
Insight into the Structure–Odor Relationship of Molecules: A Computational Study Based on Deep Learning

Abstract
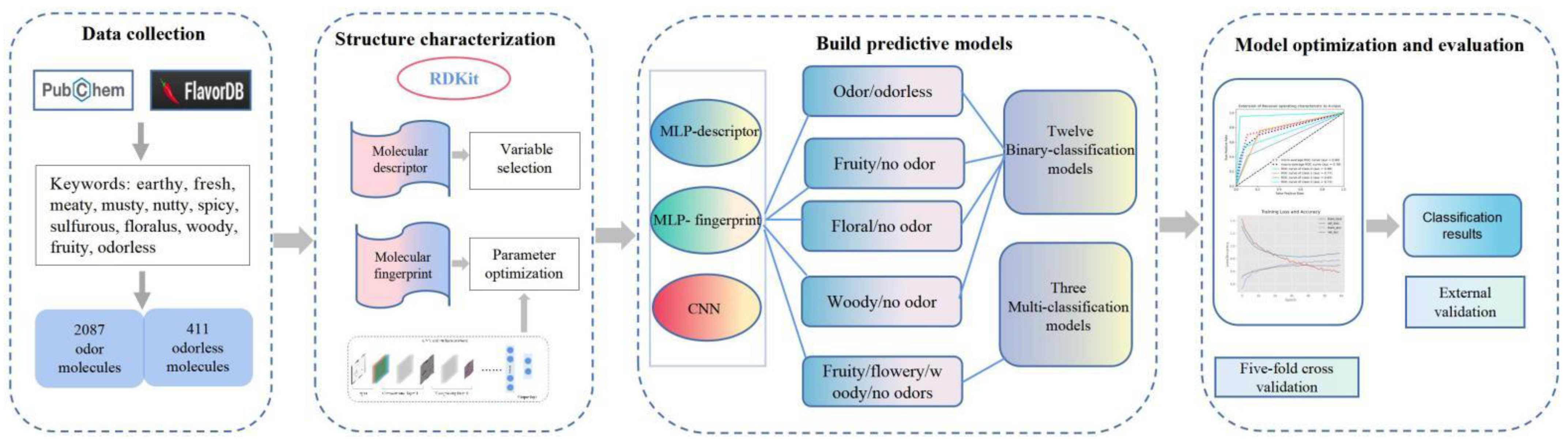
:1. Introduction
2. Methods
2.1. Dataset Collection and Preparation
2.2. Molecular Structure Characterization
2.3. Feature Selection
2.4. Multilayer Perceptron
2.5. Two-Dimensional Convolutional Neural Network
2.6. Model Evaluation
3. Results
3.1. The MLP-Des Model Exhibited the Highest Predictive Capability
3.2. The MLP-Des Model Showed Better or Equivalent Predictions Compared with Exiting Models
3.3. The Structure–Odor Relationship Derived from the MLP-Des Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Croy, I.; Nordin, S.; Hummel, T. Olfactory Disorders and Quality of Life—An Updated Review. Chem. Sens. 2014, 39, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.L.; Luo, D.H.; Wong, K.Y.; Hung, K. POP-CNN: Predicting Odor Pleasantness with Convolutional Neural Network. IEEE Sens. J. 2019, 19, 11337–11345. [Google Scholar] [CrossRef]
- Rossiter, K.J. Structure–odor relationships. Chem. Rev. 1996, 96, 3201–3240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mao, H.; Liu, L.; Du, J.; Gan, R. A machine learning based computer-aided molecular design/screening methodology for fragrance molecules. Comput. Chem. Eng. 2018, 115, 295–308. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Lin, D.; Zhu, Y.; Chen, C.; He, L.; Guo, X.; Chen, K.; Wang, R.; Liu, Z. Artificial intelligence deciphers codes for color and odor perceptions based on large-scale chemoinformatic data. GigaScience 2020, 9, giaa011. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Kumar, R.; Semwal, R.; Aier, I.; Varadwaj, P. DeepOlf: Deep neural network based architecture for predicting odorants and their interacting Olfactory Receptors. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 418–428. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, R.; Ranjta, S.; Varadwaj, P.K. SMILES to Smell: Decoding the Structure–Odor Relationship of Chemical Compounds Using the Deep Neural Network Approach. J. Chem. Inf. Model. 2021, 61, 676–688. [Google Scholar] [CrossRef]
- Tran, N.; Kepple, D.; Shuvaev, S.A.; Koulakov, A.A. DeepNose: Using artificial neural networks to represent the space of odorants. boiRxiv 2018. [Google Scholar] [CrossRef]
- Nozaki, Y.; Nakamoto, T. Predictive modeling for odor character of a chemical using machine learning combined with natural language processing. PLoS ONE 2018, 13, e0198475. [Google Scholar] [CrossRef]
- Sanchez-Lengeling, B.; Wei, J.N.; Lee, B.K.; Gerkin, R.C.; Aspuru-Guzik, A.; Wiltschko, A.B. Machine Learning for Scent: Learning Generalizable Perceptual Representations of Small Molecules. arXiv 2019, arXiv:1910.10685v2. [Google Scholar]
- Kowalewski, J.; Ray, A. Predicting Human Olfactory Perception from Activities of Odorant Receptors. iScience 2020, 23, 101361. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Sethupathy, A.; Tuwani, R.; Nk, R.; Dokania, S.; Iyer, A.; Gupta, A.; Agrawal, S.; Singh, N.; Shukla, S. FlavorDB: A database of flavor molecules. Nucleic Acids Res. 2018, 46, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, 623–633. [Google Scholar] [CrossRef]
- Mathai, N.; Chen, Y.; Kirchmair, J. Validation strategies for target prediction methods. Brief. Bioinform. 2020, 21, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Landrum, G. Rdkit: Open-source cheminformatics software. GitHub SourceForge 2016, 10, 3592822. [Google Scholar]
- Jennrich, R.I. A simple general method for oblique rotation. Psychometrika 2002, 67, 7–19. [Google Scholar] [CrossRef]
- IBM Corp. IBM SPSS Statistics for Windows, Version 20.0; IBM Corp.: Armonk, NY, USA, 2013. [Google Scholar]
- Liu, W.; Wen, Y.; Yu, Z.; Yang, M. Large-Margin Softmax Loss for Convolutional Neural Networks. arXiv 2016, arXiv:1612.02295. [Google Scholar]
- Van Erkel, A.R.; Peter, M. Receiver operating characteristic (ROC) analysis: Basic principles and applications in radiology. Eur. J. Radiol. 1998, 27, 88–94. [Google Scholar] [CrossRef]
- Wold, S.; Dunn, W.J. Multivariate quantitative structure-activity relationships (QSAR): Conditions for their applicability. J. Chem. Inf. Model. 1983, 23, 6–13. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Huang, Y.; Wang, Y.; Zhu, L.; Wu, R. A predictive model for the evaluation of flavor attributes of raw and cooked beef based on sensor array analyses. Food Res. Int. 2019, 122, 16–24. [Google Scholar] [CrossRef]
- Shang, L.; Liu, C.; Tomiura, Y.; Hayashi, K. Machine-learning-based olfactometer: Prediction of odor perception from physicochemical features of odorant molecules. Anal. Chem. 2017, 89, 11999–12005. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Gerkin, R.C.; Guan, Y.; Dhurandhar, A.; Turu, G.; Szalai, B.; Mainland, J.D.; Ihara, Y.; Yu, C.W.; Wolfinger, R. Predicting human olfactory perception from chemical features of odor molecules. Science 2017, 355, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, N.; Sayle, R.A.; Landrum, G.A. Get Your Atoms in Order—An Open-Source Implementation of a Novel and Robust Molecular Canonicalization Algorithm. J. Chem. Inf. Model. 2015, 55, 2111–2120. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Training Set | Test Set | Total | Total * | Source |
|---|---|---|---|---|---|
| Odor | 1670 | 417 | 2087 | 25,044 | FlavorDB [12] PubChem [13] |
| Fruity | 512 | 128 | 640 | 7680 | |
| Floral | 255 | 64 | 319 | 3828 | |
| Woody | 195 | 49 | 244 | 2928 | |
| Odorless | 329 | 82 | 411 | 4932 |
| Category | Models | Precision | Sensitivity | Specificity | MCC |
|---|---|---|---|---|---|
| Odor/Odorless | MLP-Des | 0.994 | 0.996 | 0.918 | 0.930 |
| MLP-Fin | 0.971 | 0.991 | 0.600 | 0.684 | |
| CNN | 0.985 | 0.994 | 0.800 | 0.836 | |
| Fruity/Odorless | MLP-Des | 0.993 | 0.979 | 0.974 | 0.931 |
| MLP-Fin | 0.917 | 0.980 | 0.640 | 0.710 | |
| CNN | 0.967 | 0.984 | 0.857 | 0.867 | |
| Floral/Odorless | MLP-Des | 0.987 | 0.974 | 0.993 | 0.962 |
| MLP-Fin | 0.891 | 0.965 | 0.765 | 0.767 | |
| CNN | 0.960 | 0.975 | 0.917 | 0.901 | |
| Woody/Odorless | MLP-Des | 0.970 | 0.983 | 0.950 | 0.938 |
| MLP-Fin | 0.899 | 0.942 | 0.835 | 0.789 | |
| CNN | 0.957 | 0.949 | 0.931 | 0.880 |
| Models | Accuracy | Precision | Sensitivity |
|---|---|---|---|
| MLP-Des | 0.800 | 0.802 | 0.800 |
| MLP-Fin | 0.700 | 0.700 | 0.701 |
| CNN | 0.704 | 0.710 | 0.703 |
| Model | Task | Input | Data Set | Accuracy | AUC |
|---|---|---|---|---|---|
| AI-RF/DBN [5] | Distinguishing color and odor | Physicochemical features | Test set | 0.93 | - |
| 0.94 | - | ||||
| GA-ANN [21] | Evaluating raw beef flavor | Sensor array | Test | 0.85 | - |
| Evaluating cooked beef flavor | Test | 0.90 | - | ||
| MILP/MINLP [4] | Screening fragrance molecules | Group contribution | Train | 0.93 | - |
| Test | 0.75 | - | |||
| DREAM-RF [23] | Predicting the perceived odor of a given molecule | Chemical features | Test | - | 0.83 |
| Olfactometer [22] | Predicting odor perception of odorant molecules | Physicochemical features | Calibration | 0.97 | - |
| 0.93 | - | ||||
| Test | 0.97 | - | |||
| SOR [7] | Predicting multi-label of odorant molecules | Fingerprint | Test | 0.97 | 0.78 |
| Descriptor | |||||
| Image | Test | 0.98 | 0.87 | ||
| CNN | Classifying odor/odorless molecules | Image | Test set | 0.98 | 0.98 |
| MLP-Des | Descriptor | 0.99 | 0.99 | ||
| MLP-Fin | Fingerprint | 0.98 | 0.99 | ||
| CNN | Classifying fruity/odorless molecules | Image | Test set | 0.96 | 0.98 |
| MLP-Des | Descriptor | 0.98 | 0.99 | ||
| MLP-Fin | Fingerprint | 0.91 | 0.95 | ||
| CNN | Classifying floral/odorless molecules | Image | Test set | 0.96 | 0.99 |
| MLP-Des | Descriptor | 0.98 | 0.99 | ||
| MLP-Fin | Fingerprint | 0.90 | 0.93 | ||
| CNN | Classifying woody/odorless molecules | Image | Test set | 0.94 | 0.98 |
| MLP-Des | Descriptor | 0.97 | 0.99 | ||
| MLP-Fin | Fingerprint | 0.90 | 0.93 | ||
| CNN | Classifying fruity/floral/woody/odorless molecules | Image | Test set | 0.71 | 0.80 |
| MLP-Des | Descriptor | 0.80 | 0.86 | ||
| MLP-Fin | Fingerprint | 0.70 | 0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bo, W.; Yu, Y.; He, R.; Qin, D.; Zheng, X.; Wang, Y.; Ding, B.; Liang, G. Insight into the Structure–Odor Relationship of Molecules: A Computational Study Based on Deep Learning. Foods 2022, 11, 2033. https://doi.org/10.3390/foods11142033
Bo W, Yu Y, He R, Qin D, Zheng X, Wang Y, Ding B, Liang G. Insight into the Structure–Odor Relationship of Molecules: A Computational Study Based on Deep Learning. Foods. 2022; 11(14):2033. https://doi.org/10.3390/foods11142033
Chicago/Turabian StyleBo, Weichen, Yuandong Yu, Ran He, Dongya Qin, Xin Zheng, Yue Wang, Botian Ding, and Guizhao Liang. 2022. "Insight into the Structure–Odor Relationship of Molecules: A Computational Study Based on Deep Learning" Foods 11, no. 14: 2033. https://doi.org/10.3390/foods11142033