
Comparative Transcriptomic Analysis Reveals Diverse Expression Pattern Underlying Fatty Acid Composition among Different Beef Cuts

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
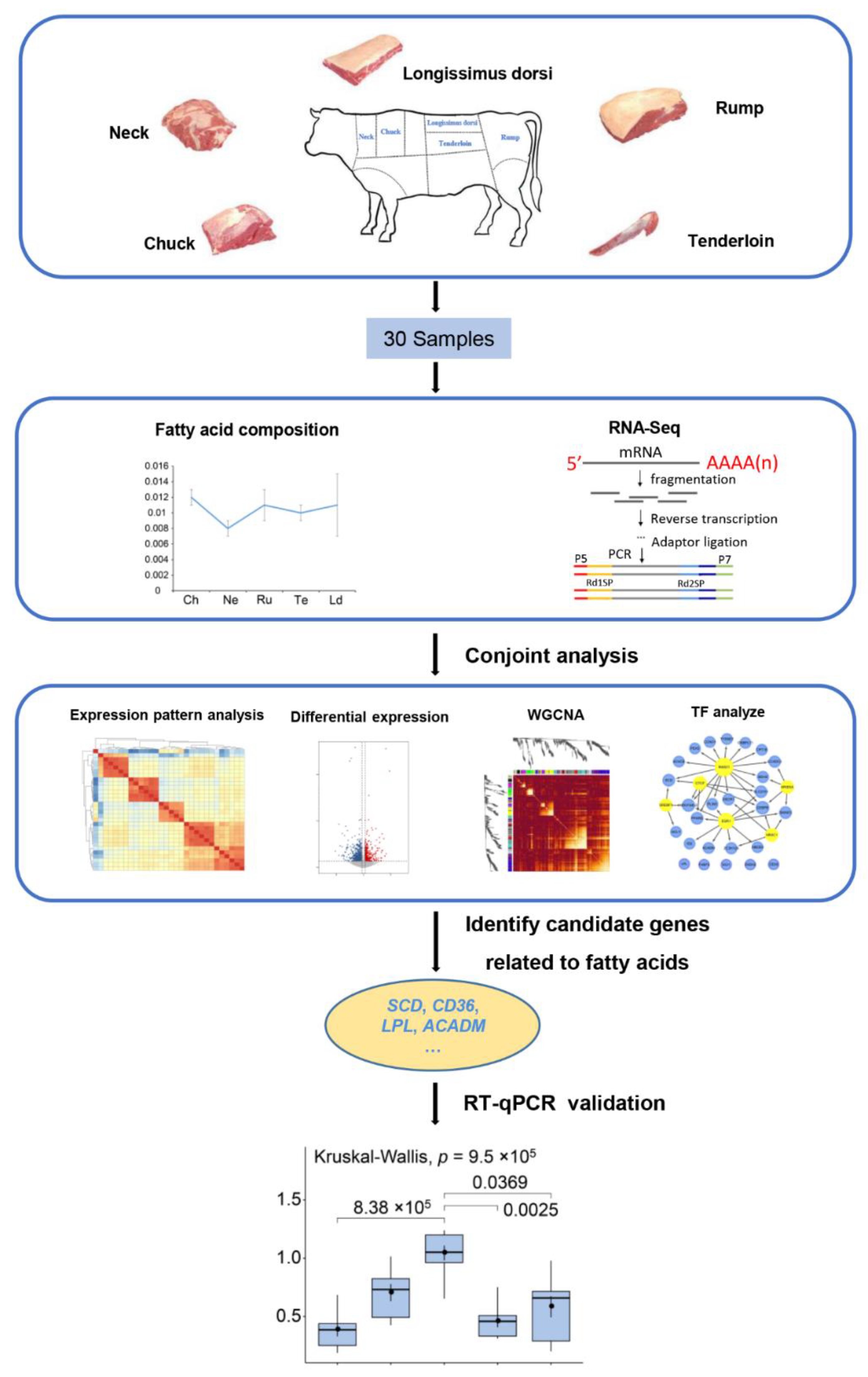
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. Fatty Acid Determination
2.4. RNA Extraction, Library Preparation, and Sequencing
2.5. Data Quality Control and Processing
2.6. Gene Expression Pattern across Tissues
2.7. Analysis and Annotation of Differentially Expressed Genes
2.8. Weighted Gene Co-Expression Network Construct
2.9. Analysis PPIs and TF of Fatty Acid Composition Candidate Genes
2.10. Real-Time Quantitative PCR (RT-qPCR) Analysis
3. Results
3.1. Difference of Fatty Acids and Fatty Acid Groups in Different Beef Cuts
3.2. Summary Statistics of Sequencing Dataset
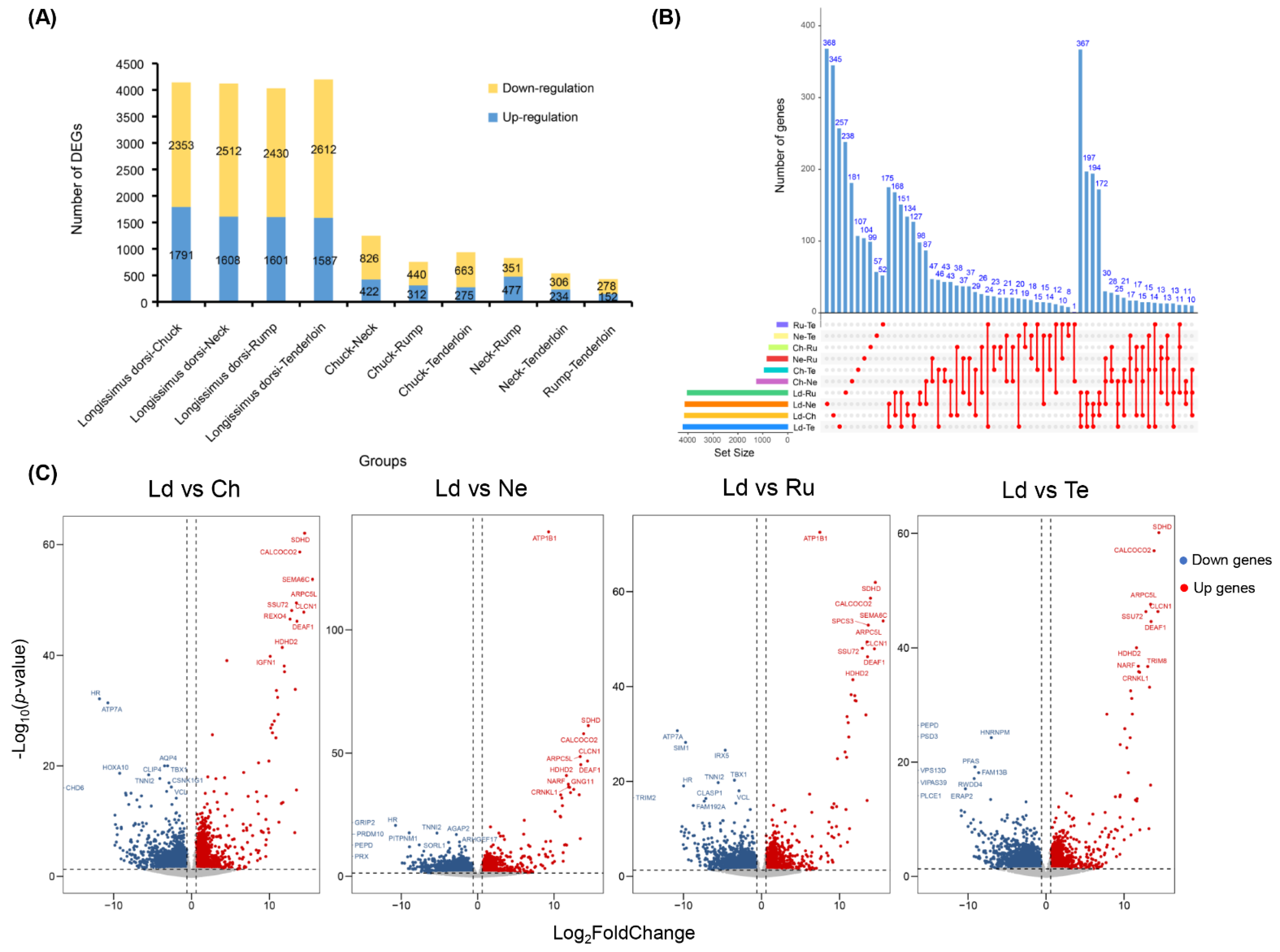
3.3. Gene Expression Profile across Tissues
3.4. Differential Expression Analysis between Five Different Beef Cuts
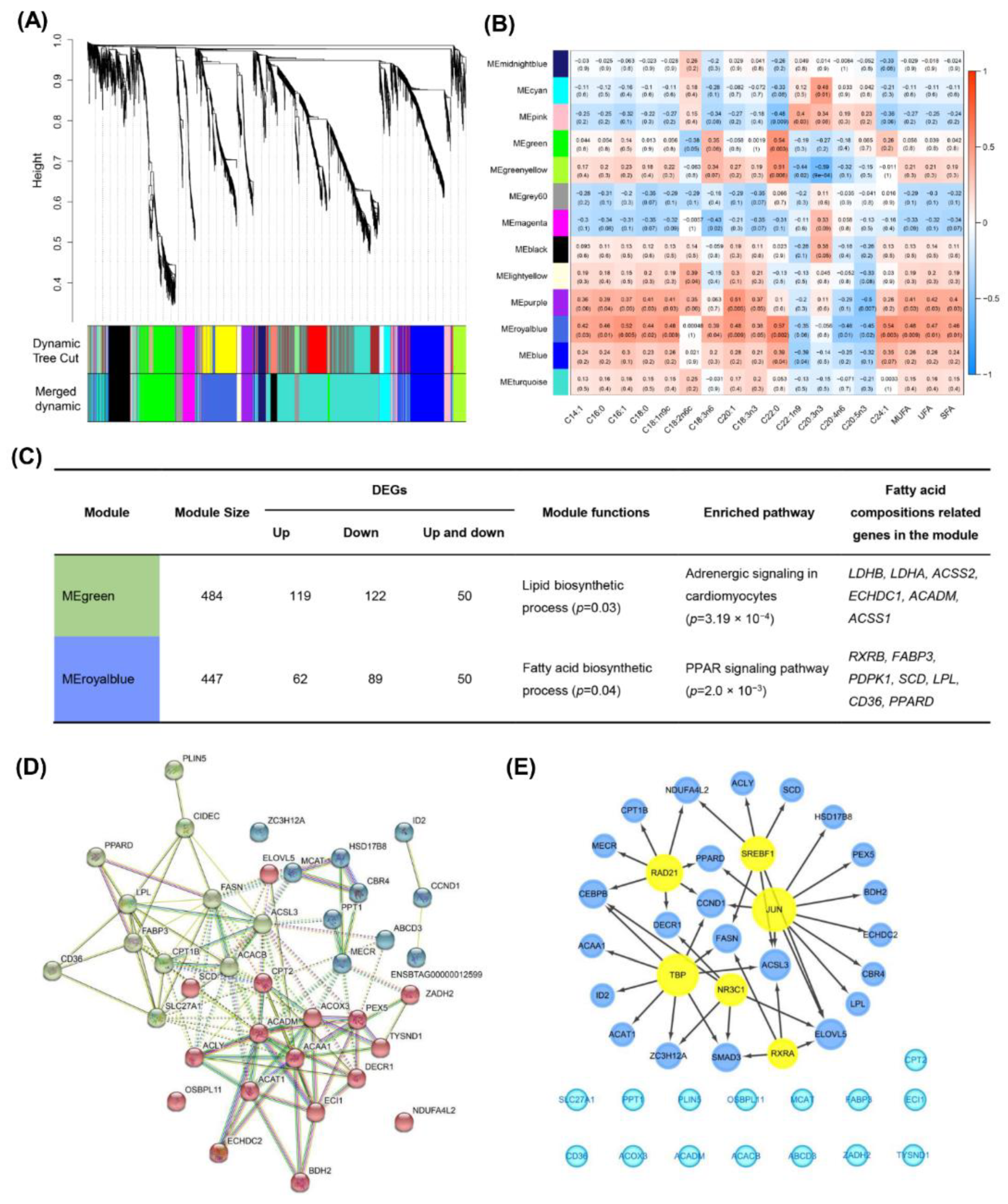
3.5. Gene Co-Expression Analysis Reveals Fatty Acid Specific Modules in Muscles
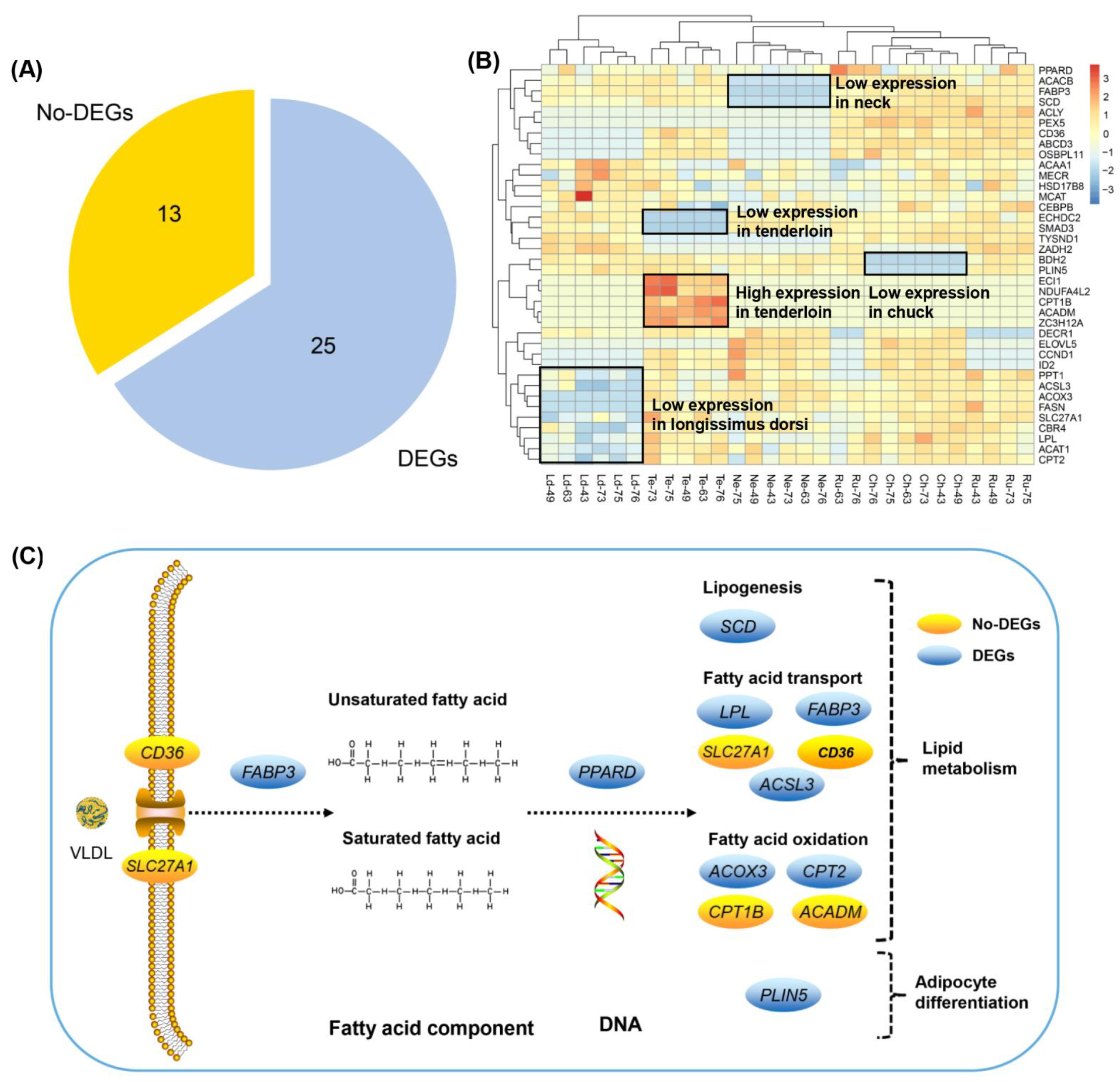
3.6. Differently Expressed Candidate Genes Related to Fatty Acid Composition across Different Beef Cuts
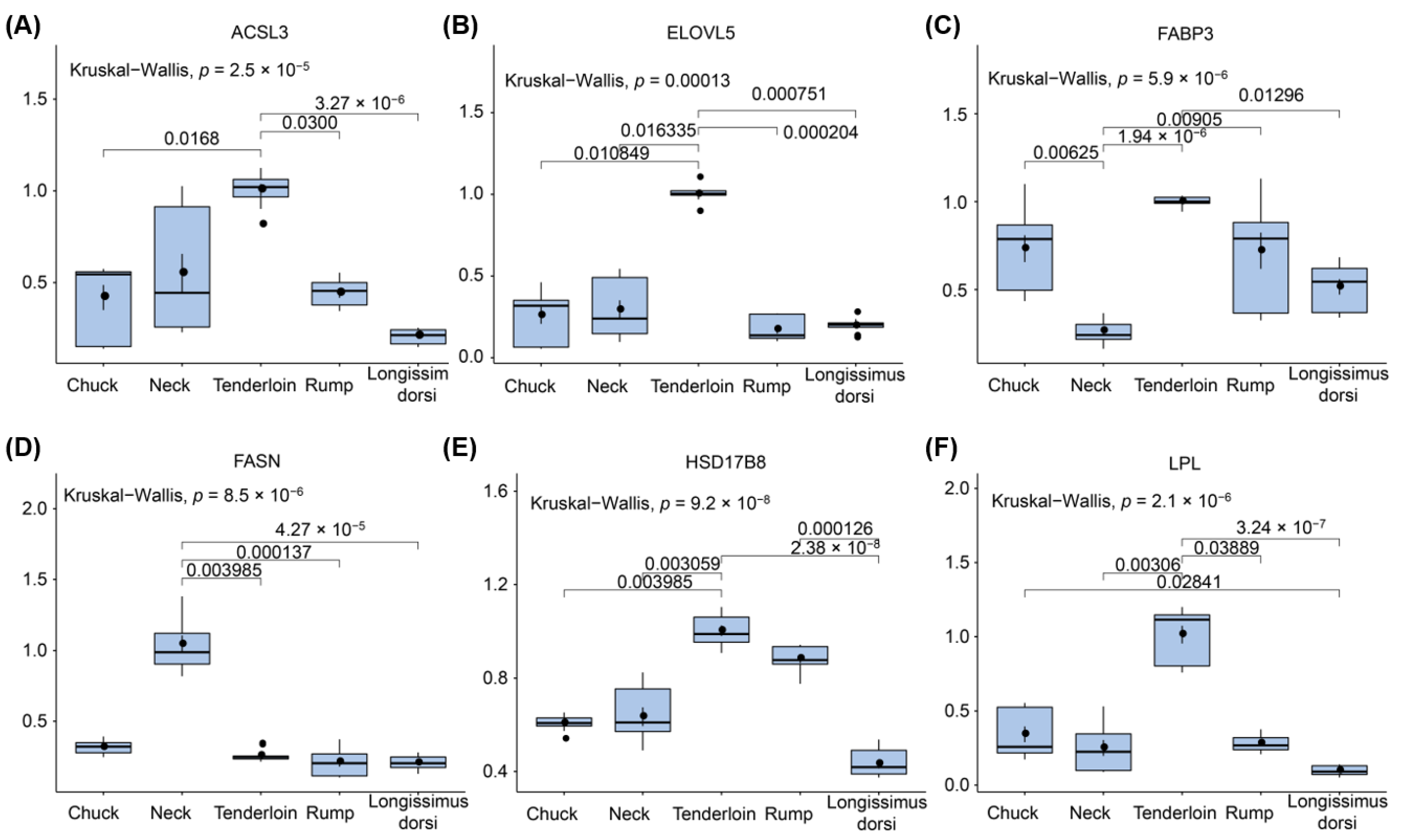
3.7. Validating Expression Profiles of Candidate Genes
4. Discussion
4.1. Detection of Fatty Acid Composition in Different Beef Cuts
4.2. Candidate Genes Involved in Fatty Acid Transport and Lipogenesis
4.3. Candidate Genes Involved in Fatty Acid Oxidation and Adipocyte Differentiation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Oh, M.; Kim, E.K.; Jeon, B.T.; Tang, Y.; Kim, M.S.; Seong, H.-J.; Moon, S.-H. Chemical compositions, free amino acid contents and antioxidant activities of Hanwoo (Bos taurus coreanae) beef by cut. Meat Sci. 2016, 119, 16–21. [Google Scholar] [CrossRef]
- Wood, J.D.; Enser, M.; Fisher, A.V.; Nute, G.R.; Sheard, P.R.; Richardson, R.I.; Hughes, S.I.; Whittington, F.M. Fat deposition, fatty acid composition and meat quality: A review. Meat Sci. 2008, 78, 343–358. [Google Scholar] [CrossRef]
- Vahmani, P.; Mapiye, C.; Prieto, N.; Rolland, D.C.; McAllister, T.A.; Aalhus, J.L.; Dugan, M.E.R. The scope for manipulating the polyunsaturated fatty acid content of beef: A review. J. Anim. Sci. Biotechnol. 2015, 1, 29. [Google Scholar] [CrossRef]
- Li, J.; Guasch-Ferré, M.; Li, Y.; Hu, F.B. Dietary intake and biomarkers of linoleic acid and mortality: Systematic review and meta-analysis of prospective cohort studies. Am. J. Clin. Nutr. 2020, 112, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Valentini, J.; Da Silva, A.S.; Fortuoso, B.F.; Reis, J.; Gebert, R.; Griss, L.; Boiago, M.; Lopes, L.; Christ-Santos, R.; Wagner, R.; et al. Chemical composition, lipid peroxidation, and fatty acid profile in meat of broilers fed with glycerol monolaurate additive. Food Chem. 2020, 330, 127187. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Niu, H.; Zhang, W.; Wang, Z.; Liang, Y.; Guan, L.; Guo, P.; Chen, Y.; Zhang, L.; Guo, Y.; et al. Genome wide association study and genomic prediction for fatty acid composition in Chinese Simmental beef cattle using high density SNP array. BMC Genom. 2017, 18, 464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, B.; Niu, H.; Zhang, W.; Xu, L.; Xu, L.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; et al. Genome wide association study identifies SNPs associated with fatty acid composition in Chinese Wagyu cattle. J. Anim. Sci. Biotechnol. 2019, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Silva-Vignato, B.; Coutinho, L.; Cesar, A.S.M.; Poleti, M.D.; Regitano, L.C.A.; Balieiro, J.C.C. Comparative muscle transcriptome associated with carcass traits of Nellore cattle. BMC Genom. 2017, 18, 506. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wu, J.-P.; Lei, Z.-M.; Zhang, M.; Gong, X.-Y.; Cheng, S.-R.; Liang, Y.; Wang, J.-F. Fatty Acid Profile of Muscles from Crossbred Angus-Simmental, Wagyu-Simmental, and Chinese Simmental Cattles. Food Sci. Anim. Resour. 2020, 40, 563–577. [Google Scholar] [CrossRef]
- Bermingham, E.N.; Reis, M.G.; Subbaraj, A.K.; Cameron-Smith, D.; Fraser, K.; Jonker, A.; Craigie, C.R. Distribution of fatty acids and phospholipids in different table cuts and co-products from New Zealand pasture-fed Wagyu-dairy cross beef cattle. Meat Sci. 2018, 140, 26–37. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 17, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 4, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 16, 2078–2079. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Janky, R.; Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Sanchez, M.N.; Potier, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- Mwangi, F.W.; Charmley, E.; Gardiner, C.P.; Malau-Aduli, B.S.; Kinobe, R.T.; Malau-Aduli, A.E.O. Diet and Genetics Influence Beef Cattle Performance and Meat Quality Characteristics. Foods 2019, 8, 648. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Tian, X.; Sun, C.; Shao, L.; Li, X.; Dai, R. Comparative transcriptomics to reveal muscle-specific molecular differences in the early postmortem of Chinese Jinjiang yellow cattle. Food Chem. 2019, 301, 125262. [Google Scholar] [CrossRef] [PubMed]
- Canto, A.C.; Costa-Lima, B.R.; Suman, S.; Monteiro, M.L.G.; Viana, F.M.; Salim, A.P.; Nair, M.N.; Silva, T.J.; Conte-Junior, C. Color attributes and oxidative stability of longissimus lumborum and psoas major muscles from Nellore bulls. Meat Sci. 2016, 121, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Hur, S.; Yang, H.; Moon, S.; Hwang, Y.; Park, G.; Joo, S. Discoloration characteristics of 3 major muscles from cattle during cold storage. J. Food Sci. 2009, 74, C1–C5. [Google Scholar] [CrossRef]
- Seyfert, M.; Mancini, R.A.; Hunt, M.C.; Tang, J.; Faustman, C.; Garcia, M. Color stability, reducing activity, and cytochrome c oxidase activity of five bovine muscles. J. Agric. Food Chem. 2006, 54, 8919–8925. [Google Scholar] [CrossRef]
- Gruffat, D.; Bauchart, D.; Thomas, A.; Parafita, E.; Durand, D. Fatty acid composition and oxidation in beef muscles as affected by ageing times and cooking methods. Food Chem. 2021, 343, 128476. [Google Scholar] [CrossRef]
- Fritzen, A.M.; Lundsgaard, A.M.; Kiens, B. Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat. Rev. Endocrinol. 2020, 16, 683–696. [Google Scholar] [CrossRef]
- Morales, P.E.; Bucarey, J.L.; Espinosa, A. Muscle Lipid Metabolism: Role of Lipid Droplets and Perilipins. J. Diabetes Res. 2017, 2017, 1789395. [Google Scholar] [CrossRef]
- Hao, J.-W.; Wang, J.; Guo, H.; Zhao, Y.-Y.; Sun, H.-H.; Li, Y.-F.; Lai, X.-Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef]
- Gimeno, R.E. Fatty acid transport proteins. Curr. Opin. Lipidol. 2007, 3, 271–276. [Google Scholar] [CrossRef]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef]
- Olivecrona, G. Role of lipoprotein lipase in lipid metabolism. Curr. Opin. Lipidol. 2016, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Eckel, R.H. Lipoprotein lipase: From gene to obesity. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E271–E288. [Google Scholar] [CrossRef] [PubMed]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tepaamorndech, S.; Kirschke, C.P.; Newman, J.W.; Keyes, W.R.; Pedersen, T.L.; Dumnil, J. Aberrant fatty acid metabolism in skeletal muscle contributes to insulin resistance in zinc transporter 7 (znt7)-knockout mice. J. Biol. Chem. 2018, 293, 7549–7563. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Liang, X.; Wu, X.; Liu, J.; Yang, S.; Tao, C.; Zhang, J.; Tian, J.; Zhao, J.; et al. Stearoyl-CoA Desaturase is Essential for Porcine Adipocyte Differentiation. Int. J. Mol. Sci. 2020, 21, 2446. [Google Scholar] [CrossRef]
- Newton, R.; Giembycz, M.A. Understanding how long-acting β(2)-adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids in asthma—An update. Br. J. Pharmacol. 2016, 173, 3405–3430. [Google Scholar] [CrossRef]
- Hang, S.; Li, X.; Wang, H.-Y.; Zheng, X.S. Beyond regulation of pol III: Role of MAF1 in growth, metabolism, aging and cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1861, 338–343. [Google Scholar]
- Niu, H.; Fujiwara, H.; di Martino, O.; Hadwiger, G.; Frederick, T.E.; Menéndez-Gutiérrez, M.P.; Ricote, M.; Bowman, G.R.; Welch, J.S. Endogenous retinoid X receptor ligands in mouse hematopoietic cells. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid. Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Zha, S.; Ferdinandusse, S.; Hicks, J.L.; Denis, S.; Dunn, T.A.; Wanders, R.J.; Luo, J.; de Marzo, A.M.; Isaacs, W.B. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate 2005, 63, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hurtado, E.; Lee, J.; Choi, J.; Wolfgang, M.J. Fatty acid oxidation is required for active and quiescent brown adipose tissue maintenance and thermogenic programing. Mol. Metab. 2018, 7, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J. Fatty Acid Oxidation in Cell Fate Determination. Trends Biochem. Sci. 2018, 43, 854–857. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Cheung, C.H.Y.; Liu, Y.-L.; Hou, C.-L.; Hsu, C.-L.; Huang, C.-T.; Yang, T.-S.; Chen, S.-F.; Chen, C.-N.; Hsu, W.-M.; et al. Quantitative Proteomics of Th-MYCN Transgenic Mice Reveals Aurora Kinase Inhibitor Altered Metabolic Pathways and Enhanced ACADM To Suppress Neuroblastoma Progression. J. Proteome Res. 2019, 18, 3850–3866. [Google Scholar] [CrossRef]
- Harris, L.-A.L.; Skinner, J.R.; Shew, T.M.; Pietka, T.A.; Abumrad, N.A.; Wolins, N.E. Perilipin 5–Driven Lipid Droplet Accumulation in Skeletal Muscle Stimulates the Expression of Fibroblast Growth Factor 21. Diabetes 2015, 64, 2757–2768. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Niu, Q.; Wang, T.; Zheng, X.; Li, H.; Gao, X.; Chen, Y.; Gao, H.; Zhang, L.; Liu, G.E.; et al. Comparative Transcriptomic Analysis Reveals Diverse Expression Pattern Underlying Fatty Acid Composition among Different Beef Cuts. Foods 2022, 11, 117. https://doi.org/10.3390/foods11010117
Zhang T, Niu Q, Wang T, Zheng X, Li H, Gao X, Chen Y, Gao H, Zhang L, Liu GE, et al. Comparative Transcriptomic Analysis Reveals Diverse Expression Pattern Underlying Fatty Acid Composition among Different Beef Cuts. Foods. 2022; 11(1):117. https://doi.org/10.3390/foods11010117
Chicago/Turabian StyleZhang, Tianliu, Qunhao Niu, Tianzhen Wang, Xu Zheng, Haipeng Li, Xue Gao, Yan Chen, Huijiang Gao, Lupei Zhang, George E. Liu, and et al. 2022. "Comparative Transcriptomic Analysis Reveals Diverse Expression Pattern Underlying Fatty Acid Composition among Different Beef Cuts" Foods 11, no. 1: 117. https://doi.org/10.3390/foods11010117