Ultra-Sensitive and Rapid Detection of Pathogenic Yersinia enterocolitica Based on the CRISPR/Cas12a Nucleic Acid Identification Platform

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Bacteria Culture and Crude Genomic DNA Extraction
2.3. Sample Preparation
2.4. Optimization of the RPA Assay
2.5. Target Cleavage Assays Based on Cas12a
2.6. Sensitivity and Specificity of CRISPR/Cas12a-RPA System
2.7. qPCR and Standard Curves
2.8. Statistical Analysis
3. Results
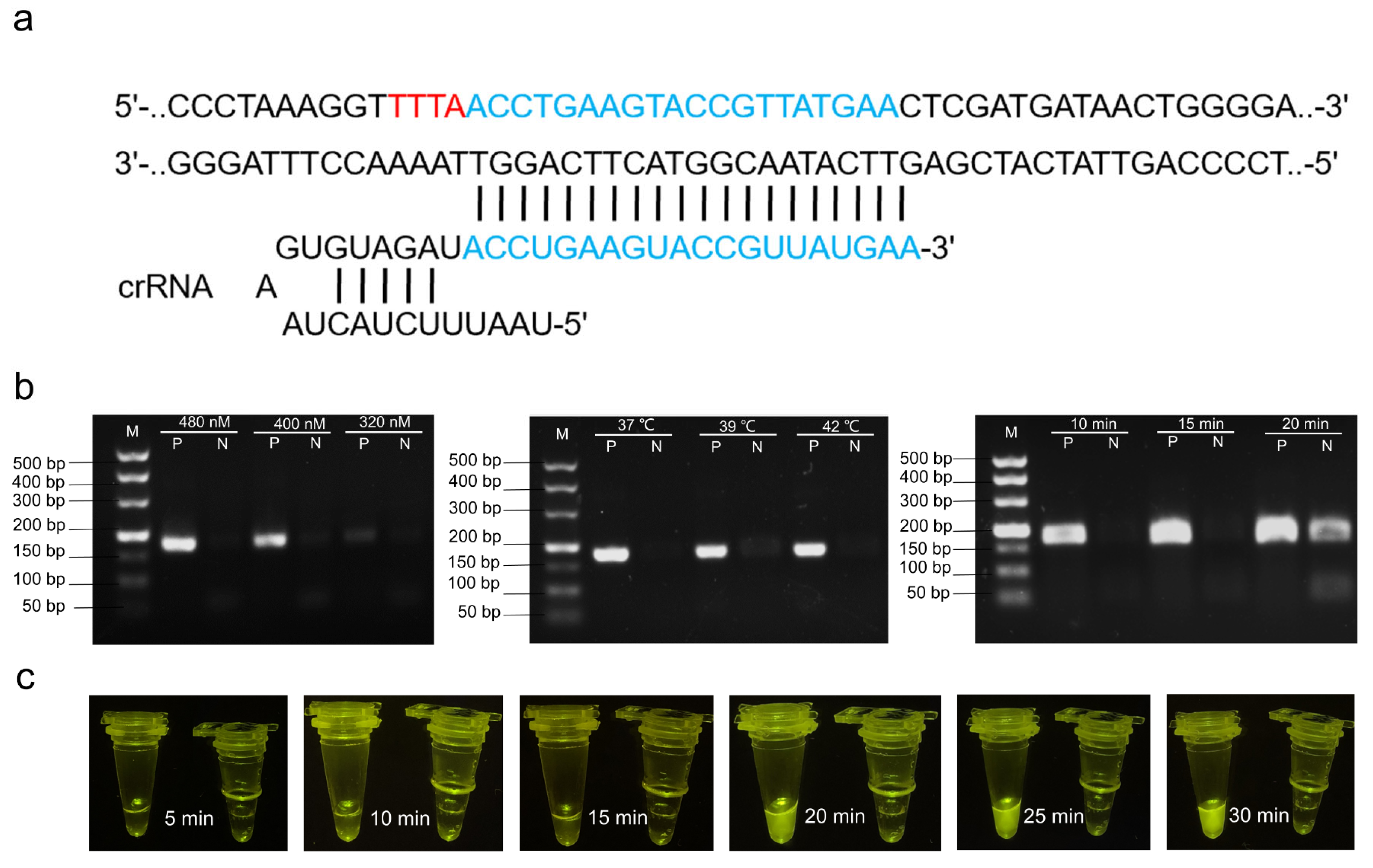
3.1. Design of Primers and crRNA
3.2. Optimization of CRISPR/Cas12a Reaction System
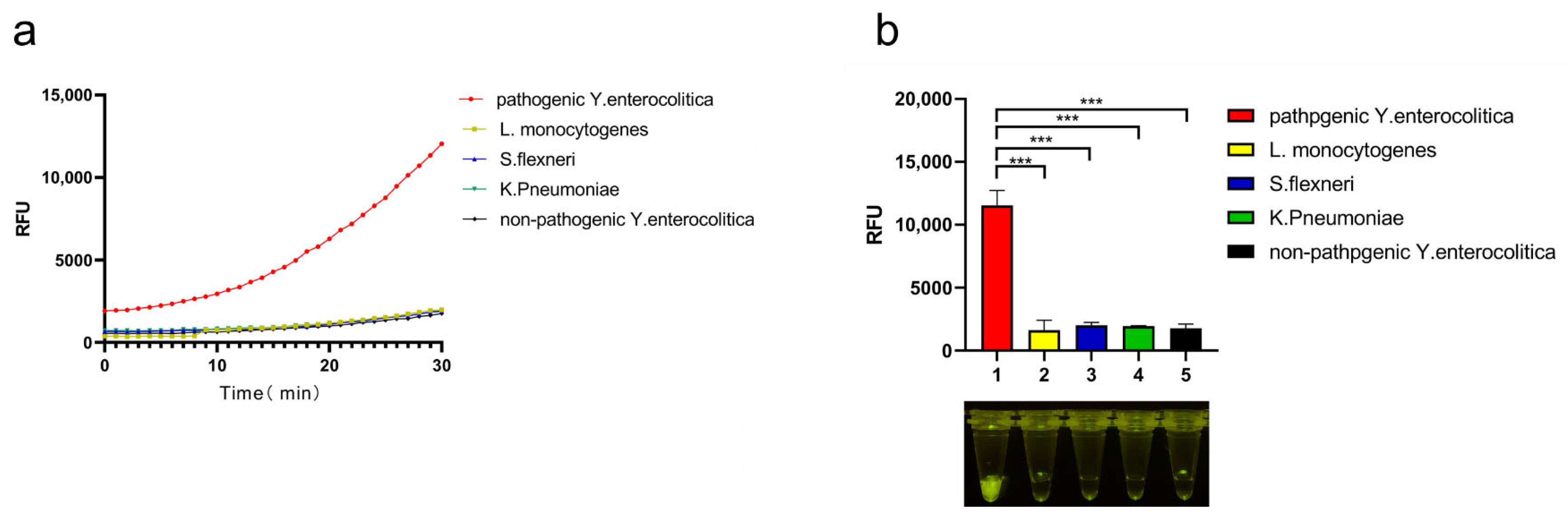
3.3. Specificity of CRISPR/Cas12a System
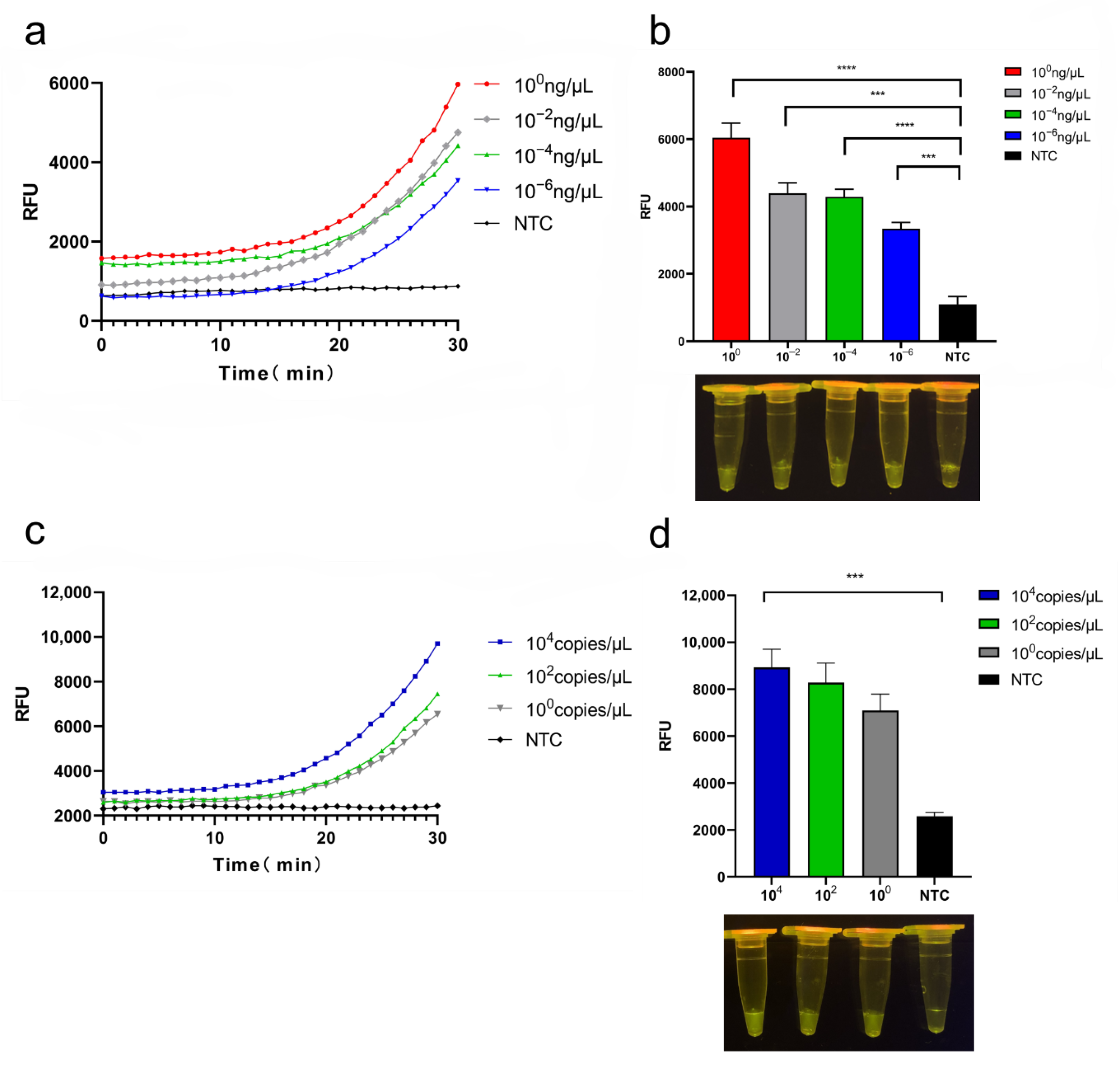
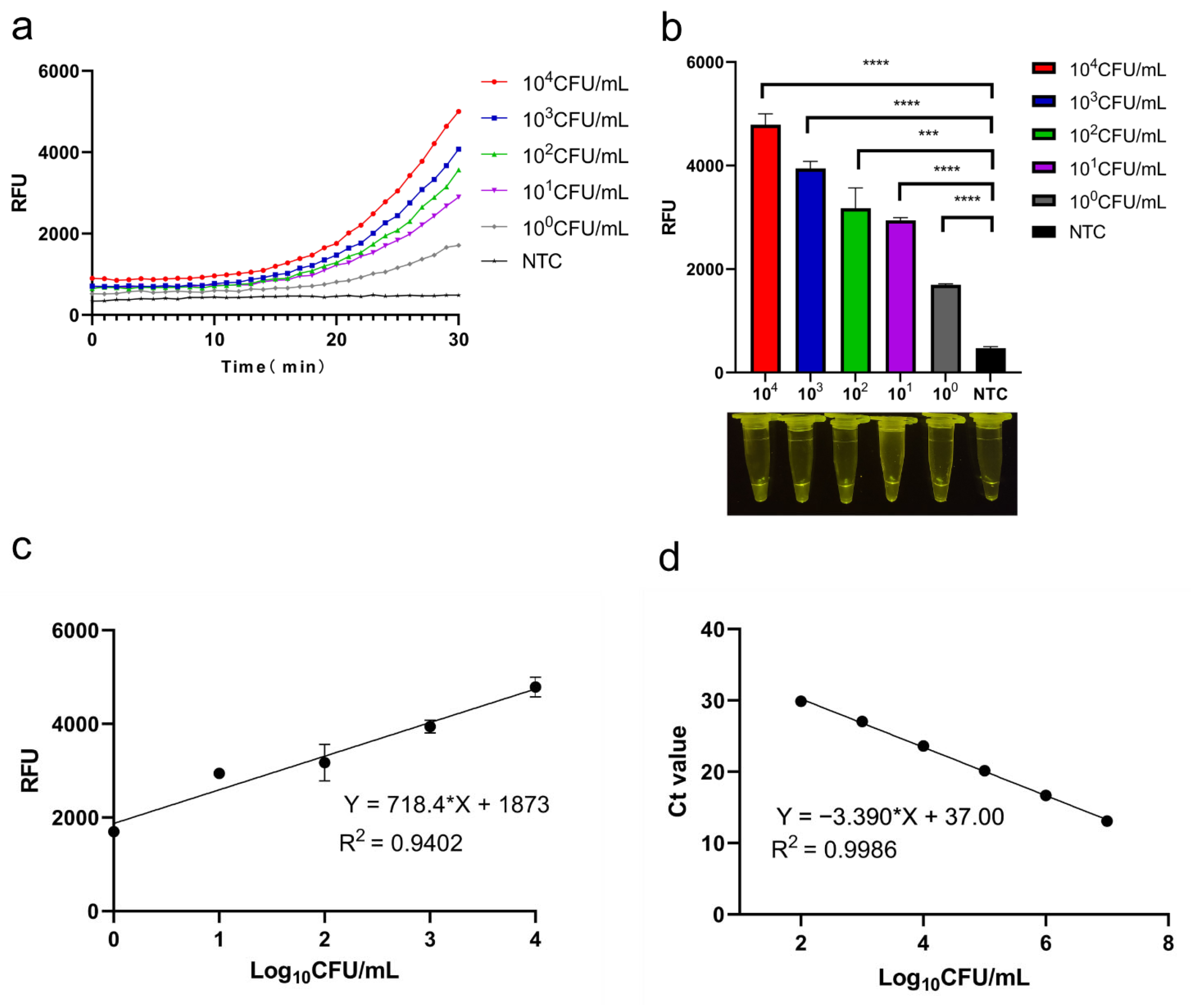
3.4. Sensitivity of CRISPR/Cas12a System
3.5. qPCR and Standard Curves
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leon-Velarde, C.G.; Jin, W.J.; Skurnik, M. Yersinia Phages and Food Safety. Viruses 2019, 11, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottone, E.J. Yersinia enterocolitica: Overview and epidemiologic correlates. Microbes Infect. 1999, 1, 323–333. [Google Scholar] [CrossRef]
- Mcnally, A.; Thomson, N.R.; Reuter, S.; Wren, B.W. ‘Add, stir and reduce’: Yersinia spp. as model bacteria for pathogen evolution. Nat. Rev. Microbiol. 2016, 14, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Gulati, P.; Bhagat, N.; Dhar, M.S.; Virdi, J.S. Detection of Yersinia enterocolitica in food: An overview. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 641. [Google Scholar] [CrossRef]
- Platt-Samoraj, A.; Syczyło, K.; Szczerba-Turek, A.; Bancerz-Kisiel, A.; Jabłoński, A.; Łabuć, S.; Pajdak, J.; Oshakbaeva, N.; Szweda, W. Presence of ail and ystB genes in Yersinia enterocolitica biotype 1A isolates from game animals in Poland. Vet. J. 2017, 221, 11–13. [Google Scholar] [CrossRef]
- Kraushaar, B.; Dieckmann, R.; Wittwer, M.; Knabner, D.; Konietzny, A.; Mäde, D.; Strauch, E. Characterization of a Yersinia enterocolitica biotype 1A strain harbouring an ail gene. J. Appl. Microbiol. 2011, 111, 997–1005. [Google Scholar] [CrossRef]
- Huai-Qi, J.; Ji-Yao, L.I.; Yu-Chun, X. PCR Detection and Investigation of Distribution of Virolence Factors for Yersinia enterocolitic Serotype O:3 and O:9. Chin. J. Vector Biol. Control 2003, 69, 1810–1816. [Google Scholar]
- Bari, M.L.; Hossain, M.A.; Isshiki, K.; Ukuku, D. Behavior of Yersinia enterocolitica in Foods. J. Pathog. 2011, 2011, 420732. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Bonny, T.S.; Stonsaovapak, S.; Ananchaipattana, C. Yersinia enterocolitica: Epidemiological Studies and Outbreaks. J. Pathog. 2011, 2011, 239391. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Qiu, H.; Dong, J.; Cui, Z.; Kan, B.; Xiao, Y.; Xu, Y.; Xia, S.; Wang, H.; Yang, J. O:8 serotype Yersinia enterocolitica strains in China. Int. J. Food Microbiol. 2008, 125, 259–266. [Google Scholar] [CrossRef]
- Wang, J.; Liu, M.; Wang, H.; Wu, Q.; Yang, R. Occurrence, Molecular Characterization, and Antimicrobial Susceptibility of Yersinia enterocolitica Isolated from Retail Food Samples in China. LWT- Food Sci. Technol. 2021, 150, 111876. [Google Scholar] [CrossRef]
- Agnieszka, C.; Śliżewska, K. Campylobacteriosis, Salmonellosis, Yersiniosis, and Listeriosis as Zoonotic Foodborne Diseases: A Review. Int. J. Environ. Res. Public Health 2018, 15, 863. [Google Scholar]
- Law, W.F.; Ab Mutalib, N.S.; Chan, K.G.; Lee, L.H. Rapid methods for the detection of foodborne bacterial pathogens: Principles, applications, advantages and limitations. Front. Microbiol. 2015, 5, 770. [Google Scholar] [CrossRef] [Green Version]
- Lantz, P.G.; Knutsson, R.; Blixt, Y.; Alsoud, W.A.; Borch, E.; Rådström, P. Detection of pathogenic Yersinia enterocolitica in enrichment media and pork by a multiplex PCR: A study of sample preparation and PCR-inhibitory components. Int. J. Food Microbiol. 1998, 45, 93–105. [Google Scholar] [CrossRef]
- Lambertz, S.T.; Nilsson, C.; Hallanvuo, S.; Lindblad, M. Real-Time PCR Method for Detection of Pathogenic Yersinia enterocolitica in Food. Appl. Environ. Microbiol. 2008, 74, 6060–6067. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Hu, P.; Ren, H.; Wang, H.; Lu, S. RPA-SYBR Green I based instrument-free visual detection for pathogenic Yersinia enterocolitica in meat. Anal. Biochem. 2021, 621, 114157. [Google Scholar] [CrossRef]
- Pakbin, B.; Brück, W.M.; Allahyari, S.; Rossen, J.W.A.; Mahmoudi, R. Antibiotic Resistance and Molecular Characterization of Cronobacter sakazakii Strains Isolated from Powdered Infant Formula Milk. Foods 2022, 11, 1093. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C. Recombinase polymerase amplification: Basics, applications and recent advances. TrAC Trends Anal. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Pabinger, S.; Thallinger, G.G.; Snajder, R.; Eichhorn, H.; Rader, R.; Trajanoski, Z. QPCR: Application for real-time PCR data management and analysis. BMC Bioinform. 2009, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, C.; Yang, R.; Williams, A.; Gardner, G.E.; Carmichael, I.; Campbell, A.J.D.; Ryan, U. Faecal shedding of pathogenic Yersinia enterocolitica determined by qPCR for yst virulence gene is associated with reduced live weight but not diarrhoea in prime lambs. Prev. Vet. Med. 2018, 152, 56–64. [Google Scholar] [CrossRef]
- Abbas, K.H. Rapid detection of Yersinia enterocolitica by using Real-Time PCR technique in some types of foods in Al-Qadisiya province. Al-Qadisiyah J. Vet. Med. Sci. 2015, 14, 7193–7197. [Google Scholar]
- Kissenktter, J.; Bhlken-Fascher, S.; Forrest, M.S.; Pieoenburg, O.; Wahed, A. Recombinase polymerase amplification assays for the identification of pork and horsemeat. Food Chem. 2020, 322, 126759. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, L.; Tong, J.; Zhao, X.; Ren, J. CRISPR-Cas12a enhanced rolling circle amplification method for ultrasensitive miRNA detection. Microchem. J. 2020, 158, 105239. [Google Scholar] [CrossRef]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef]
- Yao, Y.; Li, S.; Cao, J.; Liu, W.; Fan, K.; Xiang, W.; Yang, K.; Kong, D.; Wang, W. Development of small molecule biosensors by coupling the recognition of the bacterial allosteric transcription factor with isothermal strand displacement amplification. Chem. Commun. 2018, 54, 38. [Google Scholar]
- Świat, A.M.; Sofia, D.; den Maxime, R.; Melanie, W.; van der John, O.; Jean-Marc, D.; Pascale, D.L. FnCpf1: A novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 45, 12585–12598. [Google Scholar] [CrossRef]
- Yao, Y.; Li, S.; Cao, J.; Liu, W.; Qi, F.; Xiang, W.; Yang, K.; Wang, W.; Zhang, L. A novel signal transduction system for development of uric acid biosensors. Appl. Microbiol. Biotechnol. 2018, 102, 7489–7497. [Google Scholar] [CrossRef]
- Raschmanova, H.; Weninger, A.; De Glie, R.A.; Kovar, K.; Vogl, T. Implementing CRISPR-Cas technologies in conventional and non-conventional yeasts: Current state and future prospects. Biotechnol. Adv. 2018, 36, 641–665. [Google Scholar] [CrossRef]
- Yongmoon, J.; Hee, C.Y.; Yunsu, J.; Yu, J.; Jiyoung, G.; Gyejun, L.; Kyeong, J.Y.; Hwan, L.S.; In-San, K.; Jin-Soo, K. Direct observation of DNA target searching and cleavage by CRISPR-Cas12a. Nat. Commun. 2018, 9, 2777. [Google Scholar]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.; Abudayyeh, O.; Slaymaker, I.; Makarova, K.; Essletzbichler, P.; Volz, S.; Joung, J.; Van Der Oost, J.; Regev, A. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Liu, X.; Chen, J.; Guo, Z.; Wang, J. Development of a Rapid Test Method for Salmonella enterica Detection Based on Fluorescence Probe-Based Recombinase Polymerase Amplification. Food Anal. Methods 2019, 12, 1791–1798. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Wang, J.M.; Li, X.Y.; Zhang, Z.L.; Gao, S.; Cao, R.B.; Zhao, G.P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, eaar6245. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ke, Y.; Liu, W.; Sun, Y.; Ding, X. A one-pot toolbox based on Cas12a/crRNA enables rapid foodborne pathogen detection at attomolar level. ACS Sens. 2020, 5, 1427–1435. [Google Scholar] [CrossRef]
- Wang, S.; Fan, Y.; Feng, Z.; Song, M.; Li, Q.; Jiang, B.; Qin, F.; Liu, H.; Lan, L.; Yang, M. Rapid Nucleic Acid Detection of Escherichia coli O157:H7 Based on CRISPR/Cas12a System. Food Control 2021, 130, 108194. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, T.; Liu, C.; Xu, Q.; Fang, S.; Wu, Y.; Wu, M.; Liu, Q. An ultrasensitive and contamination-free on-site nucleic acid detection platform for Listeria monocytogenes based on the CRISPR-Cas12a system combined with recombinase polymerase amplification. LWT 2021, 152, 112166. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Primer’s Name | Sequences (5′-3′) | Product Length |
|---|---|---|---|
| RPA | ail-1F | GTCTGTTAATGTGTACGCTGCGAGTGAAAG | 180 bp |
| ail-1R | TATCCCTGATGAGTATAAGCAAACGAACCT | ||
| ail-2F | ACGCTGCGAGTGAAAGTAGT | 254 bp | |
| ail-2R | TTCGTTGATGCGGAAAGATG | ||
| ail-3F | TAATGTGTACGCTGCGAGTGAAAGTAG | 171 bp | |
| ail-3R | CCCTGATGAGTATAAGCAAACGAACCT | ||
| CRISPR/Cas12a | crRNA | UAAUUUCUACUAAGUGUAGAUACCUGAAGUACCGUUAUGAA | |
| ssDNA reporter | 6-FAM-/TTTTTT/BHQ-1 | ||
| qPCR | F | TAATGTGTACGCTGCGAGTGAAAGTAG | |
| R | CCCTGATGAGTATAAGCAAACGAACCT |
| CRISPR/Cas12a-RPA | qPCR | |
|---|---|---|
| Equipment | Blue light transilluminator | ABI 7500 Fast |
| Procedure | Easy | Moderate |
| Time | 45 min | 2 h |
| Sensitivity | High (100 CFU/mL) | Low (102 CFU/mL) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Ren, H.; Hu, P.; Wang, Y.; Wang, H.; Li, Y.; Feng, K.; Wang, C.; Cao, Q.; Guo, Y.; et al. Ultra-Sensitive and Rapid Detection of Pathogenic Yersinia enterocolitica Based on the CRISPR/Cas12a Nucleic Acid Identification Platform. Foods 2022, 11, 2160. https://doi.org/10.3390/foods11142160
Xiao Y, Ren H, Hu P, Wang Y, Wang H, Li Y, Feng K, Wang C, Cao Q, Guo Y, et al. Ultra-Sensitive and Rapid Detection of Pathogenic Yersinia enterocolitica Based on the CRISPR/Cas12a Nucleic Acid Identification Platform. Foods. 2022; 11(14):2160. https://doi.org/10.3390/foods11142160
Chicago/Turabian StyleXiao, Yiran, Honglin Ren, Pan Hu, Yang Wang, Han Wang, Yansong Li, Kai Feng, Cong Wang, Qi Cao, Yuxi Guo, and et al. 2022. "Ultra-Sensitive and Rapid Detection of Pathogenic Yersinia enterocolitica Based on the CRISPR/Cas12a Nucleic Acid Identification Platform" Foods 11, no. 14: 2160. https://doi.org/10.3390/foods11142160