolic syndrome and cancer. Eur. J. Pharmacol. 2016, 785, 70–76. [Google Scholar] [CrossRef]
- Márquez-Ruiz, G.; García-Martínez, M.C.; Holgado, F. Changes and Effects of Dietary Oxidized Lipids in the Gastrointestinal Tract. Lipid Insights 2008, 2, LPI–S904. [Google Scholar] [CrossRef] [Green Version]
- Chester, A. The apolipoprotein B mRNA editing complex performs a multifunctional cycle and suppresses nonsense-mediated decay. EMBO J. 2003, 22, 3971–3982. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhou, Y.; Rychahou, P.G.; Fan, T.W.-M.; Lane, A.N.; Weiss, H.L.; Evers, B.M. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017, 24, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, Y.; Satoh, A.; Tezuka, H.; Han, S.-I.; Takei, K.; Iwasaki, H.; Yatoh, S.; Yahagi, N.; Suzuki, H.; Iwasaki, Y.; et al. CREB3L3 controls fatty acid oxidation and ketogenesis in synergy with PPARα. Sci. Rep. 2016, 6, 39182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, A.M.; Dang, C.H. Basic review of the cytochrome p450 system. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [PubMed]
- Chang, G.W.M.; Kam, P.C.A. The physiological and pharmacological roles of cytochrome P450 isoenzymes. Anaesthesia. 1999, 54, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.-M.; Chang, L.-L.; Ying, M.-D.; Cao, J.; He, Q.-J.; Zhu, H.; Yang, B. Aldo–Keto Reductase AKR1C1–AKR1C4: Functions, Regulation, and Intervention for Anti-cancer Therapy. Front. Pharmacol. 2017, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab. Rev. 2008, 40, 553–624. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, A.M.; MacKenzie, D.A.; Olguin, S.L.; Scariano, J.K.; Rabinowitz, I.; Thompson, T.A. Antioxidants Abrogate Alpha-Tocopherylquinone-Mediated Down-Regulation of the Androgen Receptor in Androgen-Responsive Prostate Cancer Cells. Languino LR, editor. PLoS ONE 2016, 11, e0151525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolfi, S.C.; Yang, Z.; Lee, M.-J.; Guan, F.; Hong, J.; Yang, C.S. Inhibitory Effects of Different Forms of Tocopherols, Tocopherol Phosphates, and Tocopherol Quinones on Growth of Colon Cancer Cells. J. Agric. Food Chem. 2013, 61, 8533–8540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokhina, O. Antioxidants, Oxidative Damage and Oxygen Deprivation Stress: A Review. Ann. Bot. 2003, 91, 179–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Wu, D.; Koga, T.; Martin, K.R.; Meydani, M. Effect of vitamin E on human aortic endothelial cell production of chemokines and adhesion to monocytes. Atherosclerosis 1999, 147, 297–307. [Google Scholar] [CrossRef]
- Morel, S.; Didierlaurent, A.; Bourguignon, P.; Delhaye, S.; Baras, B.; Jacob, V.; Planty, C.; Elouahabi, A.; Harvengt, P.; Carlsen, H. Adjuvant System AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 2011, 29, 2461–2473. [Google Scholar] [CrossRef]
- Shi, H.; Noguchi, N.; Niki, E. Comparative study on dynamics of antioxidative action of α-tocopheryl hydroquinone, ubiquinol, and α-tocopherol against lipid peroxidation. Free Radic. Biol. Med. 1999, 27, 334–346. [Google Scholar] [CrossRef]
- Bindoli, A.; Valente, M.; Cavallini, L. Inhibition of lipid peroxidation by alpha-tocopherolquinone and alpha-tocopherolhydroquinone. Biochem. Int. 1985, 10, 753–761. [Google Scholar]
- Calviello, G. gamma-Tocopheryl quinone induces apoptosis in cancer cells via caspase-9 activation and cytochrome c release. Carcinogenesis 2003, 24, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.Y.; Massey, K.; Watson, M.A.; Hussain, T.; Volpe, G.; Buckley, C.D.; Nicolaou, A.; Badenhorst, P. Oxidised metabolites of the omega-6 fatty acid linoleic acid activate dFOXO. Life Sci. Alliance 2020, 3, e201900356. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Arango, D.; Corner, G.A.; Arañes, M.J.; Hotchkiss, K.A.; Yang, W.; Augenlicht, L.H. A gene expression profile that defines colon cell maturation in vitro. Cancer Res. 2002, 62, 4791–4804. [Google Scholar] [PubMed]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Lipid peroxidation—DNA damage by malondialdehyde. Mutat. Res. Mol. Mech. Mutagen. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Ma, Y.; Semba, S.; Khan, R.I.; Bochimoto, H.; Watanabe, T.; Fujiya, M.; Kohgo, Y.; Liu, Y.; Taniguchi, T. Focal adhesion kinase regulates intestinal epithelial barrier function via redistribution of tight junction. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.F.; Quaroni, A. Clonal analysis of sucrase-isomaltase expression in the human colon adenocarcinoma Caco-2 cells. Biochem. J. 1991, 280, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Prabhu, R.; Balasubramanian, K.A. Retinoid metabolism in the rat small intestine. Br. J. Nutr. 2005, 93, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, S.; Johnson, C.D.; Hennebelle, M.; Holtmann, T.; Taha, A.Y.; Kirpich, I.A.; Eguchi, A.; Ramsden, C.E.; Papouchado, B.G.; McClain, C.J.; et al. Oxidized linoleic acid metabolites induce liver mitochondrial dysfunction, apoptosis, and NLRP3 activation in mice. J. Lipid Res. 2018, 59, 1597–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; Beck, M.A.; Brown, D.A.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Hwang, D.H. The modulation of inflammatory gene expression by lipids: Mediation through Toll-like receptors. Mol. Cells 2006, 21, 174–185. [Google Scholar]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Kamitani, H.; Geller, M.; Eling, T. Expression of 15-Lipoxygenase by Human Colorectal Carcinoma Caco-2 Cells during Apoptosis and Cell Differentiation. J. Biol. Chem. 1998, 273, 21569–21577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodkorb, A.; Egger, L.; Alminger, M.; Alvito, P.; Assunção, R.; Ballance, S.; Bohn, T.; Bourlieu-Lacanal, C.; Boutrou, R.; Carrière, F.; et al. INFOGEST static in vitro simulation of gastrointestinal food digestion. Nat. Protoc. 2019, 14, 991–1014. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KEGG Pathway (Upregulated) | p-Value |
|---|---|
| I00140 Steroid hormone biosynthesis | 0.018 |
| I04976 Bile secretion | 0.029 |
| I04974 Protein digestion and absorption | 0.029 |
| I00980 Metabolism of xenobiotics by cytochrome P450 | 0.062 |
| I04510 Focal adhesion | 0.069 |
| I03320 PPAR signaling pathway | 0.199 |
| KEGG Pathway (Downregulated) | p-Value |
| I03040—Spliceosome | 0.0001 |
| I00240—Pyrimidine metabolism | 0.0004 |
| I03008—Ribosome biogenesis in eukaryotes | 0.0005 |
| I03013—RNA transport | 0.002 |
| I00190—Oxidative phosphorylation | 0.003 |
| I04110—Cell cycle | 0.004 |
| I00230—Purine metabolism | 0.006 |
| I03420—Nucleotide excision repair | 0.009 |
| I03020—RNA polymerase | 0.009 |
| I03050—Proteasome | 0.014 |
| I03030—DNA replication | 0.018 |
| I04120—Ubiquitin-mediated proteolysis | 0.030 |
| I03410—Base excision repair | 0.031 |
| I03010—Ribosome | 0.034 |
| KEGG Pathway (Upregulated) | p-Value |
|---|---|
| I00830—Retinol metabolism | 0.010 |
| I04080—Neuroactive ligand-receptor interaction | 0.017 |
| I00980—Metabolism of xenobiotics by cytochrome P450 | 0.017 |
| I00982—Drug metabolism—cytochrome P450 | 0.039 |
| I04974—Protein digestion and absorption | 0.047 |
| KEGG Pathway (Downregulated) | p-Value |
| I03008—Ribosome biogenesis in eukaryotes | 0.0001 |
| I03040—Spliceosome | 0.0004 |
| I03013—RNA transport | 0.0007 |
| I00240—Pyrimidine metabolism | 0.0009 |
| I00190—Oxidative phosphorylation | 0.016 |
| I03020—RNA polymerase | 0.017 |
| I03050—Proteasome | 0.020 |
| I00230—Purine metabolism | 0.037 |
| I04110—Cell cycle | 0.038 |
| I03420—Nucleotide excision repair | 0.043 |
| I04620—Toll-like receptor signaling pathway | 0.045 |
| Gene | 13-HPODE-Treated | LA-Treated |
|---|---|---|
| DKK1 | ↓↓ 3 | ↓↓ |
| CPT1A | ↑↑ 3 | ↑ 2 |
| PLIN2 | ↑↑ | ↑ |
| ODC1 | ↓ 2 | ↓ |
| CREB3L3 | ↑↑ | ↑ |
| FABP1 | ↑↑ | ↑ |
| PDK4 | ↑↑ | ↑ |
| PCK1 | ↑↑ | ↑ |
| COL7A1 | ↑↑ | ↑ |
| NOX1 | ↓ | ↓ |
| AKR1C2 | ↑↑ | ↑ |
| SOS1 | ↑ | ↑ |
| BAAT | ↑ | n 1 |
| CYP2B6 | ↑↑ | ↑ |
| CYP2C9 | ↑ | ↑ |
| INSIG1 | n | ↓ |
| DDIT4 | ↑ | ↑ |
| DUSP4 | ↓ | ↓↓ |
| UGT2B4 | ↑ | ↑ |
| TXNRD1 | ↑ | n |
| HMOX1 | ↑ | n |
| GSTP1 | ↓ | n |
| CXCL1 | ↓ | ↓ |
| GPX1 | ↓ | ↓ |
| FOXO4 | ↑ | ↑ |
| CEACAM6 | ↓ | ↓↓ |
| POLD2 | ↓ | ↓ |
| ADH6 | n | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faizo, N.; Narasimhulu, C.A.; Forsman, A.; Yooseph, S.; Parthasarathy, S. Peroxidized Linoleic Acid, 13-HPODE, Alters Gene Expression Profile in Intestinal Epithelial Cells. Foods 2021, 10, 314. https://doi.org/10.3390/foods10020314
Faizo N, Narasimhulu CA, Forsman A, Yooseph S, Parthasarathy S. Peroxidized Linoleic Acid, 13-HPODE, Alters Gene Expression Profile in Intestinal Epithelial Cells. Foods. 2021; 10(2):314. https://doi.org/10.3390/foods10020314
Chicago/Turabian StyleFaizo, Nisreen, Chandrakala Aluganti Narasimhulu, Anna Forsman, Shibu Yooseph, and Sampath Parthasarathy. 2021. "Peroxidized Linoleic Acid, 13-HPODE, Alters Gene Expression Profile in Intestinal Epithelial Cells" Foods 10, no. 2: 314. https://doi.org/10.3390/foods10020314