1. Introduction
The recent decades have witnessed an increased interest in functional food and nutrition, as well as their impact on human health. This has led companies to use them in commercial foods [
1]. As functional compounds, probiotics have been used for the development of a wide range of functional food products [
2], which has considerably increased the global consumption of probiotic foods in recent years. The world market of probiotics was estimated as USD 49.4 billion in 2018, and is projected to reach USD 69.3 billion by 2023 [
3]. Probiotics are defined by the World Health Organization as ‘‘living microorganisms which upon ingestion in certain numbers, exert health benefits beyond inherent general nutrition” [
4]. It has been recommended that in order to produce a therapeutic benefit, probiotic bacteria in food should be present at levels of least 10
6 cfu g
−1 or mL
−1 (live microorganisms per g or mL) at the time of consumption [
1]. When administered in adequate amounts, the probiotic organisms confer their health benefits through inhibition of pathogen growth by blocking the adhesion sites of pathogenic bacteria while maintaining health-promoting gut microflora [
2,
5]. These living bacteria have the capacity of controlling intestinal infection, regulating serum cholesterol levels, boosting the host’s immune system, and improving lactose utilization in persons who have suffered from lactose malabsorption. They also show a positive impact on suppressing colon cancer and irritable bowel syndrome [
6,
7].
It was found that some probiotic bacteria, such as
Lactobacillus johnsonii,
L. rhamnosus, and
Saccharomyces boulardii, provide a healthy gut flora and contribute to the host’s health [
2,
8]. Thanks to its clinically proven health-promoting effects,
L. rhamnosus is employed as the probiotic model, and it has shown a biofilm-forming ability in vitro [
5]. To confer their health benefits, the probiotic cells must retain their viability during food processing and storage, as well as gastrointestinal transit through the acidic stomach and intestine [
7,
8]. However, there are several limitations for the use of probiotics in foods and beverages. Several factors are directly associated with their growth and stability rate, including pH, storage temperature, processing conditions, and environment of the digestive system. A low pH is one of the most important factors restricting the survival rate of probiotic bacteria [
9]. Hydrogen ions ruin probiotic cells by disrupting mass transfer through the cell membrane. Fermentation, processing conditions, and storage temperature are the other factors that play an important role in probiotic stability in food products [
1,
2,
10]. Temperatures above 45–50 °C lead to a reduction in free probiotic cell viability during food processing. Moreover, transition through the gastrointestinal tract is still a major challenge to obtaining the minimum suggested concentration of viable cells to provide the aforementioned benefits [
9].
In order to improve their survival in such adverse conditions, encapsulation of probiotics in hydrocolloid beads is generally used to improve probiotics’ survival during digestion, considering the limitations of free probiotics’ survival in food processing and during gastrointestinal transit [
2,
8]. Microencapsulation is a technology of packing liquids, solids, and gaseous materials into tiny capsules that release those contents at controlled rates over long periods of time [
11,
12]. Probiotics can be encapsulated with this technique so that they can be released at a controlled rate under specific conditions [
13,
14]. It has been confirmed that encapsulated bacteria can survive better than free cells during gastric transit and harsh environmental conditions. Nevertheless, materials and methodologies, as well as the coating material used for their production, should be chosen carefully by preserving their vitality during the encapsulation process [
15].
The major methods used for probiotic encapsulation are extrusion, emulsion, and spray-drying. These techniques each have their own unique and specific characteristics that suit the encapsulation of probiotics. Spray-drying presents a great flexibility, but the process temperature is an important drawback of this technique for the encapsulation of probiotics. Despite the fact that the extrusion method utilizes a huge diversity of machines and industrial components for generating capsules from different polymer mixtures, the utilization of this technique at a large scale requires in-depth studies and a significant amount of investments. Emulsification is clearly one of the most common encapsulation techniques for producing capsules smaller than 100 µm at the laboratory scale [
2]. In this regard, some carrier matrixes have been used to cover the probiotic cells. Polysaccharides (e.g., alginate, carrageenan, and chitosan) are typically used, and effectively protect the cells from the acidic environment of the stomach and subsequently release the cells gradually into the suitable intestinal sections of the gut [
5,
16].
Considering the protection of probiotics against the harsh conditions of digestion, a wide variety and combinations of coating materials have been studied. Characteristics of the materials used play a key role because the viability of encapsulated probiotic cells depends on the physicochemical properties of the material. Several encapsulation agents, including polysaccharides derived from algae (k-carrageenan, alginate), plants (pectin and starch derivatives, gum arabic), or bacteria (gellan, xanthan) and animal proteins have been investigated for use in the probiotic microencapsulation method. In this regard, hydrocolloids are popular wall materials for the encapsulation of food ingredients. The type and concentration of the coating material, particle size, initial cell number, and bacterial strains are some other parameters to be considered [
9]. Of note, particular attention also needs to be paid when choosing the right material as food grade and approved by regulatory authorities [
2].
Among the encapsulating materials used, alginate is the most commonly employed polymer for immobilizing viable cells, due to its strong capacity to be cross-linked and the different mild gelling characteristics that change based on the molecular weight. The main reasons behind its common utilization in the microencapsulation of probiotics include its GRAS (generally regarded as safe) status worldwide as a food additive, its lack of toxicity, low cost, simplicity, and biocompatibility [
2]. However, when it comes to protecting cells from low-pH environments, there is a drawback to using alginate due to the fact that alginate beads present very porous capsules. Namely, at very low pHs, cross-linked alginate matrix systems are reduced, and this reduction in alginate molecular weight causes a faster degradation and release of active ingredients [
9]. Some works mention that alginate microbeads, without the application of a coating, have the capacity to protect probiotics during food storage, but not upon exposure to low-pH solutions, such as in gastrointestinal conditions [
2]. Mixing alginate with other polymers, such as chitosan and starch, appears to be a solution for the enhancement of microcapsules’ resistance to acidic media [
5,
9].
Xanthan gum is another coating material that has been used in combination with gellan gum to improve the protection capacity of microcapsules for probiotic bacteria (
L. plantarum and
L. rhamnosus) [
17]. The most relevant properties of xanthan gum used in probiotic microencapsulation are its ability to keep microparticles in suspension without greatly raising viscosity, its tolerance to enzyme degradation, and acid resistance [
18]. Milk proteins, such as casein and whey protein, can also be used to encapsulate probiotics because of their excellent gelation properties. Considering the extreme conditions of the stomach, these proteins are able to create a higher local pH value within the protein matrix of the capsules, thanks to the buffering capacity of the proteins. Their amphoteric feature makes it possible to mix them with negatively charged polysaccharides such as alginate, carrageenan, or pectin [
19].
In this study, we aimed to evaluate the effect of different coating materials (xanthan gum, gum acacia, sodium caseinate, chitosan, starch, carrageenan) on the efficiency and survivability of alginate-encapsulated Lactobacillus rhamnosus under different pH, temperature, NaCl, and simulated digestive system conditions. This is the first study in which such a variety of coating materials were investigated and compared for encapsulating probiotic bacteria. Moreover, the antimicrobial activity of the optimized microcapsules against some indicator microorganisms (S. aureus, E. coli, B. cereus, S. typhimurium) were also successfully evaluated.
2. Materials and Methods
2.1. Materials and Chemicals
Sodium alginate with a medium viscosity and high mannuronic acid without inulin were purchased from Hi-media (Mumbai). They were prepared in distilled water and autoclaved at 121 °C for 15 min. The low-molecular-weight chitosan (deacetylated chitin, Hi-Media, Laboratories Mumbai), was prepared in distilled water and pure hydrochloric acid (Loba Chemie, Pvt Ltd.—Mumbai, India). Xanthan gum, gum acacia, and carrageenan were purchased from Loba Chemie, Pvt Ltd.—Mumbai, India. Starch, sodium caseinate, pepsin, trypsin, calcium chloride, sodium chloride, sodium bicarbonate, trisodium citrate, sodium hydroxide, potassium chloride, phosphate buffer saline (pH 7.2), and hydrochloric acid were purchased from Hi-media Laboratories (Mumbai, India). Glacial acetic acid with molar mass of 60.05 g mol−1 was bought from Merck (Darmstadt, Germany). Pancreatin, peptone, and bile salt were obtained from Loba Chemie, Pvt Ltd. (Mumbai, India). The De Man Rogosa and Sharpes (MRS) broth and MRS agar used in this work were purchased from Hi-media Laboratories (Mumbai). Indicator microorganisms used for the antimicrobial activity; i.e., Escherichia coli (MTCC No-432), Staphylococcus aureus (MTCC No-96), and Bacillus cereus (MTCC No-430), were procured from the Institute of Microbial Technology (IMTECH; Chandigarh, India).
2.2. Bacterial Strain and Culture Preparations
Microencapsulated and free cells of the bacterial strain
L. rhamnosus isolated from indigenous fermented foods were grown in MRS broth and incubated at 37 °C in the absence of oxygen for 24–48 h. The cultures were harvested by centrifugation at 8000 rpm for 15 min at 4 °C, followed by two washes with the triple-distilled water. To achieve a suspension comprising approximately 10 log (cfu mL
−1) cells, the pellet was resuspended in 0.1% (
w/
v) peptone solution. The cell suspensions were freshly prepared for each experiment and then dissolved in 1% (
w/
v) sterile saline solution. The cell counts were calculated by the pour plate method using appropriate 10-fold dilutions onto MRS agar after 48 h of incubation at 37 °C [
20].
2.3. Microencapsulation Procedure of the Microorganisms
To produce the microcapsules, alginate beads were initially prepared as microbeads. These microbeads were then coated with a polymeric matrix such as xanthan gum, gum acacia, sodium caseinate, chitosan, starch, or carrageenan. For preparation of microbeads using the external gelation process, the sodium alginate was prepared (1%, w/v) by dissolving it in distilled water and stirring at 65 °C for 20 min. A 5 mL sample of the bacterial suspension containing a viable count of 107–108 cfu mL−1 was added to the previously prepared alginate solution (95 mL) and stirred at 65 °C for 20 min until obtaining a homogenous solution. The cross-linking solution (10%) was prepared by dissolving calcium chloride in distilled water. The alginate solution was drawn into a 3 mL syringe with a 26 G needle and dropped manually into the cross-linking medium for the formation of the alginate beads. The beads were then filtered using a strainer, rinsed with distilled water, and stored in the refrigerator until further use.
The microencapsulation of
Lactobacillus rhamnosus was performed using the method defined by Vodnar et al. [
21]. The different polymeric matrix solutions (i.e., xanthan gum solution, gum acacia solution, sodium caseinate solution, chitosan solution, starch solution, and carrageenan solution) were prepared in distilled water (2%,
w/
v) and autoclaved at 121 °C for 15 min. Each polymeric matrix solution was mixed with sodium alginate beads (1:1). The mixture was homogenized into a sunflower oil containing 0.2% (
w/
v) and emulsified for 5 min by stirring at 400 rpm. Afterwards, a solution of glacial acetic acid (900 µL) was dissolved in 10 mL of sunflower oil, and the mixture was stirred for 10 min. To stabilize the microencapsulated beads, the emulsion was lowered into the sterile solution of CaCl
2 (2%
w/
v). The oil coating on the top layer was filtered with Whatman filter paper, and the microbeads were washed in 500 mL of distilled water. The microbeads were then stored in saline water with the pH adjusted to 4.0 at 4 °C in the refrigerator until further use. As a control, the free nonencapsulated cultured bacteria were also collected and stored using the same procedure [
22].
2.4. Particle Sizes of Encapsulated Microbeads
Dynamic light scattering (DLS) studies were performed to confirm the average particle size of the beads. The particle sizes of encapsulated beads were determined at 25 °C using a Malvern Zetasizer Nano ZS system (Malvern Instruments, Ltd., Malvern, UK). For this, 2 mg of encapsulated beads was dissolved in 10 mL of distilled water, and the suspension was then filtered and used for analysis. The measurements were performed in a computer-controlled particle-size analyzer to determine the particle-size distribution. Each sample was analyzed in triplicate.
2.5. Characterization of Microbead Morphology
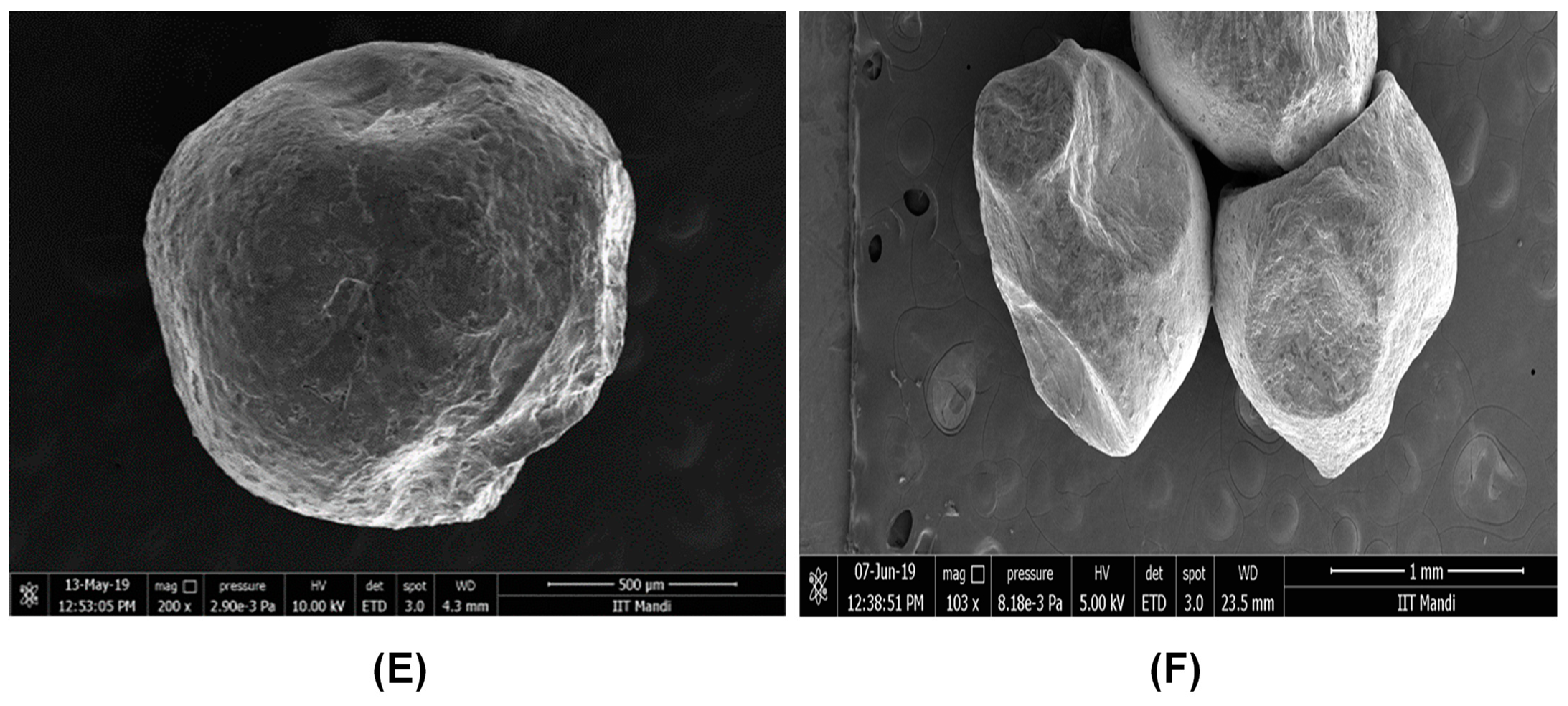
The morphologies of the encapsulated microbeads were analyzed by field emission scanning electron microscopy (FESEM) (Nova Nano SEM, 450, FEI Company, Hillsboro, OR, USA). For this, the encapsulated beads were taped to the stub and coated for 1–2 min with an Au–Pd coating using a sputter coater and then used for analysis under the FESEM at an accelerating voltage potential of 20 kV.
2.6. Microencapsulation Yield
The viable cell count of
L. rhamnosus was calculated according to a modified method [
7]. To release the entrapped bacteria from the capsules, 1 g of microbeads was dissolved in 9 mL of sterile tri-sodium citrate solution (2%,
w/
v) and vortexed at room temperature for 5 min. The samples were serially diluted with 0.1% peptone (
w/
v) and dissolved in MRS agar. The plate was incubated in anaerobic conditions at 37 °C for 48 h.
The encapsulation yield (
EY) was calculated using Equation (1):
where
N is the number of the viable entrapped bacterial cells (cfu mL
−1) released from the beads, and
N0 is the number of the free viable bacterial cells (cfu mL
−1) added to the biopolymer mixture during the preparation of microbeads.
2.7. Growth Profile of Microencapsulated Cells
Free and microencapsulated microbial cells were inoculated in a triplicate sterile MRS broth. The cell density was calculated as optical density (OD) every 2 h over a 24 h period at 600 nm by employing a UV-spectrophotometer (UV 2450, Thermo Fisher Scientific, Germany). The controls were obtained using the ODs of the bacteria-free broth and empty microbeads.
2.8. Tolerance to Bile Salts and Acid
The ability of isolated
L. rhamnosus to sustain different bile salt concentrations was studied according to the method described by Succi et al. [
23]. In sterile test tubes, 10
8 cfu mL
−1 of bacterial suspension was inoculated in 10 mL of bile solution at different concentrations of bile salt (0, 1, 1.5, and 2%). Sterile distilled water with no bile salt was kept as control. The samples were serially diluted and then plated on MRS agar plates. Afterwards, the plates were incubated aerobically at 37 °C and at different time intervals (2, 4, and 6 h), and the colony counts were recorded.
The survivability of microencapsulated
L. rhamnosus was tested by adjusting sterile MRS broth to pH 1, 2, and 3 using 1 mol HCl. Sterile distilled water at pH 7.0 was used as control. All tubes were filled with 10 mL of sterile MRS broth. The different sets of tubes were inoculated with a bacterial suspension (10
8 cfu mL
−1) at each pH (1, 2, 3, and 7). The tubes were then incubated at 37 °C for 2, 4, and 6 h, respectively. At the time of incubation, 1 mL of culture was taken from each tube and serially diluted 10-fold with 0.85% normal saline solution and poured on the MRS agar plates. The plates were then incubated at 37 °C for 24–48 h to determine the residual viable count [
24].
2.9. Growth of Encapsulated L. rhamnosus at Different Temperatures
One gram of the bacterial strain
L. rhamnosus encapsulated in alginate+xanthan, as well as 1 mL of free cells with cell density of 10
8 cfu mL
−1, were added to 10 mL of preheated sterile water and inoculated in different tubes. Distilled water was used as the suspending medium. Further, the individual tubes were incubated overnight at various temperatures of 5, 15, 37, and 45 °C, respectively. The growth of the strain at the different temperatures was compared with the control tube, which was incubated at 37 °C for 24 h. After incubation, the sample tubes were cooled to room temperature, and viable cells were counted in triplicate on MRS agar [
9].
2.10. Growth of Encapsulated L. rhamnosus at Different NaCl Concentrations
The sterilized test tubes containing 10 mL MRS broth with different NaCl concentrations (0, 2, 3, 4, and 6%) were inoculated with the bacterial suspension (100 µL) with a viable cell count (log10
8 cfu mL
−1). All test tubes were incubated at 37 °C for 24 h, after which the viability of free and microencapsulated cells was recorded in triplicate on MRS agar [
25].
2.11. Survivability of Microencapsulated Cells after Incubation in Simulated Gastric Juice
The simulated gastric juice (SGJ), which consisted of 10 mg mL
−1 of pepsin and 0.02 M phosphate buffer solution (PBS), was adjusted to pH 3.0 with 1 M HCl and sterilized by autoclaving at 121 °C for 15 min. Microencapsulated or free probiotic samples (0.5 g) were inoculated on tubes (4.5 mL, preheated to 37 °C) containing the filtered and sterilized simulated gastric juice (SGJ) and incubated at 37 °C. After incubation, the viable cell count was assessed using the surface plate count method at 0, 1, 2, and 3 h time intervals. The survival rate (%) of free and microencapsulated bacteria was calculated using Equation (2):
where
N is the number of viable cells (cfu g
−1) after exposure to the simulated gastric juice conditions, and
N0 is the number of viable cells (cfu g
−1) before exposure to the simulated gastric juice conditions.
2.12. Survival of Microencapsulated Cells after Incubation in Simulated Intestinal Juice
The simulated intestinal juice (SIJ) was prepared according to the method described by Gbassi et al. [
26]. A solution of 6.5 g L
−1 NaCl, 0.835 g L
−1 KCl, 0.22 g L
−1 CaCl
2, 1.386 g L
−1 NaHCO
3, and 3 g L
−1 bile salt was adjusted to pH 7.5 and sterilized at 121 °C for 15 min before adding pancreatin in a final concentration of 10 g L
−1. The SIJ was inoculated with 10% of the microcapsules and incubated at 37 °C. The viable cell count was determined at 0, 1, 2, and 3 h time intervals, as previously stated.
2.13. Antimicrobial Activity of Probiotic Isolate L. rhamnosus
The isolated L. rhamnosus was further investigated for its antimicrobial activity against food-borne pathogens; i.e., Escherichia coli (MTCC No-432), Staphylococcus aureus (MTCC No-96), and Bacillus cereus (MTCC No-430). A concentration of approximately 108 cfu mL−1 of the indicator strain was added to 10 mL of MRS agar and poured over the plate containing the producer after 24 h of anaerobic incubation at 37 °C. The bacterial lawns for the zones of inhibition surrounding the producer colonies were tested. Positive inhibition was described as a 5 mm or greater clear zone surrounding the producer’s colonies.
2.14. Statistical Analysis
The data obtained in this research were expressed as mean ± SD of triplicates and were analyzed using one-way analysis of variance (ANOVA); 0.05 was chosen as the level of statistical meaning. The mean and standard deviation (SD) were calculated by subjecting the values to SPSS 16.020 statistical analyses when required using Microsoft Excel 2007. The graphs were created using GraphPad Prism.








{kind=link}
{kind=link}
{kind=link}
{kind=link}