Epicatechin Gallate as Xanthine Oxidase Inhibitor: Inhibitory Kinetics, Binding Characteristics, Synergistic Inhibition, and Action Mechanism


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments and Methods
2.2.1. XO Activity Assay
2.2.2. Kinetic Analysis of Enzyme-Catalyzed Reaction
2.2.3. DPPH Radical Scavenging Activity Assay
2.2.4. O2− Radical Scavenging Activity Assay
2.2.5. The Scavenging Activity Assay of O2− Radical Generated by XO
2.2.6. Fluorescence Titration Experiments
2.2.7. Measurements of CD Spectra
2.2.8. Microstructure Analysis
2.2.9. Molecular Docking
2.2.10. Molecular Dynamics (MD) Simulation
2.2.11. Combined Inhibition Analysis
2.2.12. Statistical Analysis
3. Results
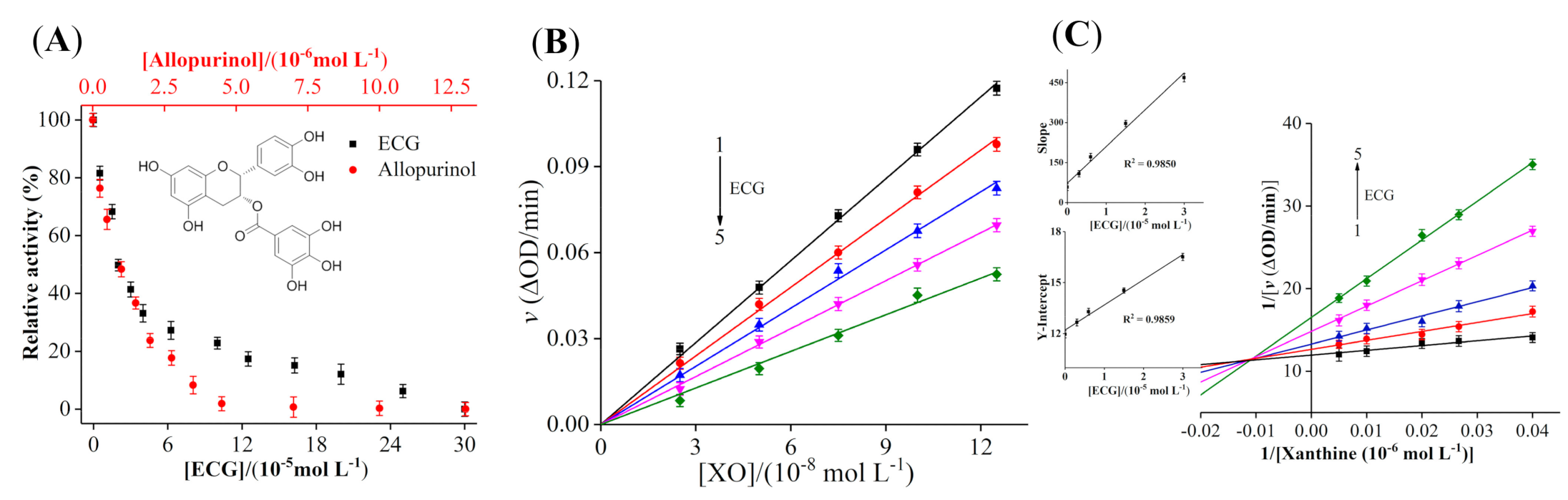
3.1. Inhibitory Activity of ECG on XO
3.2. Inhibition Kinetic Analysis of ECG
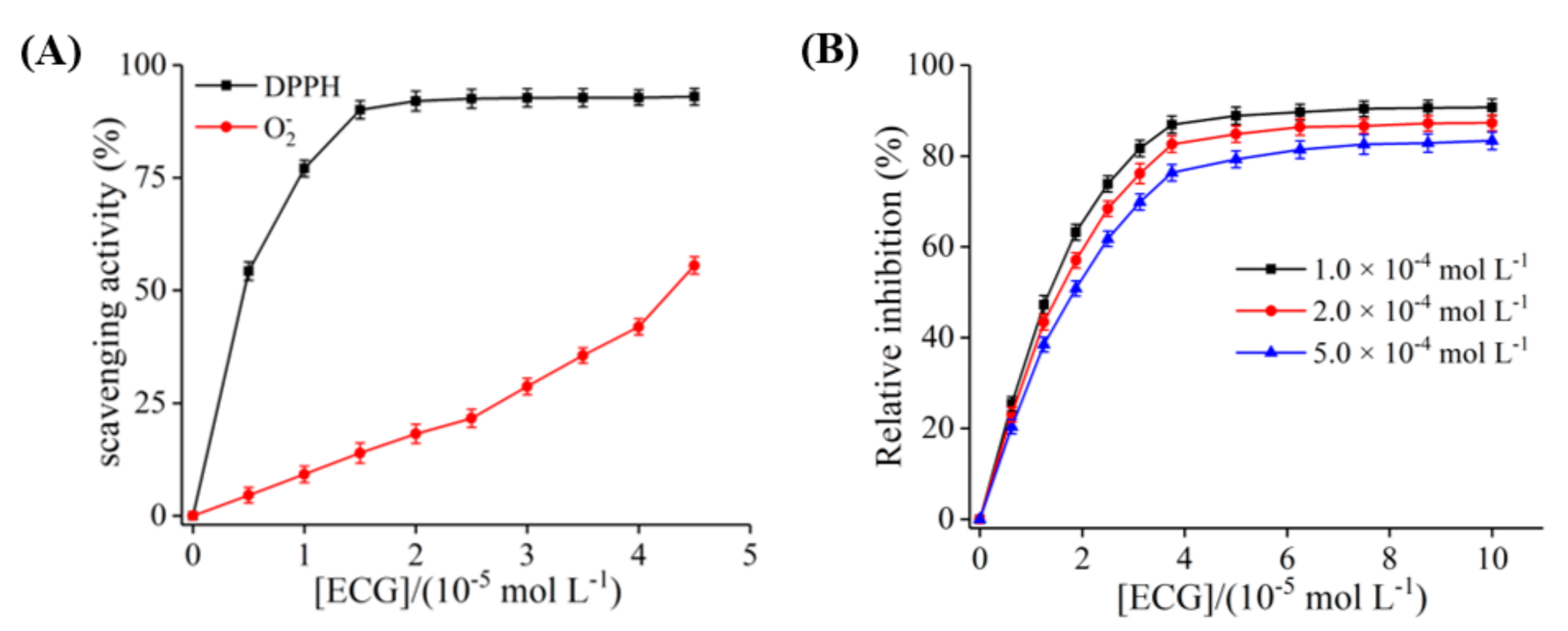
3.3. The Inhibition of ECG on O2− Radical Produced by XO
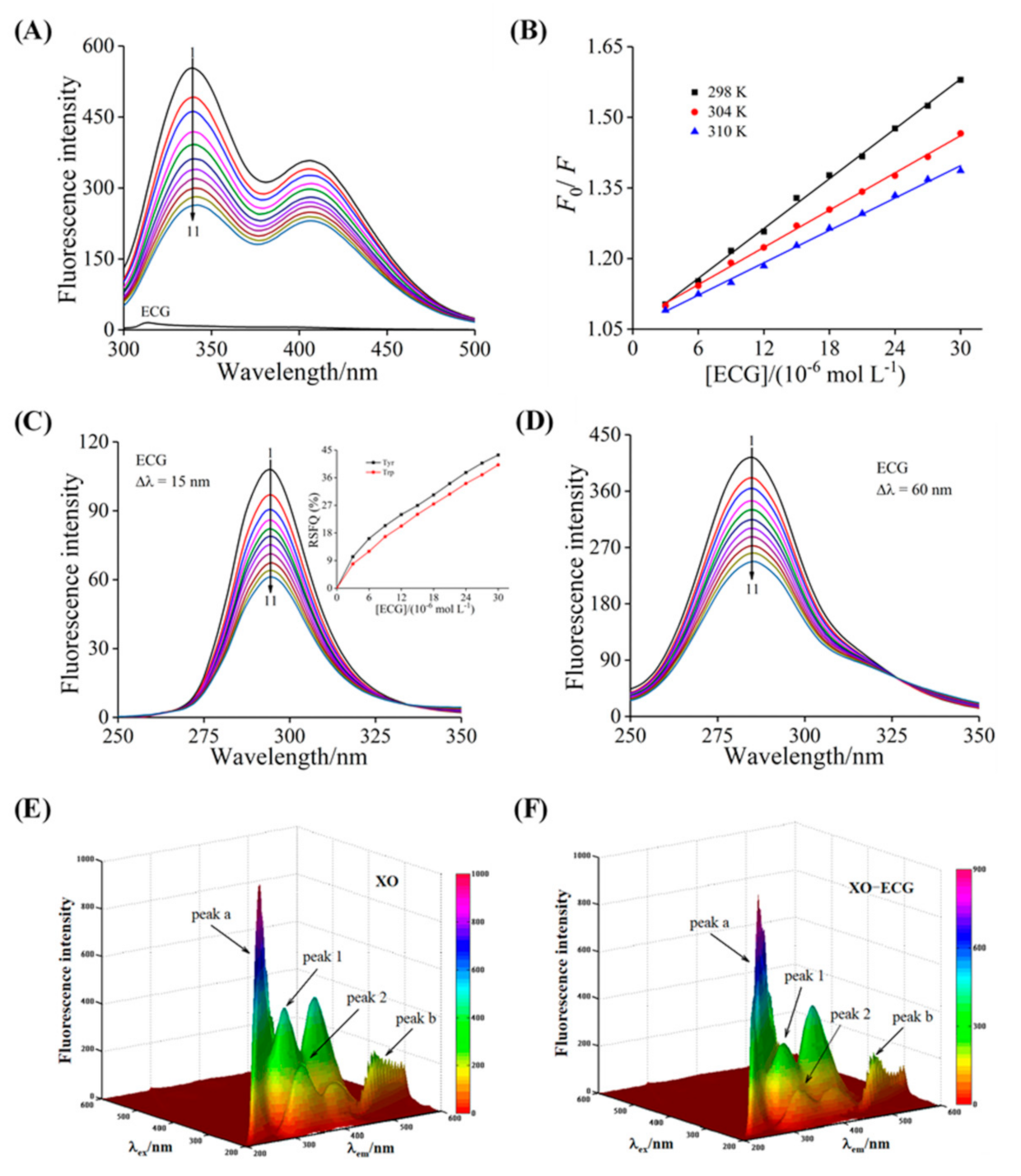
3.4. Fluorescence Quenching and Thermodynamic Analysis
3.5. Synchronous Fluorescence Analysis
3.6. The 3D Fluorescence Analysis
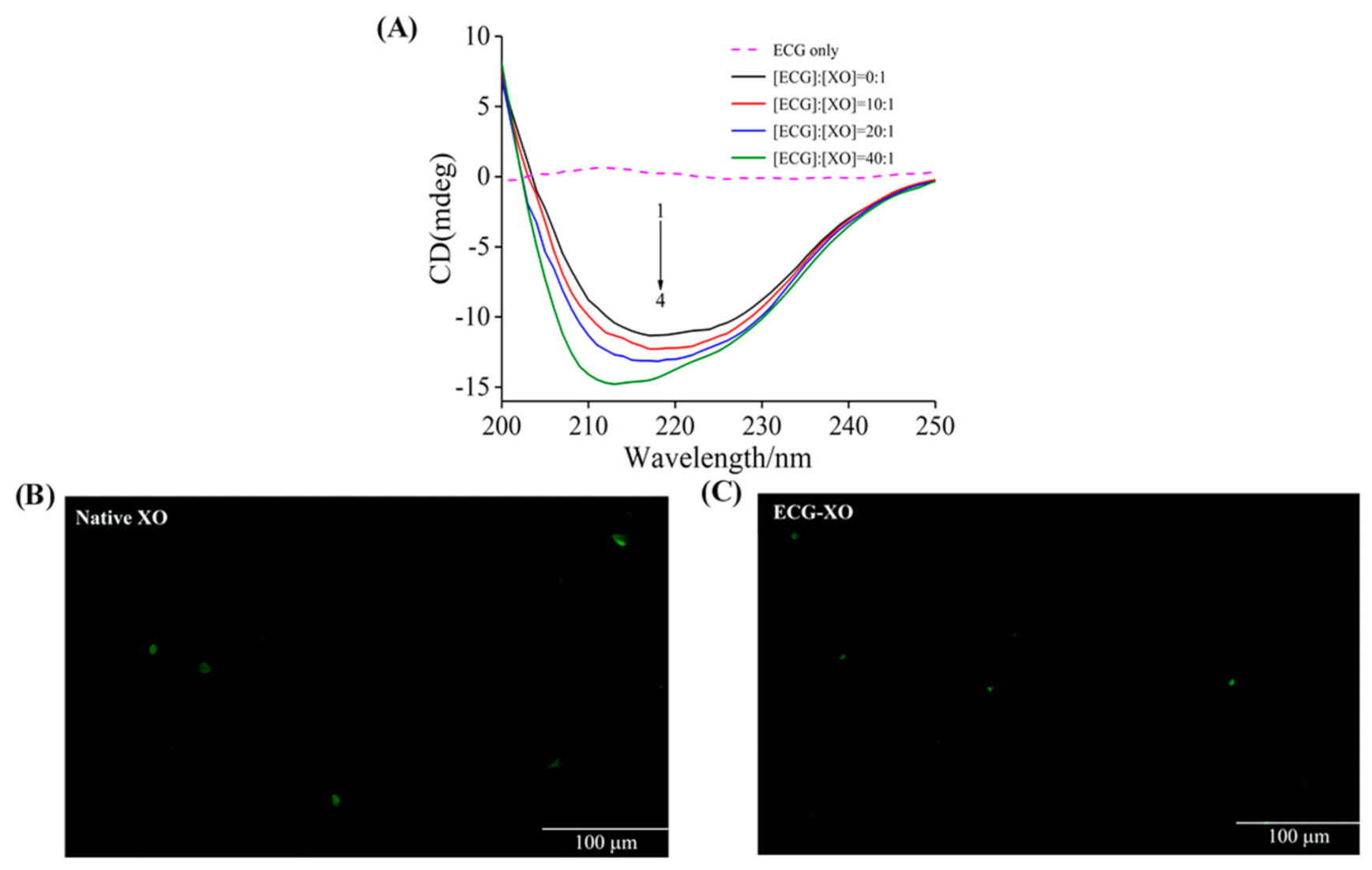
3.7. CD Spectra Studies
3.8. Fluorescence Microscopy Imaging
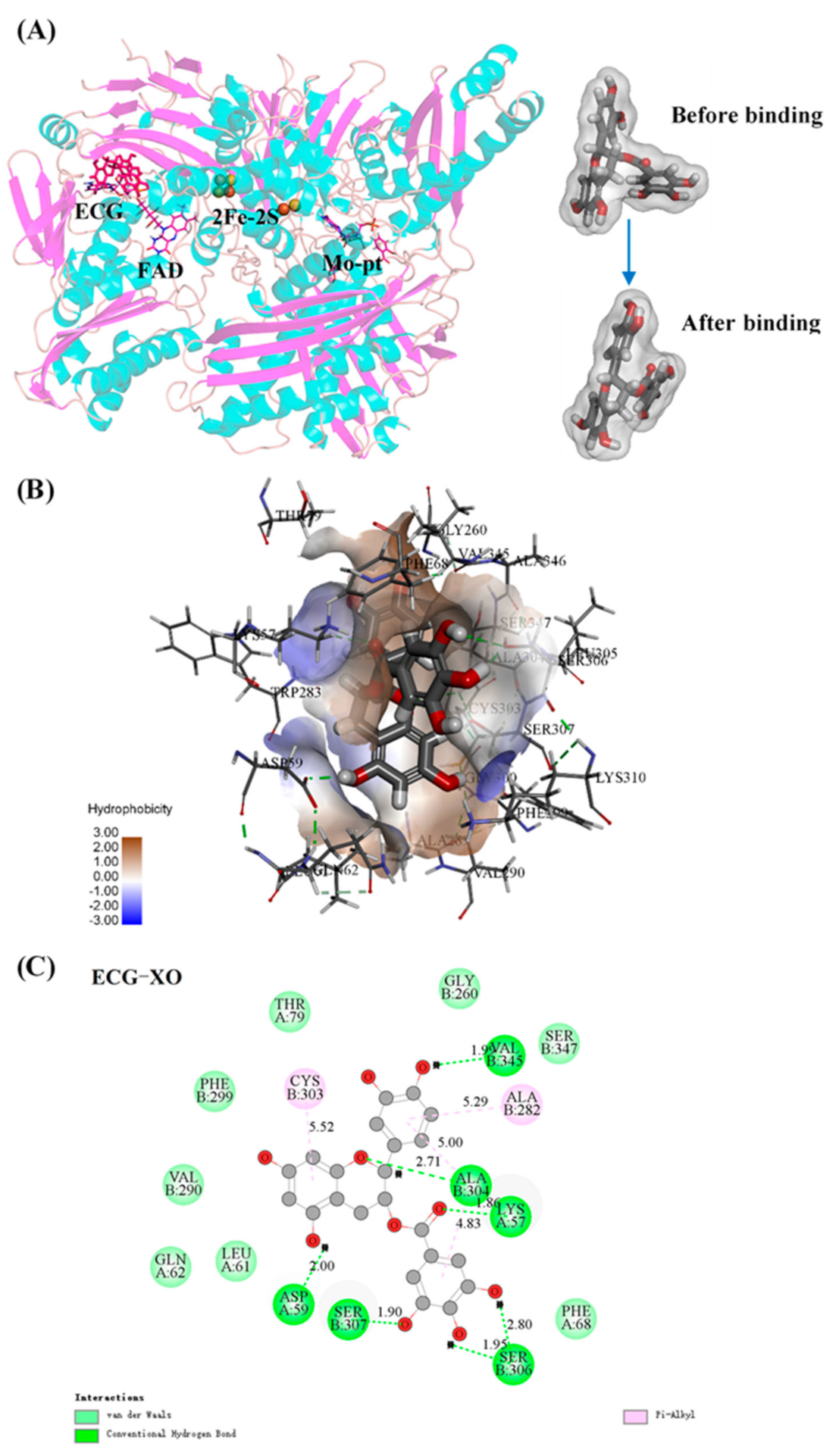
3.9. Molecular Docking
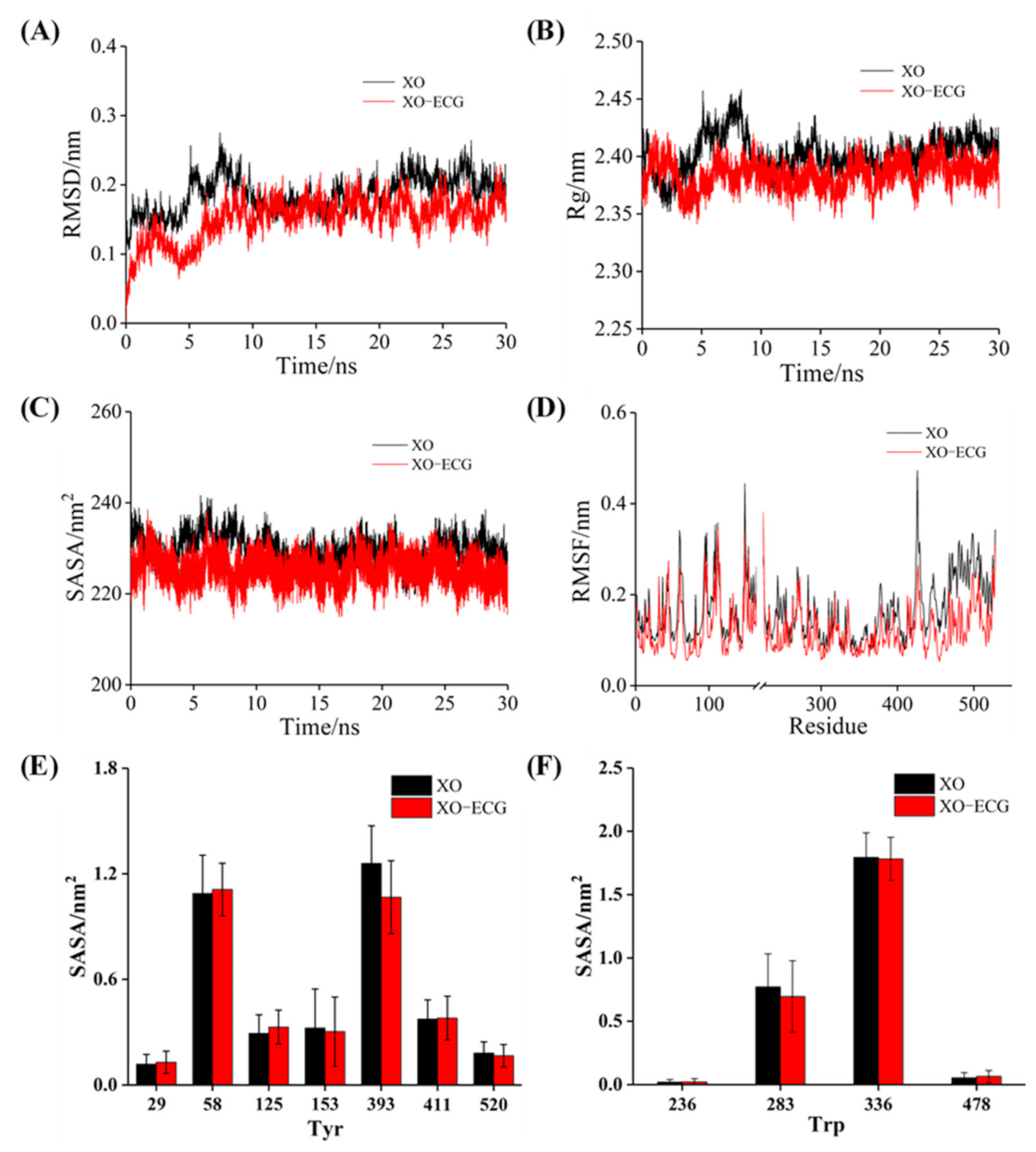
3.10. MD Simulation
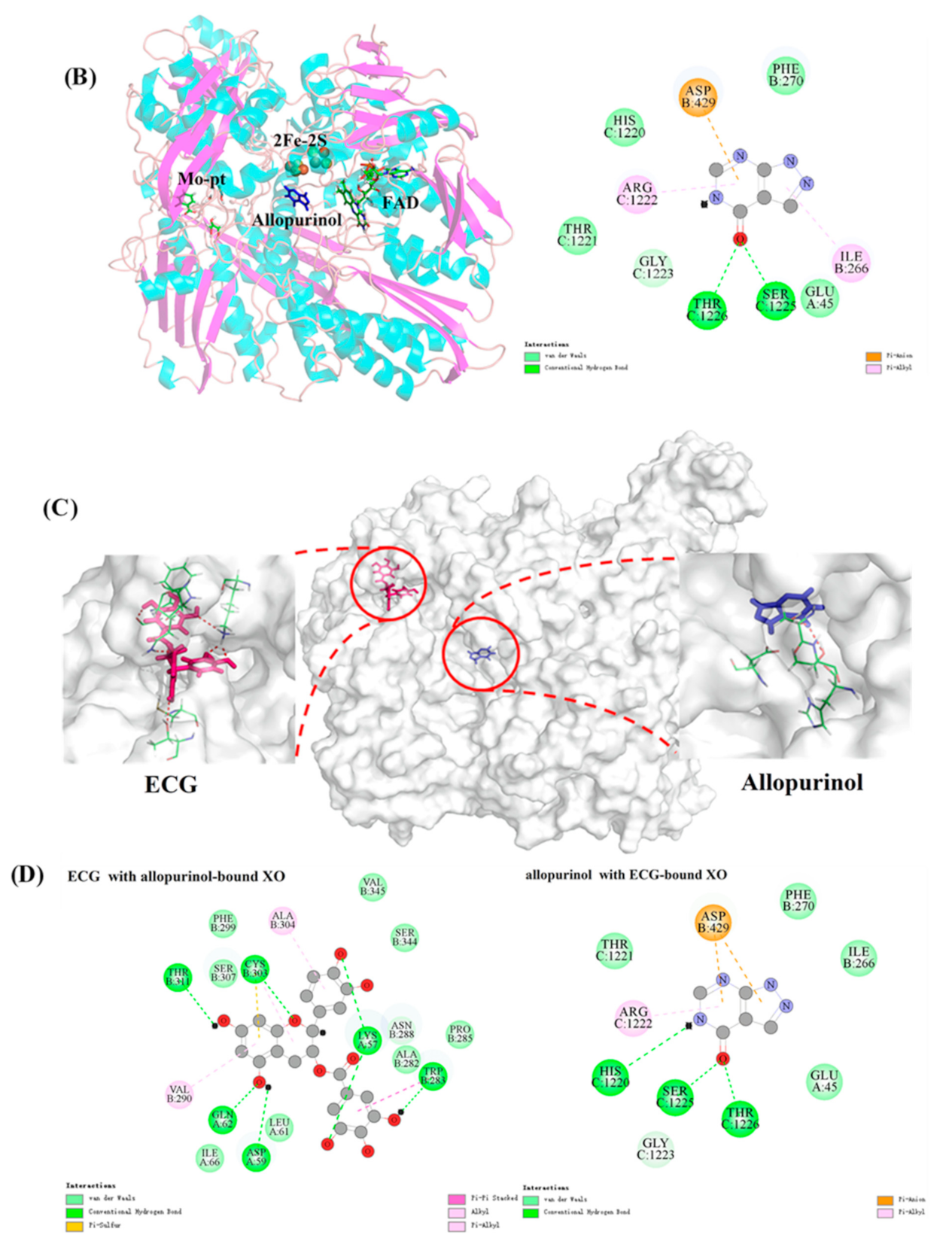
3.11. Joint Inhibitory Effect and Mechanism Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singh, J.A.; Gaffo, A. Gout epidemiology and comorbidities. Semin. Arthritis Rheum. 2020, 50, S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Dehlin, M.; Jacobsson, L.; Roddy, E. Global epidemiology of gout: Prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 2020, 16, 380–390. [Google Scholar] [CrossRef]
- Yokose, C.; Chen, M.; Berhanu, A.; Pillinger, M.H.; Krasnokutsky, S. Gout and Osteoarthritis: Associations, Pathophysiology, and Therapeutic Implications. Curr. Rheumatol. Rep. 2016, 18, 65. [Google Scholar] [CrossRef]
- Pillinger, M.H.; Mandell, B.F. Therapeutic approaches in the treatment of gout. Semin. Arthritis Rheum. 2020, 50, S24–S30. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Pauff, J.M.; Hille, R. X-ray Crystal Structure of a Xanthine Oxidase Complex with the Flavonoid Inhibitor Quercetin. J. Nat. Prod. 2014, 77, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.N.; Gupta, G.; Sharma, P. A comprehensive review of free radicals, antioxidants, and their relationship with human ailments. Crit. Rev. Eukaryot. Gene Expr. 2018, 28, 139–154. [Google Scholar] [CrossRef]
- Lindsay, A.; McCourt, P.M.; Karachunski, P.; Lowe, D.A.; Ervasti, J.M. Xanthine oxidase is hyper-active in Duchenne muscular dystrophy. Free Radic. Biol. Med. 2018, 129, 364–371. [Google Scholar] [CrossRef]
- Schmidt, H.M.; Kelley, E.E.; Straub, A.C. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019, 21, 101072. [Google Scholar] [CrossRef]
- Baldissera, M.D.; Souza, C.F.; Doleski, P.H.; Moreira, K.L.S.; da Rocha, M.I.U.M.; da Veiga, M.L.; Santos, R.C.V.; Baldisserotto, B. Xanthine oxidase activity exerts a pro-oxidant and pro-inflammatory profile in gills of experimentally infected silver catfish with Streptococcus agalactiae. Aquaculture 2017, 477, 71–75. [Google Scholar] [CrossRef]
- White, W.B. Gout, Xanthine Oxidase Inhibition, and Cardiovascular Outcomes. Circulation 2018, 138, 1127–1129. [Google Scholar] [CrossRef]
- Stamp, L.K.; Chapman, P.T. Allopurinol hypersensitivity: Pathogenesis and prevention. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101501. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Liu, Y.; Xiao, A.; Leng, J.; Liao, L.; Ma, L.; Liu, L. The interaction of dietary flavonoids with xanthine oxidase in vitro: Molecular property-binding affinity relationship aspects. RSC Adv. 2019, 9, 10781–10788. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Huang, H.; Zhao, M.; Sun-Waterhouse, D.; Lin, L.; Xiao, C. Mechanisms underlying the xanthine oxidase inhibitory effects of dietary flavonoids galangin and pinobanksin. J. Funct. Foods 2016, 24, 26–36. [Google Scholar] [CrossRef]
- Ooi, K.L.; Zakaria, R.; Tan, M.L.; Sulaiman, S.F. The influence of chemical composition of potent inhibitors in the hydrolyzed extracts of anti-hyperuricemic plants to their xanthine oxidase activities. J. Ethnopharmacol. 2021, 278, 114294. [Google Scholar] [CrossRef]
- Lin, L.; Yang, Q.; Zhao, K.; Zhao, M. Identification of the free phenolic profile of Adlay bran by UPLC-QTOF-MS/MS and inhibitory mechanisms of phenolic acids against xanthine oxidase. Food Chem. 2018, 253, 108–118. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hussaini, R.; White, R.; Atwi, D.; Fried, A.; Sampat, S.; Piao, L.; Pan, Q.; Banerjee, P. TriCurin, a synergistic formulation of curcumin, resveratrol, and epicatechin gallate, repolarizes tumor-associated macrophages and triggers an immune response to cause suppression of HPV+ tumors. Cancer Immunol. Immunother. 2018, 67, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Gao, Y.; Granato, D. Effects of epigallocatechin gallate, epigallocatechin and epicatechin gallate on the chemical and cell-based antioxidant activity, sensory properties, and cytotoxicity of a catechin-free model beverage. Food Chem. 2021, 339, 128060. [Google Scholar] [CrossRef] [PubMed]
- Rameshrad, M.; Razavi, B.M.; Hosseinzadeh, H. Protective effects of green tea and its main constituents against natural and chemical toxins: A comprehensive review. Food Chem. Toxicol. 2017, 100, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, G.; Martel, F. The effect of dietary polyphenols on intestinal absorption of glucose and fructose: Relation with obesity and type 2 diabetes. Food Rev. Int. 2019, 35, 390–406. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, C.; Zhang, B.; Huang, Q. The inhibitory effects of flavonoids on α-amylase and α-glucosidase. Crit. Rev. Food Sci. Nutr. 2020, 60, 695–708. [Google Scholar] [CrossRef]
- Janssens, P.L.H.R.; Hursel, R.; Westerterp-Plantenga, M.S. Nutraceuticals for body-weight management: The role of green tea catechins. Physiol. Behav. 2016, 162, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Min, Y.; Xiao, J.; Feng, N.; Chen, Y.; Luo, Q.; Zhou, M.; Li, D.; Hu, Z.; Wang, C. Liquid state fermentation vinegar enriched with catechin as an antiglycative food product. Food Funct. 2019, 10, 4877–4887. [Google Scholar] [CrossRef]
- Irondi, E.A.; Olanrewaju, S.; Oboh, G.; Olasupo, F.; Boligon, A.A. Inhibitory potential of cocoa leaves polyphenolics-rich extract on xanthine oxidase and angiotensin 1-converting enzyme. J. Biol. Act. Prod. Nat. 2017, 7, 39–51. [Google Scholar] [CrossRef]
- Peluso, I.; Serafini, M. Antioxidants from black and green tea: From dietary modulation of oxidative stress to pharmacological mechanisms. Br. J. Pharmacol. 2017, 174, 1195–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, G.; Pan, J.; Gong, D. Novel insights into the inhibitory mechanism of kaempferol on xanthine oxidase. J. Agric. Food Chem. 2015, 63, 526–534. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Z.C.; Zhou, Q.; Yan, J.X.; Zhang, J.L.; Su, G.H. Hypouricemic effect in hyperuricemic mice and xanthine oxidase inhibitory mechanism of dietary anthocyanins from purple sweet potato (Ipomoea batatas L.). J. Funct. Foods 2020, 73, 104151. [Google Scholar] [CrossRef]
- Miranda, H.F.; Puig, M.M.; Prieto, J.C.; Pinardi, G. Synergism between paracetamol and nonsteroidal anti-inflammatory drugs in experimental acute pain. Pain 2006, 121, 22–28. [Google Scholar] [CrossRef]
- Djouahri, A.; Saka, B.; Boudarene, L.; Benseradj, F.; Aberrane, S.; Aitmoussa, S.; Chelghoum, C.; Lamari, L.; Sabaou, N.; Baaliouamer, A. In vitro synergistic/antagonistic antibacterial and anti-inflammatory effect of various extracts/essential oil from cones of Tetraclinis articulata (Vahl) Masters with antibiotic and anti-inflammatory agents. Ind. Crops Prod. 2014, 56, 60–66. [Google Scholar] [CrossRef]
- Mehmood, A.; Zhao, L.; Wang, C.; Hossen, I.; Nadeem, M. Stevia residue extract alone and combination with allopurinol attenuate hyperuricemia in fructose-PO-induced hyperuricemic mice. J. Food Biochem. 2020, 44, e13087. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, G.; Lin, S.; Gong, D. Inhibitory mechanism of apigenin on α-glucosidase and synergy analysis of flavonoids. J. Agric. Food Chem. 2016, 64, 6939–6949. [Google Scholar] [CrossRef]
- Zeng, N.; Zhang, G.; Hu, X.; Pan, J.; Gong, D. Mechanism of fisetin suppressing superoxide anion and xanthine oxidase activity. J. Funct. Foods 2019, 58, 1–10. [Google Scholar] [CrossRef]
- Aalim, H.; Belwal, T.; Jiang, L.; Huang, H.; Meng, X.; Luo, Z. Extraction optimization, antidiabetic and antiglycation potentials of aqueous glycerol extract from rice (Oryza sativa L.) bran. LWT Food Sci. Technol. 2019, 103, 147–154. [Google Scholar] [CrossRef]
- Masuoka, N.; Tamsampaoloet, K.; Chavasiri, W.; Kubo, I. Superoxide anion scavenging activity of alk(en)yl phenol compounds by using PMS-NADH system. Heliyon 2016, 2, e00169. [Google Scholar] [CrossRef] [Green Version]
- Fan, M.; Zhang, G.; Hu, X.; Xu, X.; Gong, D. Quercetin as a tyrosinase inhibitor: Inhibitory activity, conformational change and mechanism. Food Res. Int. 2017, 100, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Chen, J.; McClements, D.J.; Hu, P.; Ye, X.; Liu, C.; Li, T. Protein-polyphenol interactions enhance the antioxidant capacity of phenolics: Analysis of rice glutelin-procyanidin dimer interactions. Food Funct. 2019, 10, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, B.; Tan, C.; Huang, Q. α-Glucosidase inhibitors: Consistency of in silico docking data with in vitro inhibitory data and inhibitory effect prediction of quercetin derivatives. Food Funct. 2019, 10, 6312–6321. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, G.; Liao, Y.; Gong, D. Myricetin inhibits the generation of superoxide anion by reduced form of xanthine oxidase. Food Chem. 2017, 221, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Warren, F.J.; Netzel, G.; Gidley, M.J. 3 or 3′-Galloyl substitution plays an important role in association of catechins and theaflavins with porcine pancreatic α-amylase: The kinetics of inhibition of α-amylase by tea polyphenols. J. Funct. Foods 2016, 26, 144–156. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, L.; Sun, C.; Zhao, D.; Tang, H. Studies on the structure-activity relationship and interaction mechanism of flavonoids and xanthine oxidase through enzyme kinetics, spectroscopy methods and molecular simulations. Food Chem. 2020, 323, 126807. [Google Scholar] [CrossRef]
- Sae-Leaw, T.; Benjakul, S.; Simpson, B.K. Effect of catechin and its derivatives on inhibition of polyphenoloxidase and melanosis of Pacific white shrimp. J. Food Sci. Technol. 2017, 54, 1098–1107. [Google Scholar] [CrossRef]
- Masuoka, N.; Kubo, I. Suppression of superoxide anion generation catalyzed by xanthine oxidase with alkyl caffeates and the scavenging activity. Int. J. Food Sci. Nutr. 2016, 67, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Belwal, T.; He, Y.; Xu, Y.; Li, L.; Luo, Z. Interaction and binding mechanism of cyanidin-3-O-glucoside to ovalbumin in varying pH conditions: A spectroscopic and molecular docking study. Food Chem. 2020, 320, 126616. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Hu, X.; Zhang, Y.; Pan, J.; Zhang, G. Revealing the groove binding characteristics of plant growth regulator 3-indoleacetic acid with calf thymus DNA. J. Mol. Liq. 2021, 326, 115265. [Google Scholar] [CrossRef]
- Meng, Y.; Hao, L.; Tan, Y.; Yang, Y.; Liu, L.; Li, C.; Du, P. Noncovalent interaction of cyanidin-3-O-glucoside with whey protein isolate and β-lactoglobulin: Focus on fluorescence quenching and antioxidant properties. LWT Food Sci. Technol. 2021, 137, 110386. [Google Scholar] [CrossRef]
- Ni, M.; Hu, X.; Gong, D.; Zhang, G. Inhibitory mechanism of vitexin on α-glucosidase and its synergy with acarbose. Food Hydrocoll. 2020, 105, 105824. [Google Scholar] [CrossRef]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, Y.; Ye, X.; Wang, L.; Shao, J.; Jing, H.; Jiang, C.; Wang, H.; Ma, C. Three flavanols delay starch digestion by inhibiting α-amylase and binding with starch. Int. J. Biol. Macromol. 2021, 172, 503–514. [Google Scholar] [CrossRef]
- Song, X.; Ni, M.; Zhang, Y.; Zhang, G.; Pan, J.; Gong, D. Comparing the inhibitory abilities of epigallocatechin-3-gallate and gallocatechin gallate against tyrosinase and their combined effects with kojic acid. Food Chem. 2021, 349, 129172. [Google Scholar] [CrossRef] [PubMed]
- Tayyab, S.; Sam, S.E.; Kabir, M.Z.; Ridzwan, N.F.W.; Mohamad, S.B. Molecular interaction study of an anticancer drug, ponatinib with human serum albumin using spectroscopic and molecular docking methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 214, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zang, F.; Luo, W.; Zhao, Z.; Wang, Y.; Xu, X.; Wang, C. Spectroscopic study on the interaction between mononaphthalimide spermidine (MINS) and bovine serum albumin (BSA). J. Photochem. Photobiol B Biol. 2015, 142, 103–109. [Google Scholar] [CrossRef]
- Vijeesh, V.; Jisha, N.; Vysakh, A.; Latha, M.S. Interaction of eugenol with xanthine oxidase: Multi spectroscopic and in silico modelling approach. Spectrochim. Acta A 2021, 258, 119843. [Google Scholar] [CrossRef]
- Wang, J.; Sun, S.; Zhao, K.; Shi, H.; Fan, J.; Wang, H.; Liu, Y.; Liu, X.; Wang, W. Insights into the inhibitory mechanism of purpurogallin on xanthine oxidase by multiple spectroscopic techniques and molecular docking. J. Mol. Struct. 2021, 1228, 129772. [Google Scholar] [CrossRef]
- Ni, M.; Song, X.; Pan, J.; Gong, D.; Zhang, G. Vitexin Inhibits Protein Glycation through Structural Protection, Methylglyoxal Trapping, and Alteration of Glycation Site. J. Agric. Food Chem. 2021, 69, 2462–2476. [Google Scholar] [CrossRef]
- Wu, X.; Hu, M.; Hu, X.; Ding, H.; Gong, D.; Zhang, G. Inhibitory mechanism of epicatechin gallate on α-amylase and α-glucosidase and its combinational effect with acarbose or epigallocatechin gallate. J. Mol. Liq. 2019, 290, 111202. [Google Scholar] [CrossRef]
- Qi, C.; Zhang, R.; Liu, F.; Zheng, T.; Wu, W. Molecular mechanism of interactions between inhibitory tripeptide GEF and angiotensin-converting enzyme in aqueous solutions by molecular dynamic simulations. J. Mol. Liq. 2018, 249, 389–396. [Google Scholar] [CrossRef]
- Yu, Q.; Fan, L. Understanding the combined effect and inhibition mechanism of 4-hydroxycinnamic acid and ferulic acid as tyrosinase inhibitors. Food Chem. 2021, 352, 129369. [Google Scholar] [CrossRef] [PubMed]
- Ou, R.; Lin, L.; Zhao, M.; Xie, Z. Action mechanisms and interaction of two key xanthine oxidase inhibitors in galangal: Combination of in vitro and in silico molecular docking studies. Int. J. Biol. Macromol. 2020, 162, 1526–1535. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T (K) | Ksv (×104 L mol−1) | Ra | Ka (×104 L mol−1) | Rb | n | ΔH° (kJ mol−1) | ΔG° (kJ mol−1) | ΔS° (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|---|
| 298 | 1.76 ± 0.02 | 0.9982 | 1.82 ± 0.05 | 0.9934 | 0.91 ± 0.01 | −25.05 ± 0.20 | −24.32 ± 0.40 | −2.44 ± 0.07 |
| 304 | 1.32 ± 0.03 | 0.9984 | 1.51 ± 0.02 | 0.9928 | 0.95 ± 0.02 | −24.31 ± 0.35 | ||
| 310 | 1.15 ± 0.01 | 0.9964 | 1.23 ± 0.04 | 0.9985 | 0.87 ± 0.02 | −24.29 ± 0.28 |
| Molar Ratio [ECG]:[XO] | α-Helix (%) | β-Sheet (%) | β-Turn (%) | Random Coil (%) |
|---|---|---|---|---|
| 0:1 | 8.41 ± 0.21 | 41.59 ± 0.36 | 22.27 ± 0.36 | 27.73 ± 0.36 |
| 10:1 | 10.83 ± 0.51 | 42.51 ± 0.41 | 21.65 ± 0.94 | 25.12 ± 0.69 |
| 20:1 | 10.96 ± 0.62 | 43.41 ± 0.74 | 21.59 ± 0.13 | 24.03 ± 0.31 |
| 40:1 | 11.83 ± 0.93 | 45.02 ± 0.22 | 21.39 ± 0.25 | 22.51 ± 0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, M.; Pan, J.; Hu, X.; Zhang, G. Epicatechin Gallate as Xanthine Oxidase Inhibitor: Inhibitory Kinetics, Binding Characteristics, Synergistic Inhibition, and Action Mechanism. Foods 2021, 10, 2191. https://doi.org/10.3390/foods10092191
Zhu M, Pan J, Hu X, Zhang G. Epicatechin Gallate as Xanthine Oxidase Inhibitor: Inhibitory Kinetics, Binding Characteristics, Synergistic Inhibition, and Action Mechanism. Foods. 2021; 10(9):2191. https://doi.org/10.3390/foods10092191
Chicago/Turabian StyleZhu, Miao, Junhui Pan, Xing Hu, and Guowen Zhang. 2021. "Epicatechin Gallate as Xanthine Oxidase Inhibitor: Inhibitory Kinetics, Binding Characteristics, Synergistic Inhibition, and Action Mechanism" Foods 10, no. 9: 2191. https://doi.org/10.3390/foods10092191