
Novel Thermostable Heparinase Based on the Genome of Bacteroides Isolated from Human Gut Microbiota

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmid, Culture Conditions, and Chemicals
2.2. Analysis of the Heparin-Degrading Enzyme Strains
2.3. Genome Sequencing, Data Assembly, and CAZymes Annotation
2.4. Sequence Alignment and Evolutionary Tree Construction
2.5. Utilization of Heparin by Bacteroides and Growth Curves
2.6. Determination of Heparin Consumption
2.7. Cloning, Expression, and Purification of Heparinase
2.8. Effects of Temperature and pH on Enzyme Activity
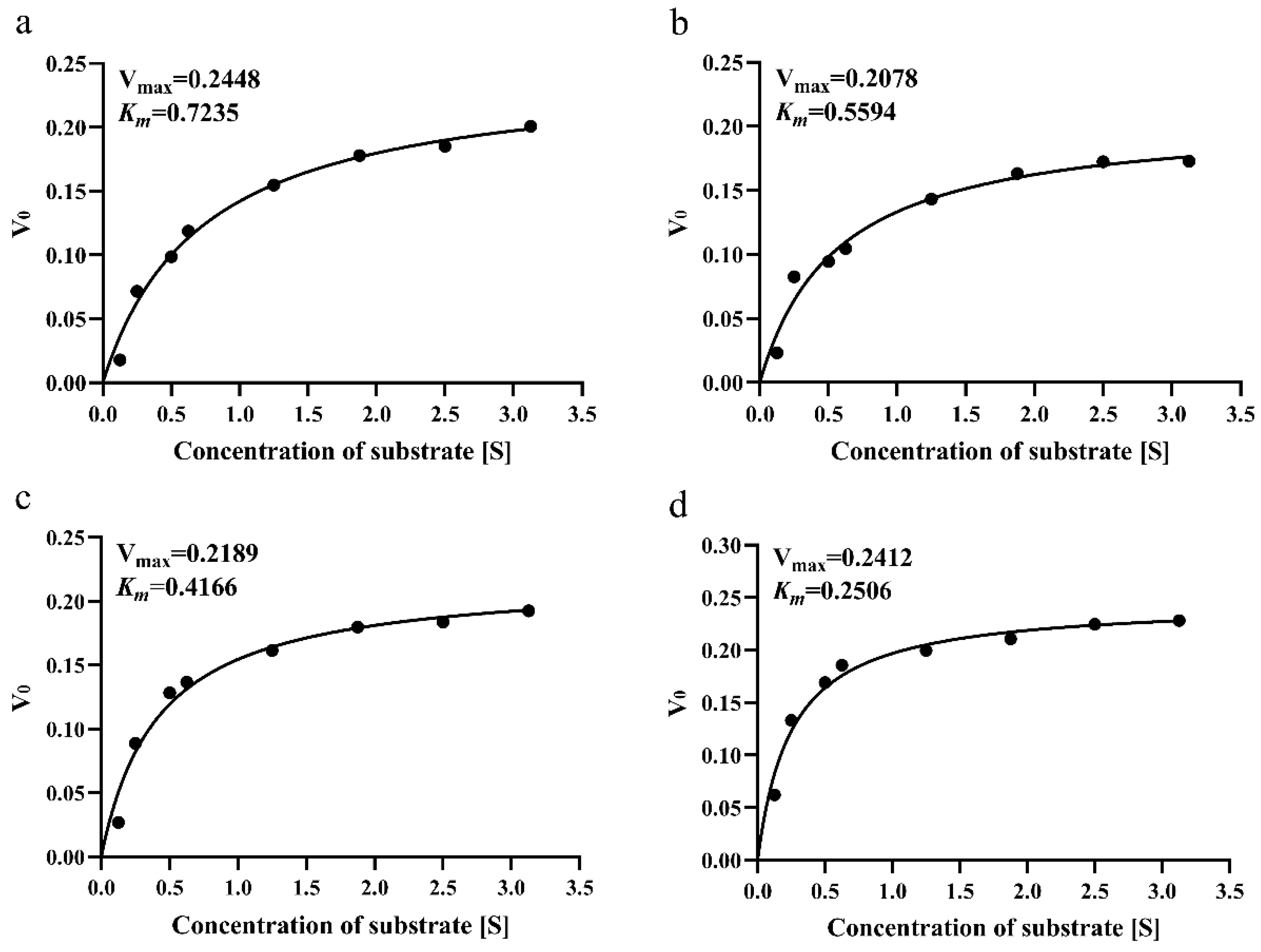
2.9. Enzyme Activity Determination and Kinetic Parameters
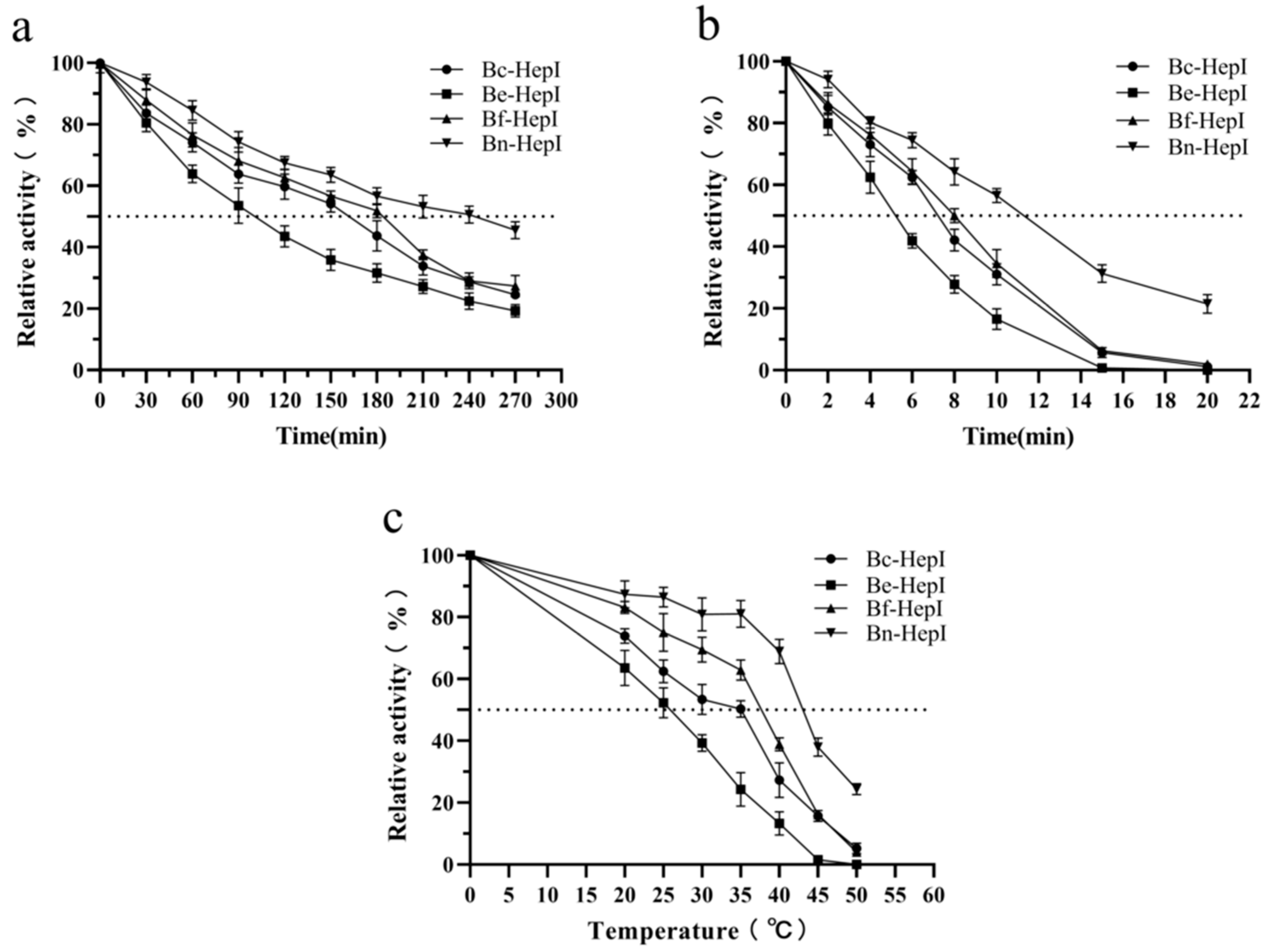
2.10. Thermostability of HepI
2.10.1. Determination of Half-Life
2.10.2. Determination of
2.10.3. Determination of Melting Temperature (Tm)
2.10.4. Determination of the Secondary Structure Using Circular Dichroism Spectroscopy
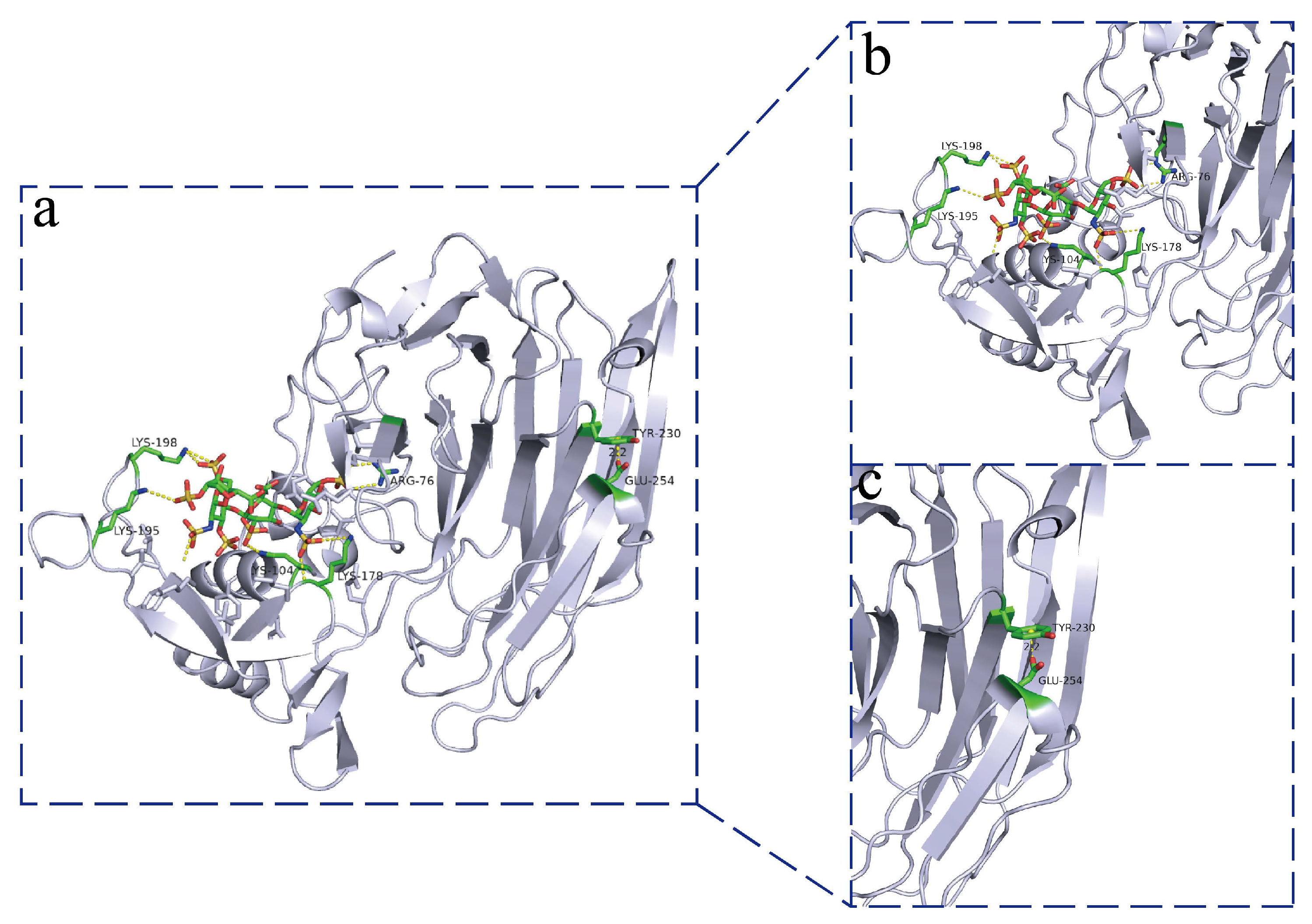
2.11. Homology Modeling and Molecular Docking
2.12. Statistical Analysis
3. Results
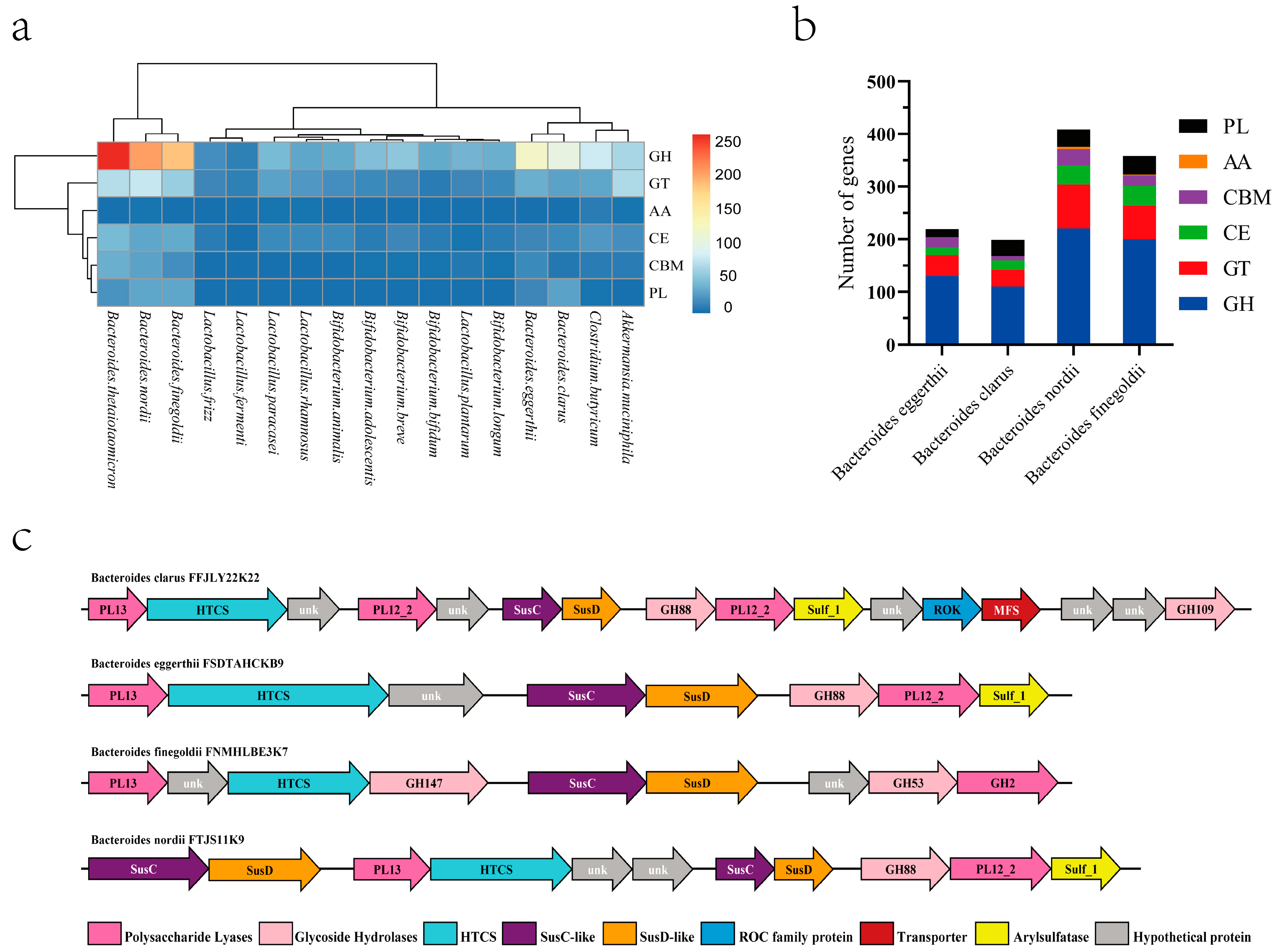
3.1. Analysis of Genomes, CAZymes, and PULs
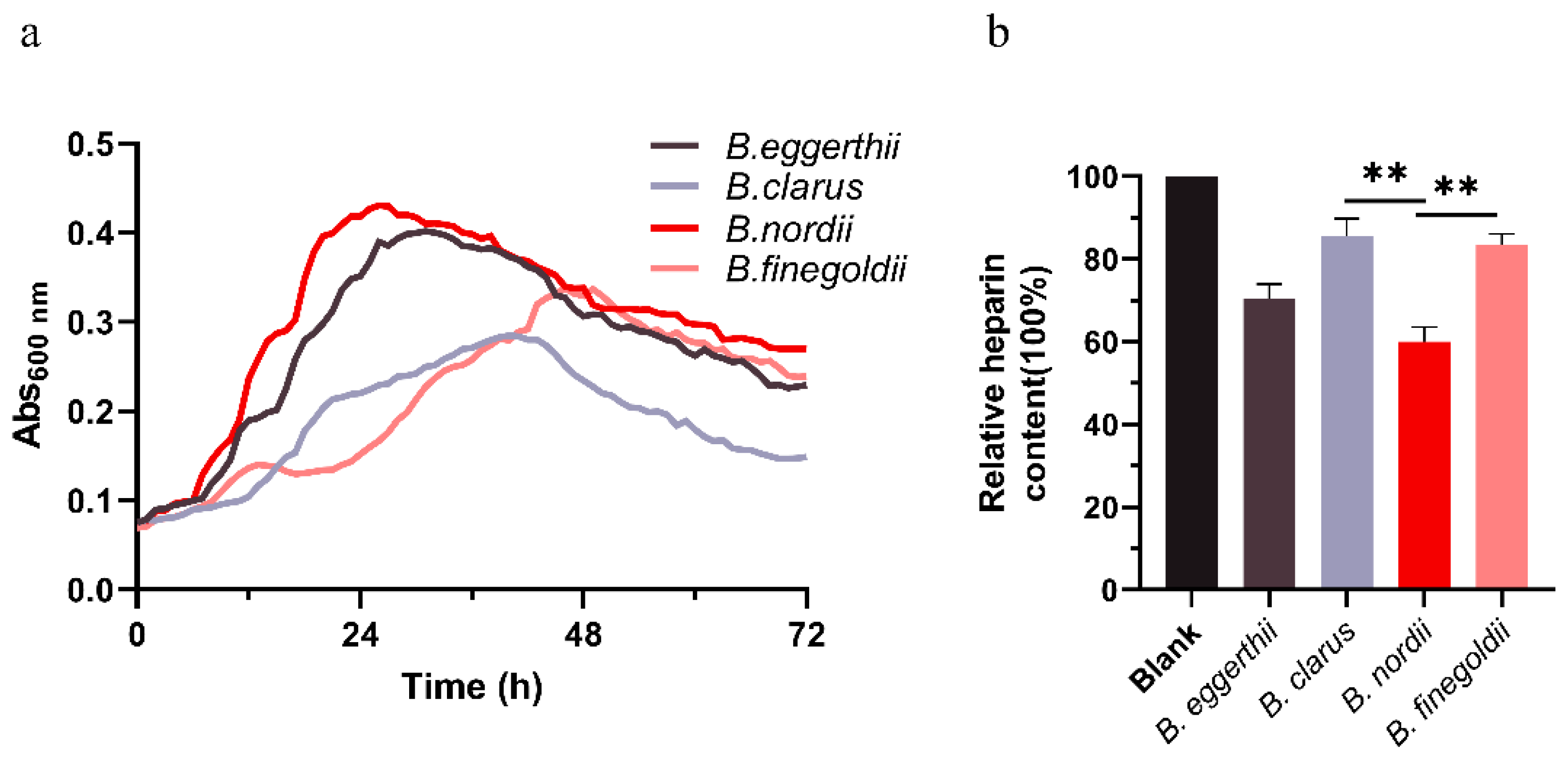
3.2. In Vitro Fermentation of Heparin Polysaccharide
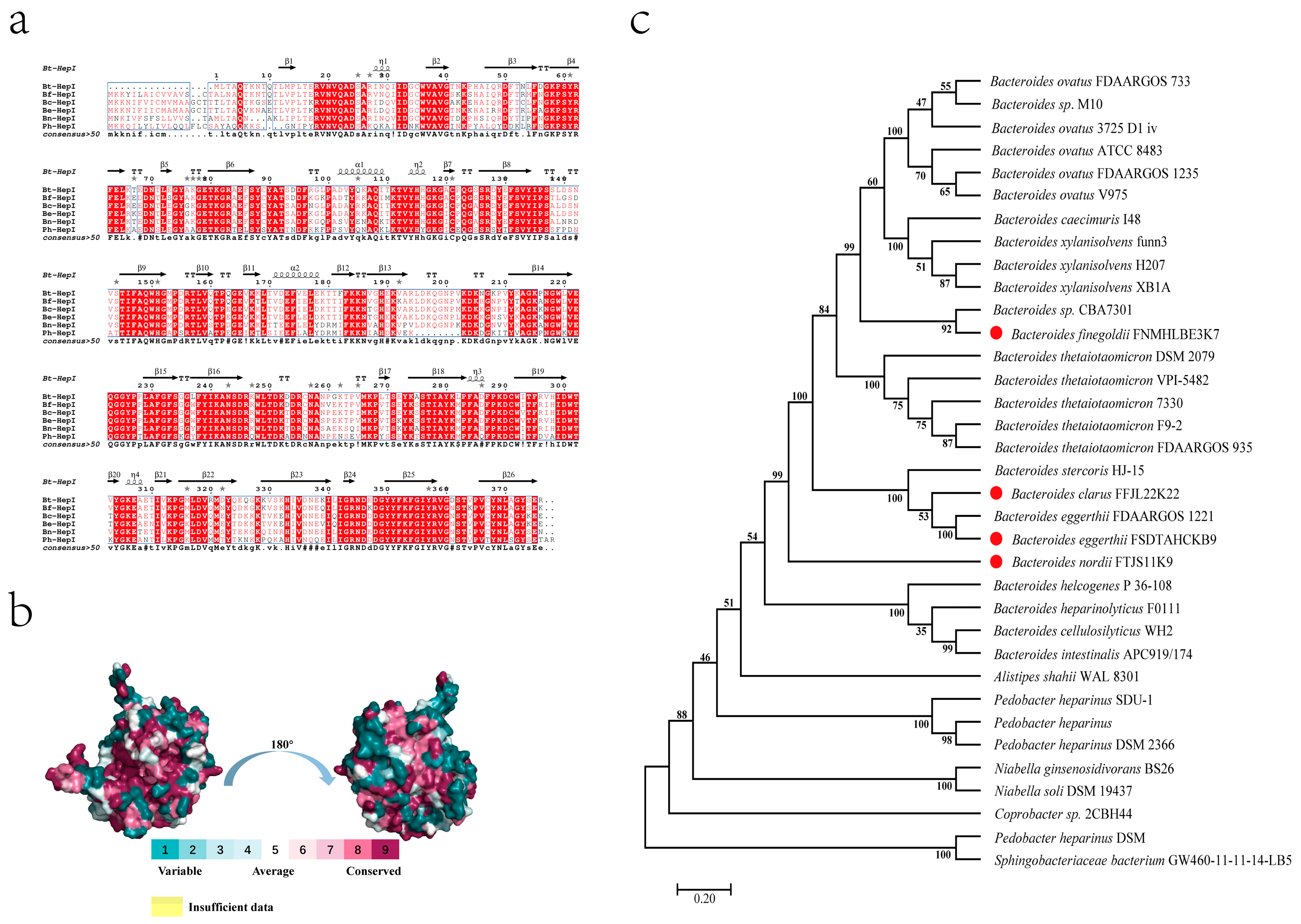
3.3. Multiple Sequence Alignment and Evolutionary Tree
3.4. Fusion Expression and Purification of Heparinase
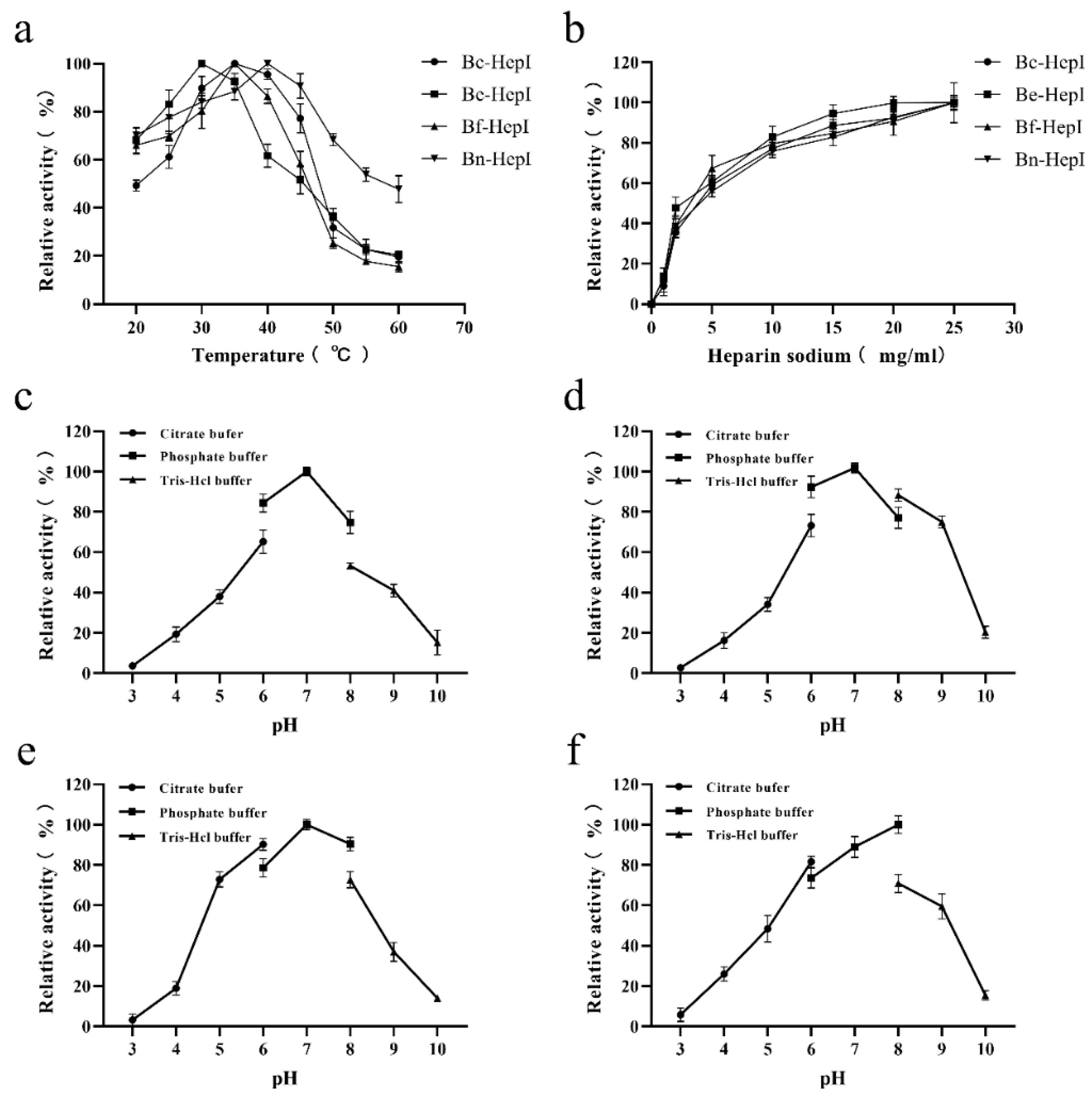
3.5. Enzymatic Characterization
3.6. Thermal Stability and Enzyme Kinetics
3.7. Structural Analysis of Heparinase
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lewis, C.M.; Obregón-Tito, A.; Tito, R.Y.; Foster, M.W.; Spicer, P.G. The Human Microbiome Project: Lessons from Human Genomics. Trends Microbiol. 2012, 20, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohajeri, M.H.; Brummer, R.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.; Faas, M.; Eggersdorfer, M. The Role of the Microbiome for Human Health: From Basic Science to Clinical Applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndeh, D.; Baslé, A.; Strahl, H.; Yates, E.A.; Cartmell, A. Metabolism of Multiple Glycosaminoglycans by Bacteroides Thetaiotaomicron Is Orchestrated by a Versatile Core Genetic Locus. Nat. Commun. 2020, 11, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; He, D.; Xing, Y.; Liu, J.; Xing, X. Internal Connections between Dietary Intake and Gut Microbiota Homeostasis in Disease Progression of Ulcerative Colitis: A Review. Food Sci. Hum. Wellness 2021, 10, 119–130. [Google Scholar] [CrossRef]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How Glycan Metabolism Shapes the Human Gut Microbiota. Nat. Rev. Microbiol. 2012, 10, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Tramontano, M.; Andrejev, S.; Pruteanu, M.; Klünemann, M.; Kuhn, M.; Galardini, M.; Jouhten, P.; Zelezniak, A.; Zeller, G.; Bork, P.; et al. Nutritional Preferences of Human Gut Bacteria Reveal Their Metabolic Idiosyncrasies. Nat. Microbiol. 2018, 3, 514–522. [Google Scholar] [CrossRef]
- Cockburn, D.W.; Koropatkin, N.M. Polysaccharide Degradation by the Intestinal Microbiota and Its Influence on Human Health and Disease. J. Mol. Biol. 2016, 428, 3230–3252. [Google Scholar] [CrossRef]
- Harris, H.C.; Morrison, D.J.; Edwards, C.A. Impact of the Source of Fermentable Carbohydrate on SCFA Production by Human Gut Microbiota in Vitro—A Systematic Scoping Review and Secondary Analysis. Crit. Rev. Food Sci. Nutr. 2021, 61, 3892–3903. [Google Scholar] [CrossRef]
- Luis, A.; Martens, E. Interrogating Gut Bacterial Genomes for Discovery of Novel Carbohydrate Degrading Enzymes. Curr. Opin. Chem. Biol. 2018, 47, 126–133. [Google Scholar] [CrossRef]
- CAZypedia Consortium. Ten Years of CAZypedia: A Living Encyclopedia of Carbohydrate-Active Enzymes. Glycobiology 2018, 28, 3–8. [Google Scholar] [CrossRef]
- Cartmell, A.; Lowe, E.C.; Baslé, A.; Firbank, S.J.; Bolam, D.N. How Members of the Human Gut Microbiota Overcome the Sulfation Problem Posed by Glycosaminoglycans. Proc. Natl. Acad. Sci. USA 2017, 114, 7037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pudlo, N.A.; Urs, K.; Kumar, S.S.; German, J.B.; Mills, D.A.; Martens, E.C. Symbiotic Human Gut Bacteria with Variable Metabolic Priorities for Host Mucosal Glycans. mBio 2015, 6, e01282-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndeh, D.; Rogowski, A.; Cartmell, A.; Luis, A.S.; Baslé, A.; Gray, J.; Venditto, I.; Briggs, J.; Zhang, X.; Labourel, A.; et al. Corrigendum: Complex Pectin Metabolism by Gut Bacteria Reveals Novel Catalytic Functions. Nature 2017, 548, 612. [Google Scholar] [CrossRef] [PubMed]
- Rogowski, A.; Briggs, J.A.; Mortimer, J.C.; Tryfona, T.; Terrapon, N.; Lowe, E.C.; Baslé, A.; Morland, C.; Day, A.M.; Zheng, H. Glycan Complexity Dictates Microbial Resource Allocation in the Large Intestine. Nat. Commun. 2016, 7, 10705. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin-Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Laremore, T.N.; Zhang, F.; Dordick, J.S.; Jian, L.; Linhardt, R.J. Recent Progress and Applications in Glycosaminoglycan and Heparin Research. Curr. Opin. Chem. Biol. 2009, 13, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Linhardt, R.J. Hudson Award Address in Carbohydrate Chemistry. Heparin: Structure and Activity. J. Med. Chem. 2003, 46, 2551–2564. [Google Scholar] [CrossRef]
- Lee, J.; Wee, S.; Gunaratne, J.; Chua, R.; Smith, R.; Ling, L.; Fernig, D.G.; Swaminathan, K.; Nurcombe, V.; Cool, S.M. Structural Determinants of Heparin–Transforming Growth Factor-Β1 Interactions and Their Effects on Signaling. Glycobiology 2015, 25, 1491–1504. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Linask, K.L.; Mallon, B.; Johnson, K.; Klein, M.; Beers, J.; Xie, W.; Du, Y.; Liu, C.; Lai, Y. Heparin Promotes Cardiac Differentiation of Human Pluripotent Stem Cells in Chemically Defined Albumin-Free Medium, Enabling Consistent Manufacture of Cardiomyocytes. Stem Cells Transl. Med. 2016, 6, 527–538. [Google Scholar] [CrossRef]
- Sepuru, K.M.; Nagarajan, B.; Desai, U.R.; Rajarathnam, K. Structural Basis, Stoichiometry, and Thermodynamics of Binding of the Chemokines KC and MIP2 to the Glycosaminoglycan Heparin. J. Biol. Chem. 2018, 293, 17817–17828. [Google Scholar] [CrossRef] [Green Version]
- Linhardt, R.J.; Wang, H.M.; Ampofo, S.A. New Methodologies in Heparin Structure Analysis and the Generation of LMW Heparins. Adv. Exp. Med. Biol. 1992, 313, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Galliher, P.M.; Cooney, C.L. Polysaccharide Lyases. Appl. Biochem. Biotechnol. 1987, 12, 135–176. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Garron, M.L.; Kim, H.Y.; Kim, W.S.; Zhang, Z.; Ryu, K.S.; Shaya, D.; Xiao, Z.; Cheong, C.; Kim, Y.S. Structural Snapshots of Heparin Depolymerization by Heparin Lyase I. J. Biol. Chem. 2009, 284, 34019–34027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.W.; Zhu, H.T.; Liu, C.Y.; Lv, Z.X.; Zhang, Y.W. A Highly Active Heparinase I from Bacteroides Cellulosilyticus: Cloning, High Level Expression, and Molecular Characterization. PLoS ONE 2020, 15, e0240920. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.S.; Sirk, S.J.; Diaz, C.; Klein, A.P.; Sattely, E.S. A Metabolic Pathway for Activation of Dietary Glucosinolates by a Human Gut Symbiont. Cell 2020, 180, 717–728.e19. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Wang, J. SOAPdenovo2: An Empirically Improved Memory-Efficient Short-Read de Novo Assembler. GigaScience 2015, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.; Xu, Y.; Yin, Y. DbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. DbCAN: A Web Resource for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Vincent, L.; Hemalatha, G.R.; Elodie, D.; Coutinho, P.M.; Bernard, H. The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar] [CrossRef]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple Sequence Alignment with the Clustal Series of Programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouet, P.; Courcelle, E.; Stuart, D.I.; Métoz, F. ESPript: Analysis of Multiple Sequence Alignments in PostScript. Bioinformatics 1999, 15, 305–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, Y.; Ai, C.; Wen, C.; Dong, X.; Sun, X.; Cao, C.; Zhang, X.; Zhu, B.; Song, S. Gut Microbiota Response to Sulfated Sea Cucumber Polysaccharides in a Differential Manner Using an in Vitro Fermentation Model. Food Res. Int. 2021, 148, 110562. [Google Scholar] [CrossRef] [PubMed]
- Moens, F.; Rivière, A.; Selak, M.; Vuyst, L.D. Inulin-Type Fructan Degradation Capacity of Interesting Butyrate-Producing Colon Bacteria and Cross-Feeding Interactions of Faecalibacterium prausnitzii DSM 17677T with Bifidobacteria. Arch. Public Health 2014, 72, O6. [Google Scholar] [CrossRef] [Green Version]
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.T.; Smith, F. Colorimetric Method for Determination of Sugars and Related Substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Pan, L.; Sun, W.; Shang, Q.; Niu, Q.; Liu, C.; Li, G.; Yu, G. In Vitro Fermentation and Isolation of Heparin-Degrading Bacteria from Human Gut Microbiota. Anaerobe 2021, 68, 102289. [Google Scholar] [CrossRef]
- Yang, B.C.; Zhang, C.; Wang, C.; Zhou, H.; Li, Z.Y.; Song, Y.J.; Zhang, T.C.; Luo, X.G. Soluble Expression and Purification of Heparinase I in Escherichia coli Using a Hexahistidine-Tagged Small Ubiquitin-like Modifier as a Fusion Partner. Biotechnol. Biotechnol. Equip. 2017, 31, 1040–1045. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Bernstein, H.; Yang, V.C.; Cooney, C.L.; Langer, R. Immobilized Heparin Lyase System for Blood Deheparinization. Methods Enzym. 1988, 137, 515–529. [Google Scholar] [CrossRef]
- Bramucci, E.; Paiardini, A.; Bossa, F.; Pascarella, S. PyMod: Sequence Similarity Searches, Multiple Sequence-Structure Alignments, and Homology Modeling within PyMOL. BMC Bioinform. 2012, 13, S2. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Quince, C.; Faith, J.J.; Mchardy, A.C.; Yatsunenko, T.; Niazi, F.; Affourtit, J.; Egholm, M.; Henrissat, B.; Knight, R. Organismal, Genetic, and Transcriptional Variation in the Deeply Sequenced Gut Microbiomes of Identical Twins. Proc. Natl. Acad. Sci. USA 2010, 107, 7503–7508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.; Midtvedt, T.; Gordon, J. How Host-Microbial Interactions Shape the Nutrient Environment of the Mammalian Intestine. Annu. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef] [Green Version]
- Martens, E.; Chiang, H.; Gordon, J. Mucosal Glycan Foraging Enhances Fitness and Transmission of a Saccharolytic Human Gut Bacterial Symbiont. Cell Host Microbe 2008, 4, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Charles, A.; Todd, A. Observations on the Structure of the Barium Salt of Heparin. Biochem. J. 1940, 34, 112–118. [Google Scholar] [CrossRef]
- Sasisekharan, R.; Venkataraman, G. Heparin and Heparan Sulfate: Biosynthesis, Structure and Function. Curr. Opin. Chem. Biol. 2000, 4, 626–631. [Google Scholar] [CrossRef]
- Ahn, M.Y.; Shin, K.H.; Kim, D.H.; Jung, E.A.; Toida, T.; Linhardt, R.J.; Kim, Y.S. Characterization of a Bacteroides Species from Human Intestine That Degrades Glycosaminoglycans. Can. J. Microbiol. 1998, 44, 423–429. [Google Scholar] [CrossRef]
- Pan, L.; Ai, X.; Fu, T.; Ren, L.; Shang, Q.; Li, G. In Vitro Fermentation of Hyaluronan by Human Gut Microbiota: Changes in Microbiota Community and Potential Degradation Mechanism. Carbohydr. Polym. 2021, 269, 118313. [Google Scholar] [CrossRef]
- Kaoutari, A.E.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The Abundance and Variety of Carbohydrate-Active Enzymes in the Human Gut Microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Kim, B.-T.; Park, S.-Y.; Kim, N.-Y.; Han, M.J.; Shin, K.-H.; Kim, W.-S.; Kim, Y.-S. Degradation of Acharan Sulfate and Heparin by Bacteroides stercoris HJ-15, a Human Intestinal Bacterium. Arch. Pharm. Res. 1998, 21, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Chen, X.; Wang, F.; Zhang, T.; Ling, P. Oral Administration of Heparin or Heparosan Increases the Lactobacillus Population in Gut Microbiota of Rats. Carbohydr. Polym. 2013, 94, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ye, F.; Yang, C.; Chen, Y.; Zhao, H.; Yatsunami, R.; Nakamura, S.; Arisaka, F.; Xing, X.H. Biochemical Analysis and Kinetic Modeling of the Thermal Inactivation of MBP-Fused Heparinase I: Implications for a Comprehensive Thermostabilization Strategy. Biotechnol. Bioeng. 2011, 108, 1841–1851. [Google Scholar] [CrossRef]
- Saha, A.; Saha, B.K. Metal−π Interactions in Light of Geometry-Corrected Statistical Analysis. Cryst. Growth Des. 2019, 19, 7264–7270. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Sherrill, C.D. High-Accuracy Quantum Mechanical Studies of π-π Interactions in Benzene Dimers. J. Phys. Chem. A 2006, 110, 10656–10668. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. no. | Enzyme | Primer Sequences a | Tm (°C) |
|---|---|---|---|
| 1 | Bc-HepI | F: 5′-GAACAGATTGGAGGTGGATCCACCGCGCAGACCAAAGGC-3′ R: 5′-GTGGTGGTGGTGGTGCTCGAGTTATTCTTCTTTATAGCCCGCCAGG-3′ | 58–63 |
| 2 | Be-HepI | F: 5′-GAACAGATTGGAGGTGGATCCACCGCGCAAGTGAAAAACGC-3′ R: 5′-GTGGTGGTGGTGGTGCTCGAGTTATTCTTCTTTATAGCCCGCCAGG-3′ | 58–62 |
| 3 | Bf-HepI | F: 5′-GAACAGATTGGAGGTGGATCCAACGCGCAGACCAAAAACACG-3′ R: 5′-GTGGTGGTGGTGGTGCTCGAGTTATTTTTCGCTATAGCCCGCCAGG-3′ | 59–62 |
| 4 | Bn-HepI | F: 5′-GAACAGATTGGAGGTGGATCCCAGAACGCGAAACTGATTCCGC-3′ R: 5′-GTGGTGGTGGTGGTGCTCGAGTTAGTTTTCGCTATAGCCCGCCAGG-3′ | 60–62 |
| Enzyme | TP * (mg/mL) | TEA * (IU/mL) | SA * (IU/mg) |
|---|---|---|---|
| Bc-HepI | 0.667 ± 0.02 | 181.58 ± 3.96 | 272.23 |
| Be-HepI | 0.550 ± 0.04 | 178.84 ± 4.21 | 325.16 |
| Bf-HepI | 0.705 ± 0.01 | 214.21 ± 5.44 | 303.84 |
| Bn-HepI | 0.667 ± 0.03 | 240.42 ± 10.61 | 360.45 |
| Bt-HepI | 0.613 ± 0.02 | 192.44 ± 1.35 | 313.93 |
| Enzyme | Number of Amino Acids (aa) | Molecular Weight (KDa) | Theoretical pI |
|---|---|---|---|
| Bc-HepI | 374 | 42.4 | 8.57 |
| Be-HepI | 374 | 42.4 | 9.23 |
| Bf-HepI | 374 | 42.3 | 9.09 |
| Bn-HepI | 369 | 41.8 | 8.48 |
| SUMO-Tag | 108 | 12.4 | 5.71 |
| Enzyme | Km(mM) | Vmax(mM/min) | kcat (/s) | kcat/Km (/s/mM) | t1/2(40 °C) (min) | t1/2(50 °C) (min) | Tm(°C) | |
|---|---|---|---|---|---|---|---|---|
| Bc-HepI | 0.72 ± 0.1 | 0.24 ± 0.04 | 201.1 ± 4.9 | 279.3 ± 3.8 | 173 ± 5.4 | 4.9 ± 0.5 | 39.42 | 34.1 ± 1.1 |
| Be-HepI | 0.56 ± 0.09 | 0.21 ± 0.06 | 206.9 ± 6.3 | 369.5 ± 6.3 | 101 ± 3.8 | 6.5 ± 0.4 | 38.97 | 25.8 ± 1.5 |
| Bf-HepI | 0.42 ± 0.1 | 0.22 ± 0.06 | 169.8 ± 5.1 | 404.3 ± 8.5 | 184 ± 4.7 | 7.9 ± 0.3 | 41.79 | 37.8 ± 1.8 |
| Bn-HepI | 0.25 ± 0.07 | 0.24 ± 0.05 | 199.9 ± 4.6 | 799.6 ± 0.1 | 244 ± 6.3 | 10.6 ± 0.4 | 44.79 | 43.8 ± 1.7 |
| Bt-HepI | 0.26 ± 0.02 | 0.20 ± 0.05 | 179.9 ± 4.6 | 692.0 ± 0.1 | 148 ± 1.2 | 6.6 ± 0.3 | 40.36 | 39.5 ± 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Yu, L.; Zhai, Q.; Zhao, R.; Wang, C.; Zhao, J.; Zhang, H.; Chen, W.; Tian, F. Novel Thermostable Heparinase Based on the Genome of Bacteroides Isolated from Human Gut Microbiota. Foods 2022, 11, 1462. https://doi.org/10.3390/foods11101462
Zhang C, Yu L, Zhai Q, Zhao R, Wang C, Zhao J, Zhang H, Chen W, Tian F. Novel Thermostable Heparinase Based on the Genome of Bacteroides Isolated from Human Gut Microbiota. Foods. 2022; 11(10):1462. https://doi.org/10.3390/foods11101462
Chicago/Turabian StyleZhang, Chuan, Leilei Yu, Qixiao Zhai, Ruohan Zhao, Chen Wang, Jianxin Zhao, Hao Zhang, Wei Chen, and Fengwei Tian. 2022. "Novel Thermostable Heparinase Based on the Genome of Bacteroides Isolated from Human Gut Microbiota" Foods 11, no. 10: 1462. https://doi.org/10.3390/foods11101462