
Comparative Transcriptome Analysis Reveals Gene Expression Differences in Eggplant (Solanum melongena L.) Fruits with Different Brightness

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Construction, and Illumina Sequencing
2.3. RNA-Seq Data Analyses and DEG Identification
2.4. Annotation and Classification of DEGs
2.5. Validation of Gene Expression Profile by qRT-PCR
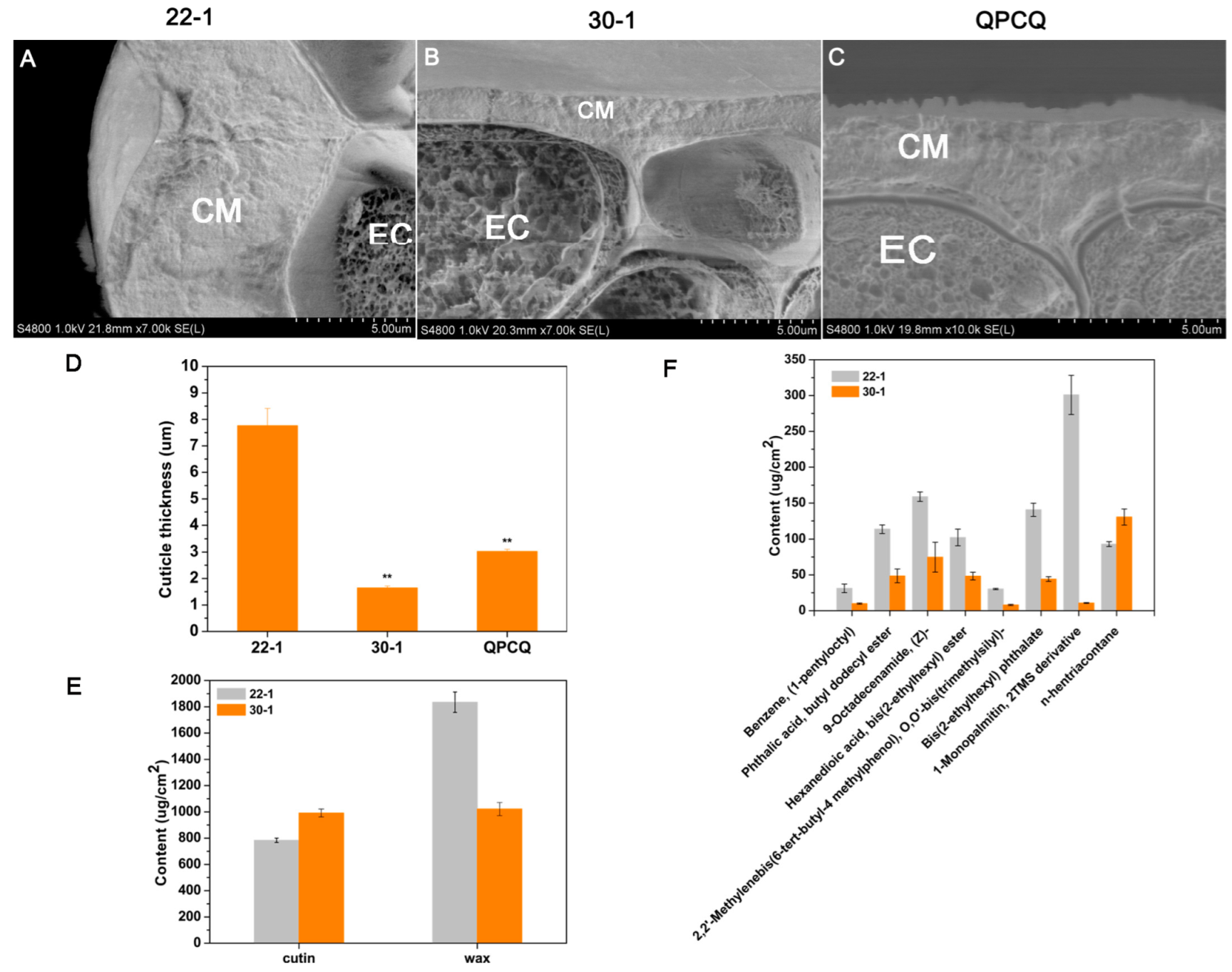
2.6. Cutin Monomer and Wax Analysis
2.7. Cryo-SEM
2.8. Identification of KCS Family in Eggplant
2.9. Phylogenetic Analysis
2.10. Structure and Motif Analysis
2.11. Data Analysis
3. Results
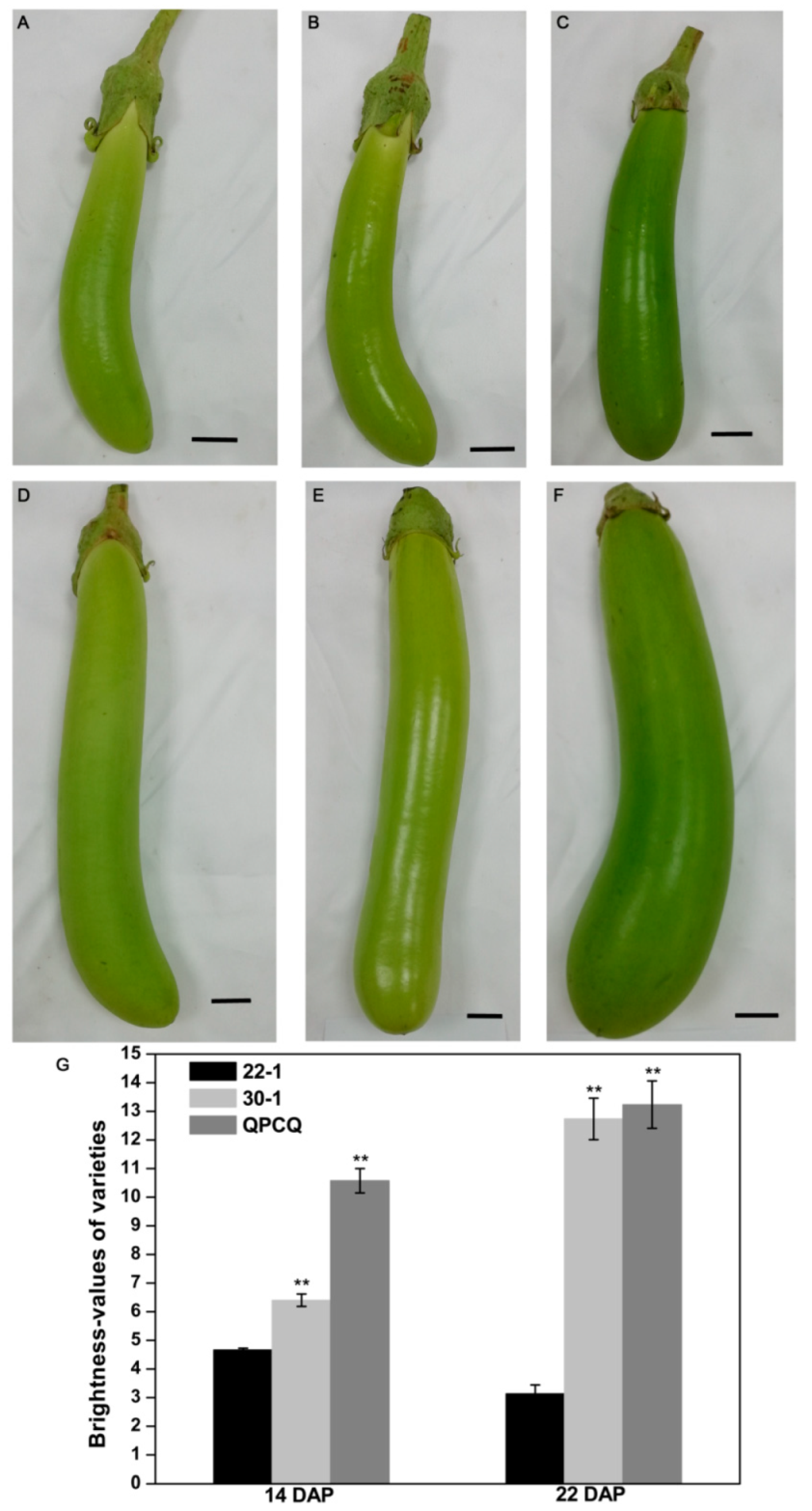
3.1. Identification of Three Independent Eggplant Varieties with Different Fruit Brightness
3.2. Fruit Transcriptome Sequencing and Analyses
3.3. Prediction of New Transcripts
3.4. Single Nucleotide Polymorphisms and INDEL Detection
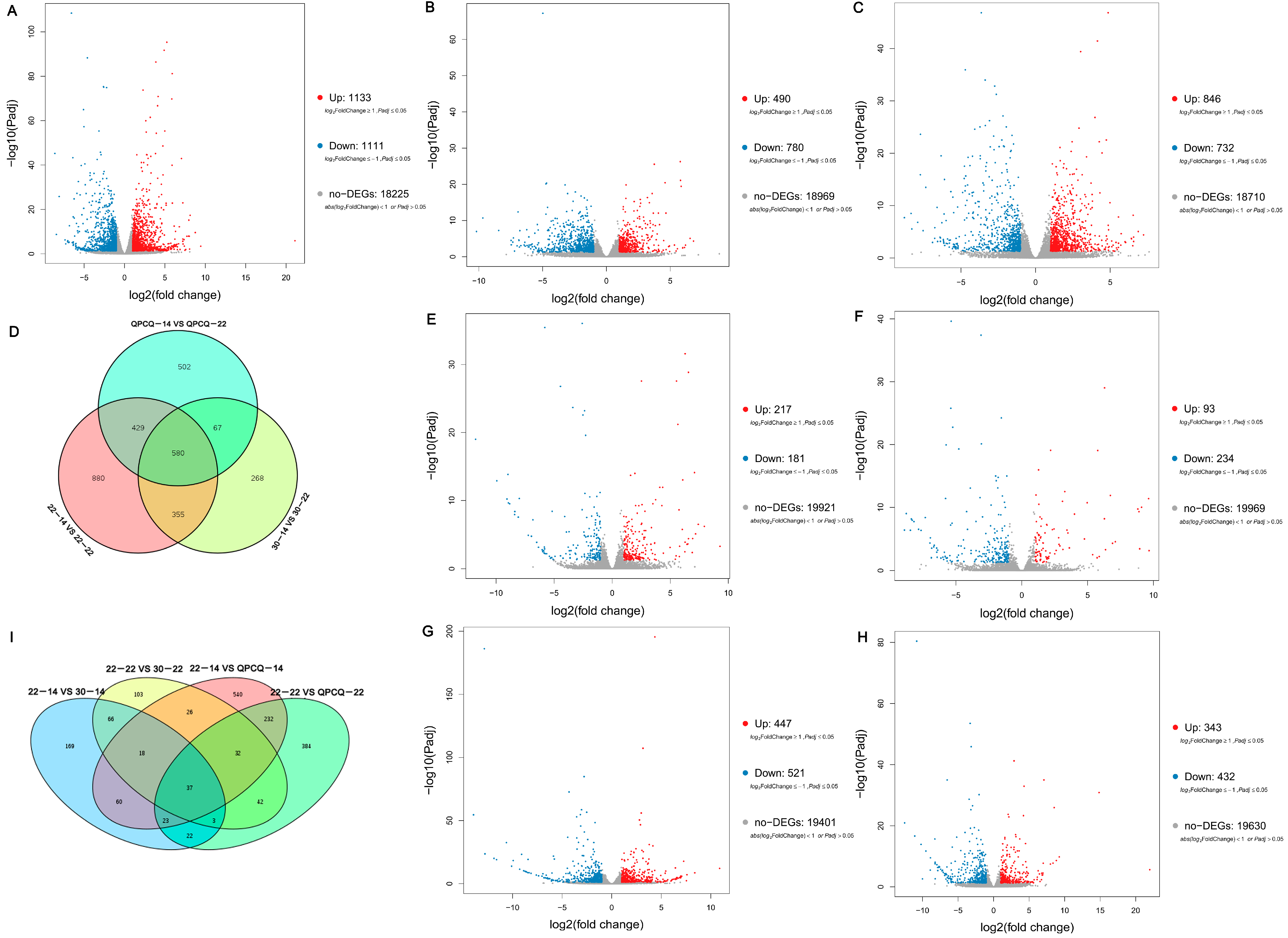
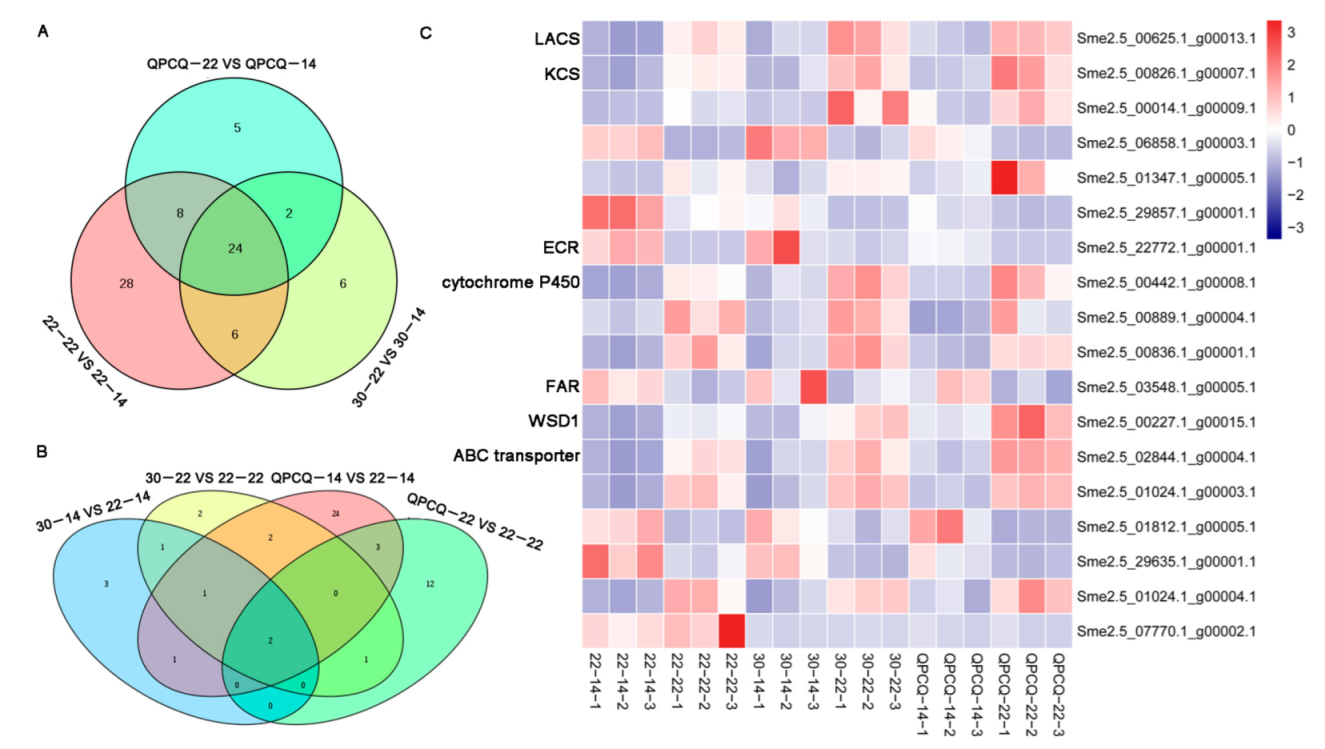
3.5. Analyses of DEGs
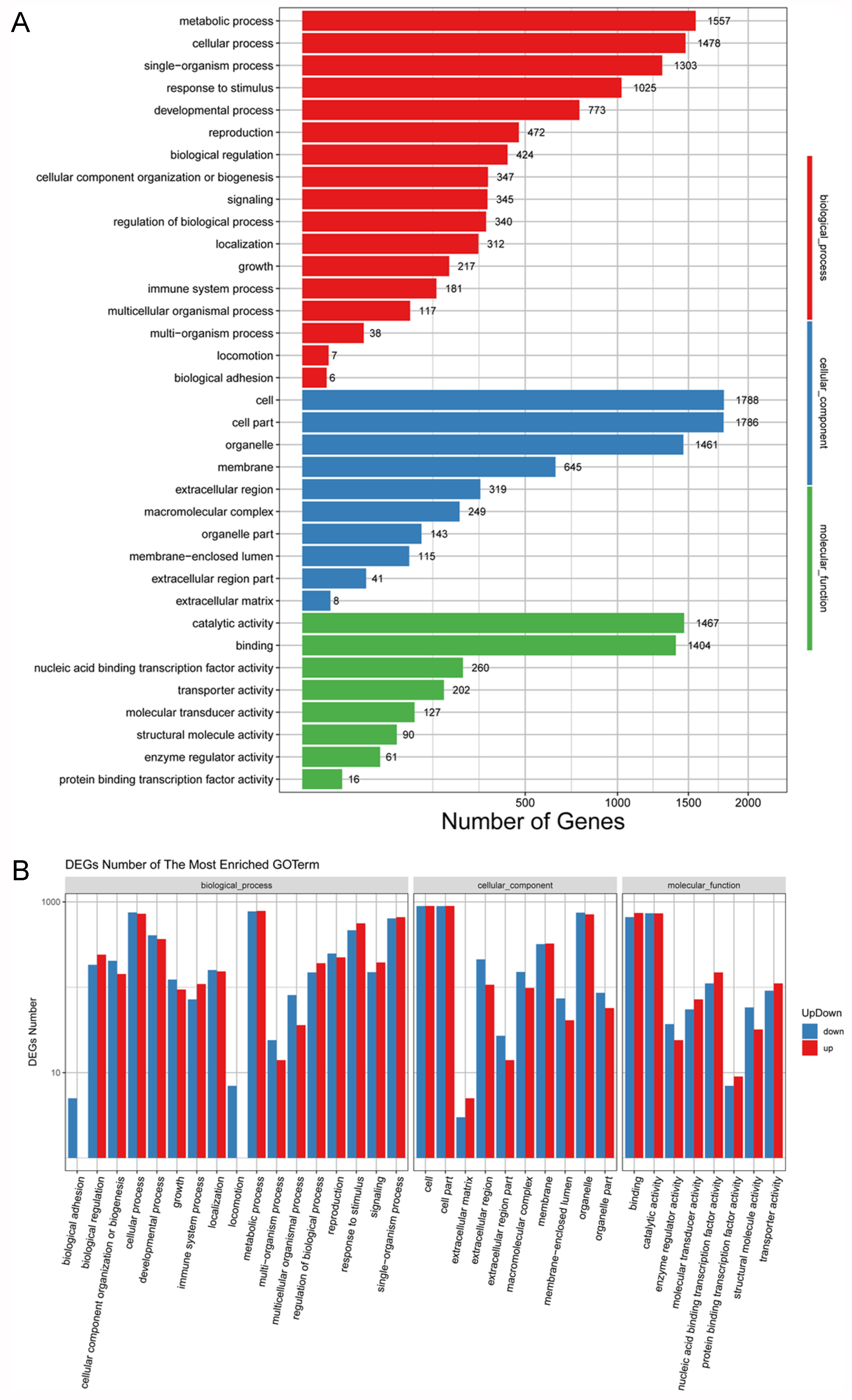
3.6. GO Functional Enrichment Analyses of DEGs
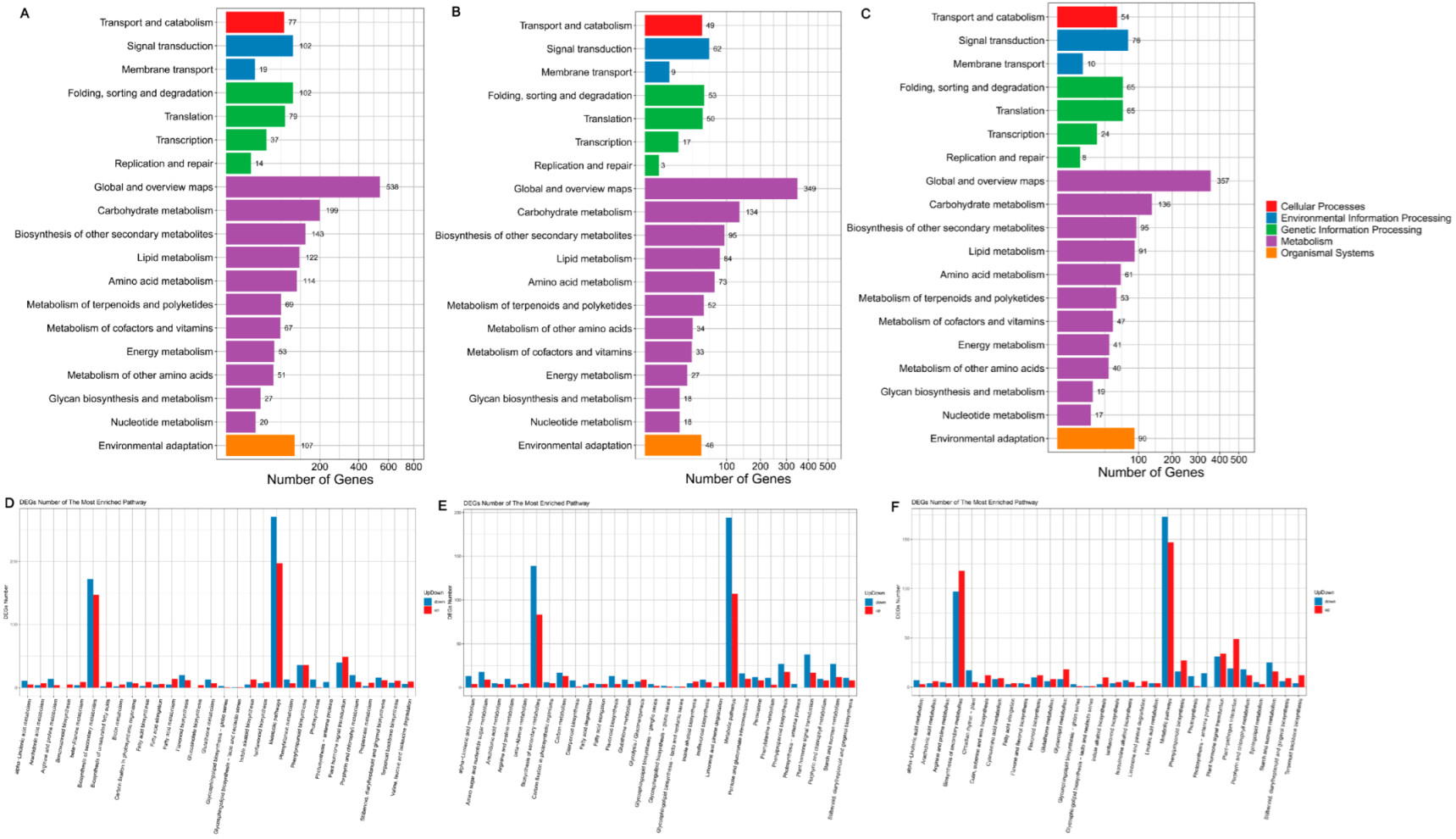
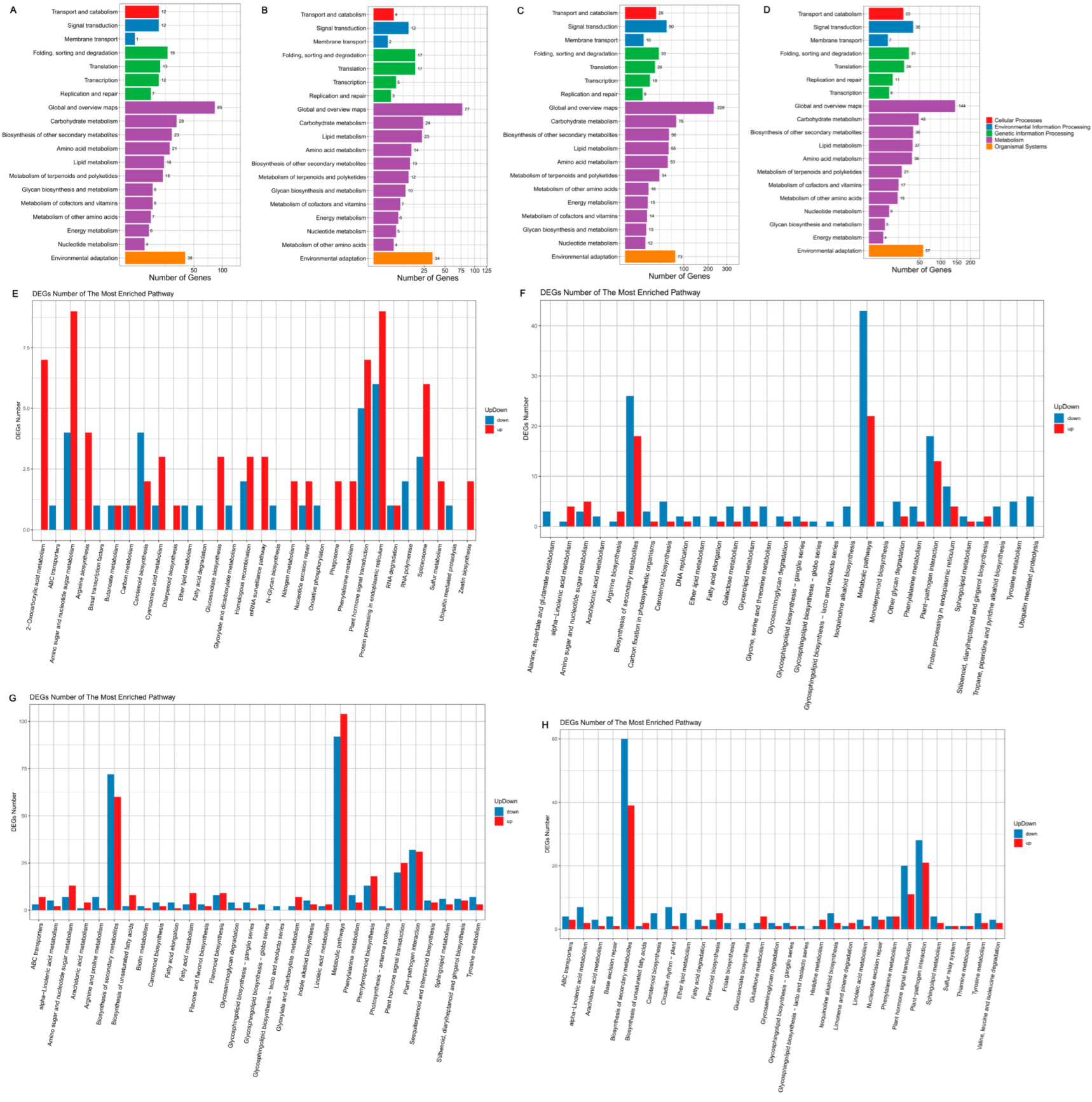
3.7. KEGG Pathway Database Enrichment Analyses of DEGs
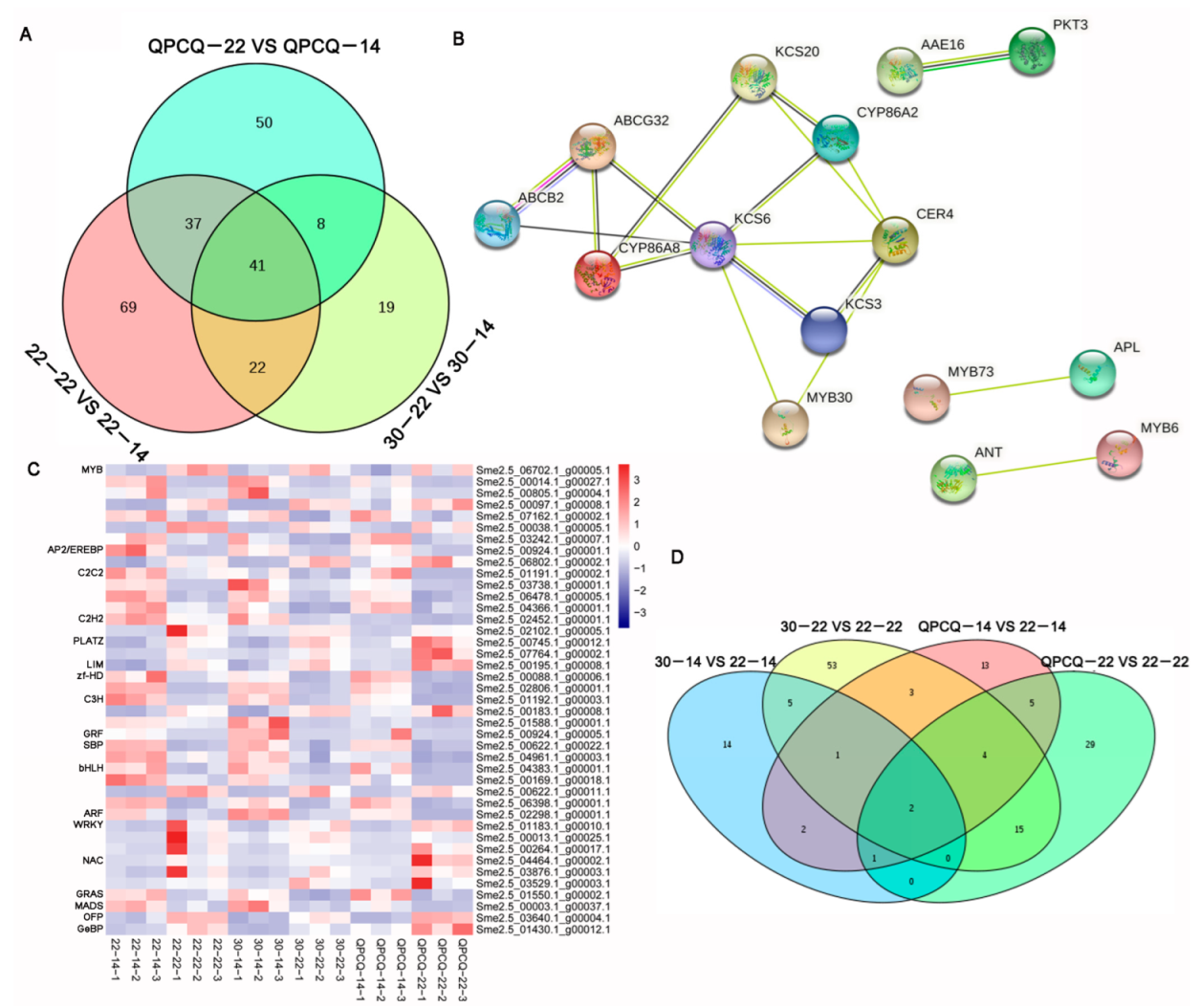
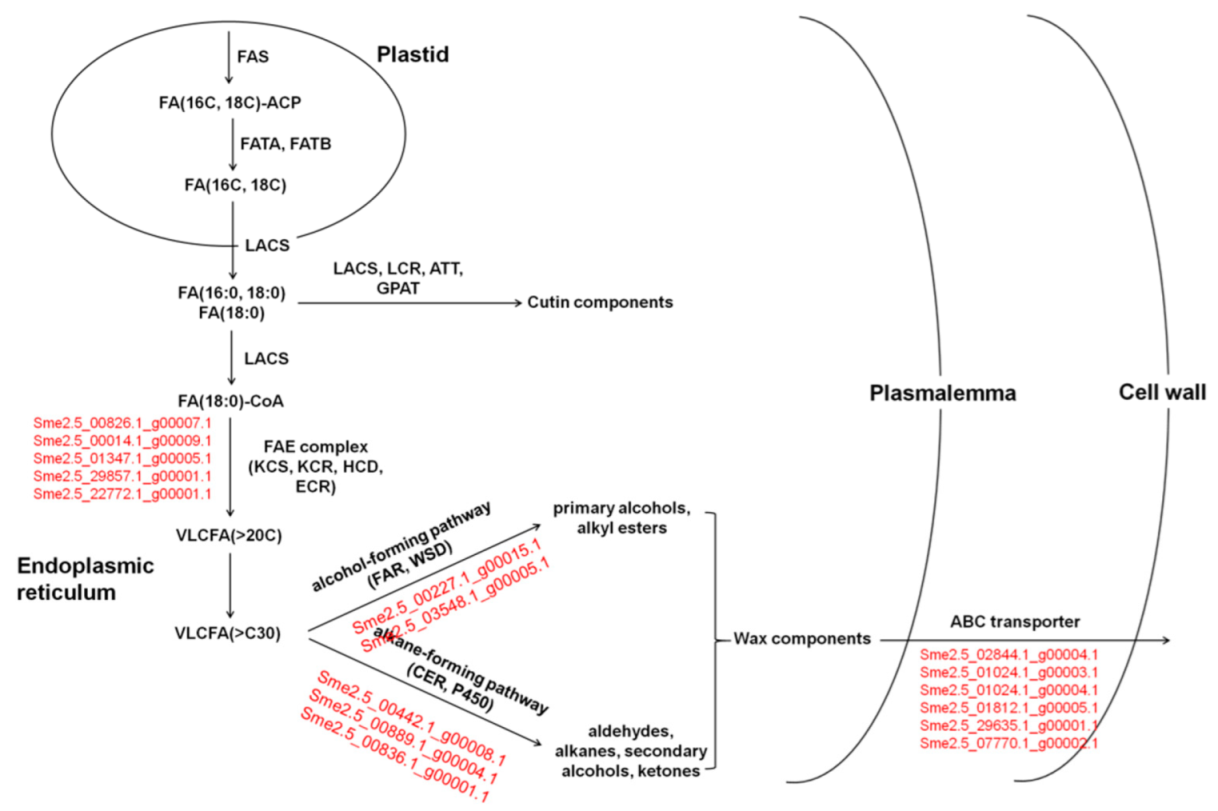
3.8. DEGs Related to Cutin and Wax in Eggplants of Different Brightness
3.9. DEGs Identified as Transcription Factors in Eggplants of Different Brightness
3.10. Cuticle Condition of Eggplants of Different Brightness
3.11. Identification of SmKCS Gene Family in Eggplant
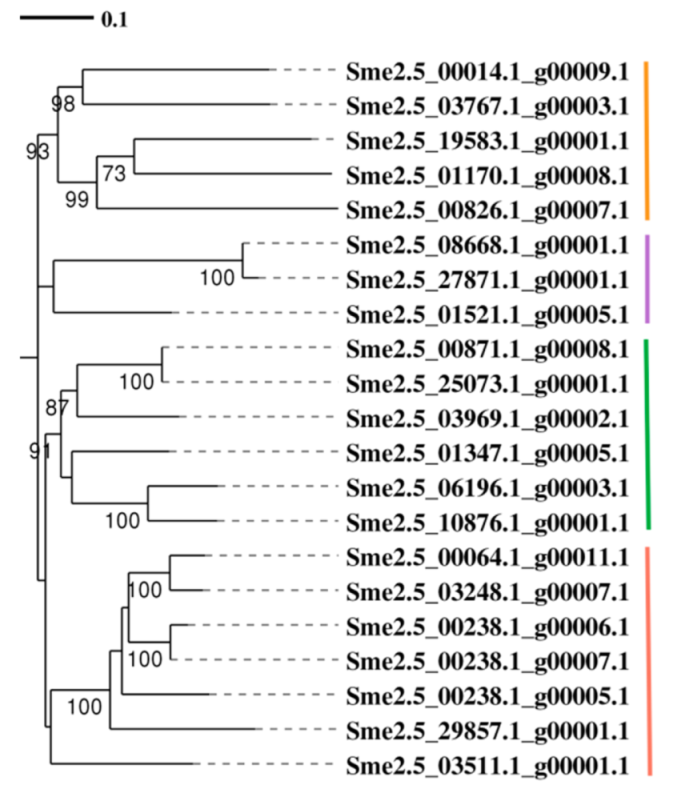
3.12. Phylogenetic Analysis of SmKCS Proteins
3.13. Structure and Conserved Motif Analyses of the SmKCS Gene Family
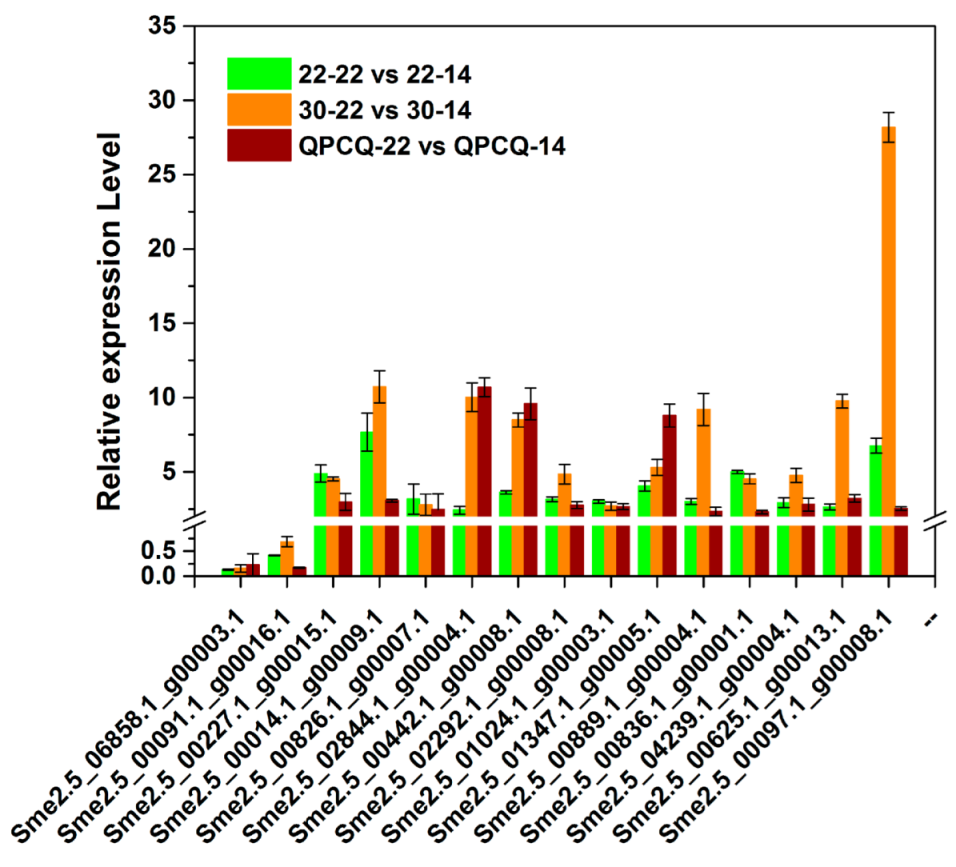
3.14. qPCR Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, G.L.; Qin, Z.W.; Zhou, X.Y.; Zhao, Z.Y. Genetic analysis and SSR markers of tuberculate trait in Cucumis sativus. Chin. Bull. Bot. 2007, 24, 168–172. [Google Scholar]
- Pollack, S.L. Consumer demand for fruit and vegetables: The US example. In Changing Structure of Global Food Consumption and Trade, Economic Research Service USDA; Regmi, A., Ed.; Diane: Collingdale, PA, USA, 2001; pp. 49–54. [Google Scholar]
- Du Plooy, G.W.; Van der Merwe, C.F.; Korsten, L. Lenticel discolouration in mango (Mangifera indica L.) fruit—A cytological study of mesophyll cells from affected tissue. J. Hortic. Sci. Biotechnol. 2006, 81, 869–873. [Google Scholar] [CrossRef]
- Strong, W.J. Breeding experiments with the cucumber (Cucumis sativus L.). Sci. Agric. 1931, 11, 333–346. [Google Scholar]
- Yang, X.; Zhang, W.; Li, Y.; He, H.; Bie, B.; Ren, G.; Zhao, J.; Wang, Y.; Nie, J.; Pan, J.; et al. High-resolution mapping of the dull fruit skin gene D in cucumber (Cucumis sativus L.). Mol. Breed. 2014, 33, 15–22. [Google Scholar] [CrossRef]
- Yuan, X.; Pan, J.; Cai, R.; Guan, Y.; Liu, L.; Zhang, W.; Li, Z.; He, H.; Zhang, C.; Si, L.; et al. Genetic mapping and QTL analysis of fruit and flower related traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Euphytica 2008, 164, 473–491. [Google Scholar] [CrossRef]
- Miao, H.; Zhang, S.; Wang, X.; Zhang, Z.; Li, M.; Mu, S.; Cheng, Z.; Zhang, R.; Huang, S.; Xie, B.; et al. A linkage map of cultivated cucumber (Cucumis sativus L.) with 248 microsatellite marker loci and seven genes for horticulturally important traits. Euphytica 2011, 182, 167–176. [Google Scholar] [CrossRef]
- Girard, A.L.; Mounet, F.; Lemaire-Chamley, M.; Gaillard, C.; Elmorjani, K.; Vivancos, J.; Runavot, J.L.; Quemener, B.; Petit, J.; Germain, V.; et al. Tomato GDSL1 is required for cutin deposition in the fruit cuticle. Plant Cell 2012, 24, 3119–3134. [Google Scholar] [CrossRef]
- Nadakuduti, S.S.; Pollard, M.; Kosma, D.K.; Allen, C., Jr.; Ohlrogge, J.B.; Barry, C.S. Pleiotropic phenotypes of the sticky peel mutant provide new insight into the role of CUTIN DEFICIENT 2 in epidermal cell function in tomato. Plant Physiol. 2012, 159, 945–960. [Google Scholar] [CrossRef]
- Shi, J.X.; Adato, A.; Alkan, N.; He, Y.; Lashbrooke, J.; Matas, A.J.; Meir, S.; Malitsky, S.; Isaacson, T.; Prusky, D.; et al. The tomato SlSHINE3 transcription factor regulates fruit cuticle formation and epidermal patterning. New Phytol. 2013, 197, 468–480. [Google Scholar] [CrossRef]
- Petit, J.; Bres, C.; Just, D.; Garcia, V.; Mauxion, J.P.; Marion, D.; Bakan, B.; Joubès, J.; Domergue, F.; Rothan, C. Analyses of tomato fruit brightness mutants uncover both cutin-deficient and cutin-abundant mutants and a new hypomorphic allele of GDSL lipase. Plant Physiol. 2014, 164, 888–906. [Google Scholar] [CrossRef]
- Jenks, M.A.; Eigenbrode, S.D.; Lemieux, B. Cuticular waxes of Arabidopsis. Arab. Book 2002, 1, e0016. [Google Scholar] [CrossRef]
- Liu, D.; Zeng, Q.; Ji, Q.; Liu, C.; Liu, S.; Liu, Y. A comparison of the ultrastructure and composition of fruits’ cuticular wax from the wild-type ‘Newhall’ navel orange (Citrus sinensis [L.] Osbeck cv. Newhall) and its glossy mutant. Plant Cell Rep. 2012, 31, 2239–2246. [Google Scholar] [CrossRef]
- Nawrath, C. Unraveling the complex network of cuticular structure and function. Curr. Opin. Plant Biol. 2006, 9, 281–287. [Google Scholar] [CrossRef]
- Schreiber, L. Transport barriers made of cutin, suberin and associated waxes. Trends Plant Sci. 2010, 15, 546–553. [Google Scholar] [CrossRef]
- Li-Beisson, Y.; Pollard, M.; Sauveplane, V.; Pinot, F.; Ohlrogge, J.; Beisson, F. Nanoridges that characterize the surface morphology of flowers require the synthesis of cutin polydster. Proc. Natl. Acad. Sci. USA 2009, 106, 22008–22013. [Google Scholar] [CrossRef]
- Zhang, A.; Zhu, Z.; Shang, J.; Zhang, S.; Shen, H.; Wu, X.; Zha, D. Transcriptome profiling and gene expression analyses of eggplant (Solanum melongena L.) under heat stress. PLoS ONE 2020, 15, e0236980. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Roberts, A.; Trapnell, C.; Donaghey, J.; Rinn, J.L.; Pachter, L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential Expression of RNA-Seq Data at the Gene Level—The DeSeq Package; European Molecular Biology Laboratory: Heidelberg, Germany, 2013. [Google Scholar]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, L.; Raboanatahiry, N.; Lu, G.; Zhang, X.; Xiang, J.; Gan, J.; Fu, C.; Li, M. Transcriptome analysis of stem and globally comparison with other tissues in brassica napus. Front. Plant Sci. 2016, 7, 1403. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Punta, M.; Coggill, P.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2000, 30, 276–280. [Google Scholar] [CrossRef]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Isaacson, T.; Kosma, D.K.; Matas, A.J.; Buda, G.J.; He, Y.; Yu, B.; Pravitasari, A.; Batteas, J.D.; Stark, R.E.; Jenks, M.A.; et al. Cutin deficiency in the tomato fruit cuticle consistently affects resistance to microbial infection and biomechanical properties, but not transpirational water loss. Plant J. 2009, 60, 363–377. [Google Scholar] [CrossRef]
- Kohlwein, S.D.; Eder, S.; Oh, C.S.; Martin, C.E.; Gable, K.; Bacikova, D.; Dunn, T. Tsc13p Is Required for Fatty Acid Elongation and Localizes to a Novel Structure at the Nuclear-vacuolar Interface in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 109. [Google Scholar] [CrossRef]
- Rowland, O.; Lee, R.; Franke, R.; Schreiber, L.; Kunst, L. The CER3 wax biosynthetic gene from Arabidopsis thaliana is allelic to WAX2/YRE/FLP1. FEBS Lett. 2007, 581, 3538–3544. [Google Scholar] [CrossRef]
- Bourdenx, B.; Bernard, A.; Domergue, F.; Pascal, S.; Léger, A.; Roby, D.; Pervent, M.; Vile, D.; Haslam, R.P.; Napier, J.A.; et al. Overexpression of Arabidopsis ECERIFERUM1 promotes wax very-long-chain alkane biosynthesis and influences plant response to biotic and abiotic stresses. Plant Physiol. 2011, 156, 29–45. [Google Scholar] [CrossRef]
- Bernard, A.; Domergue, F.; Pascal, S.; Jetter, R.; Renne, C.; Faure, J.D.; Haslam, R.P.; Napier, J.A.; Lessire, R.; Joubès, J. Reconstitution of plant alkane biosynthesis in yeast demonstrates that Arabidopsis ECERIFERUM1 and ECERIFERUM3 are core components of a very-long-chain alkane synthesis complex. Plant Cell 2012, 24, 3106–3118. [Google Scholar] [CrossRef]
- Millar, A.A.; Kunst, L. Very-long-chain fatty acid biosynthesis is controlled through the expression and specificity of the condensing enzyme. Plant J. 1997, 12, 121–131. [Google Scholar] [CrossRef]
- Millar, A.A.; Clemens, S.; Zachgo, S.; Giblin, E.M.; Taylor, D.C.; Kunst, L. CUT1, an Arabidopsis gene required for cuticular wax biosynthesis and pollen fertility, encodes a very-long-chain fatty acid condensing enzyme. Plant Cell 1999, 11, 825–838. [Google Scholar] [CrossRef]
- Todd, J.; Post-Beittenmiller, D.; Jaworski, J.G. KCS1 encodes a fatty acid elongase 3-ketoacyl-CoA synthase affecting wax biosynthesis in Arabidopsis thaliana. Plant J. 1999, 17, 119–130. [Google Scholar] [CrossRef]
- Fiebig, A.; Mayfield, J.A.; Miley, N.L.; Chau, S.; Fischer, R.L.; Preuss, D. Alterations in CER6, a gene identical to CUT1, differentially affect long-chain lipid content on the surface of pollen and stems. Plant Cell 2000, 12, 2001–2008. [Google Scholar] [CrossRef]
- Pruitt, R.E.; Vielle-Calzada, J.P.; Ploense, S.E.; Grossniklaus, U.; Lolle, S.J. FIDDLEHEAD, a gene required to suppress epidermal cell interactions in Arabidopsis, encodes a putative lipid biosynthetic enzyme. Proc. Natl. Acad. Sci. USA 2000, 97, 1311–1316. [Google Scholar] [CrossRef]
- Kim, J.; Jung, J.H.; Lee, S.B.; Go, Y.S.; Kim, H.J.; Cahoon, R.; Markham, J.E.; Cahoon, E.B.; Suh, M.C. Arabidopsis 3-ketoacyl-coenzyme A synthase9 is involved in the synthesis of tetracosanoic acids as precursors of cuticular waxes, suberins, sphingolipids, and phospholipids. Plant Physiol. 2013, 162, 567–580. [Google Scholar] [CrossRef]
- Hooker, T.S.; Millar, A.A.; Kunst, L. Significance of the expression of the CER6 condensing enzyme for cuticular wax production in Arabidopsis. Plant Physiol. 2002, 129, 1568–1580. [Google Scholar] [CrossRef]
- Aarts, M.G.; Keijzer, C.J.; Stiekema, W.J.; Pereira, A. Molecular characterization of the CER1 gene of arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 1995, 7, 2115–2127. [Google Scholar] [CrossRef]
- Greer, S.; Wen, M.; Bird, D.; Wu, X.; Samuels, L.; Kunst, L.; Jetter, R. The cytochrome P450 enzyme CYP96A15 is the midchain alkane hydroxylase responsible for formation of secondary alcohols and ketones in stem cuticular wax of Arabidopsis. Plant physiol. 2007, 145, 653–667. [Google Scholar] [CrossRef]
- Broun, P.; Poindexter, P.; Osborne, E.; Jiang, C.; Riechmann, J.L. WIN1, a transcriptional activator of epidermal wax accumulation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 4706–4711. [Google Scholar] [CrossRef]
- Oshima, Y.; Mitsuda, N. The MIXTA-like transcription factor MYB16 is a major regulator of cuticle formation in vegetative organs. Plant Signal. Behav. 2013, 8, e26826. [Google Scholar] [CrossRef]
- Lee, S.B.; Kim, H.U.; Suh, M.C. MYB94 and MYB96 Additively Activate Cuticular Wax Biosynthesis in Arabidopsis. Plant Cell Physiol. 2016, 57, 2300–2311. [Google Scholar] [CrossRef]
- Cominelli, E.; Sala, T.; Calvi, D.; Gusmaroli, G.; Tonelli, C. Over-expression of the Arabidopsis AtMYB41 gene alters cell expansion and leaf surface permeability. Plant J. 2008, 53, 53–64. [Google Scholar] [CrossRef]
- Raffaele, S.; Vailleau, F.; Léger, A.; Joubès, J.; Miersch, O.; Huard, C.; Blée, E.; Mongrand, S.; Domergue, F.; Roby, D. A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 2008, 20, 752–767. [Google Scholar] [CrossRef]
- Wu, R.; Li, S.; He, S.; Wassmann, F.; Yu, C.; Qin, G.; Schreiber, L.; Qu, L.; Gu, H. CFL1, a WW domain protein, regulates cuticle development by modulating the function of HDG1, a class IV homeodomain transcription factor, in rice and Arabidopsis. Plant Cell 2011, 23, 3392–3411. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | A-G | C-T | Transition | A-C | A-T | C-G | G-T | Transversion | Total |
|---|---|---|---|---|---|---|---|---|---|
| 22-1-14-1 | 18,464 | 18,369 | 36,833 | 5698 | 6145 | 4105 | 5747 | 21,695 | 58,528 |
| 22-1-14-2 | 19,034 | 18,866 | 37,900 | 5841 | 6319 | 4208 | 5816 | 22,184 | 60,084 |
| 22-1-14-3 | 17,944 | 17,728 | 35,672 | 5464 | 5948 | 3973 | 5524 | 20,909 | 56,581 |
| 22-1-22-1 | 19,262 | 19,241 | 38,503 | 5913 | 6516 | 4364 | 5962 | 22,755 | 61,258 |
| 22-1-22-2 | 17,514 | 17,474 | 34,988 | 5409 | 5914 | 3998 | 5452 | 20,773 | 55,761 |
| 22-1-22-3 | 20,370 | 20,289 | 40,659 | 6255 | 6810 | 4492 | 6344 | 23,901 | 64,560 |
| 30-1-14-1 | 16,194 | 16,451 | 32,645 | 4951 | 5597 | 3756 | 5077 | 19,381 | 52,026 |
| 30-1-14-2 | 16,661 | 16,602 | 33,263 | 5062 | 5737 | 3777 | 5222 | 19,798 | 53,061 |
| 30-1-14-3 | 17,317 | 17,187 | 34,504 | 5195 | 5929 | 3848 | 5378 | 20,350 | 54,854 |
| 30-1-22-1 | 17,060 | 17,071 | 34,131 | 5183 | 5872 | 3764 | 5351 | 20,170 | 54,301 |
| 30-1-22-2 | 17,316 | 17,299 | 34,615 | 5178 | 5871 | 3814 | 5313 | 20,176 | 54,791 |
| 30-1-22-3 | 18,755 | 18,678 | 37,433 | 5686 | 6322 | 4094 | 5825 | 21,927 | 59,360 |
| QPCQ-14-1 | 13,173 | 13,135 | 26,308 | 4058 | 4481 | 2862 | 4125 | 15,526 | 41,834 |
| QPCQ-14-2 | 14,314 | 14,021 | 28,335 | 4378 | 4772 | 3086 | 4448 | 16,684 | 45,019 |
| QPCQ-14-3 | 14,479 | 14,465 | 28,944 | 4419 | 4931 | 3135 | 4528 | 17,013 | 45,957 |
| QPCQ-22-1 | 14,146 | 13,985 | 28,131 | 4304 | 4826 | 3063 | 4352 | 16,545 | 44,676 |
| QPCQ-22-2 | 15,405 | 15,446 | 30,851 | 4696 | 5252 | 3351 | 4759 | 18,058 | 48,909 |
| QPCQ-22-3 | 15,608 | 15,552 | 31,160 | 4762 | 5283 | 3398 | 4876 | 18,319 | 49,479 |
| Pathways | 22-22 vs. 22-14 | 30-22 vs. 30-14 | QPCQ-22 vs. QPCQ-14 | |||
|---|---|---|---|---|---|---|
| UP | DOWN | UP | DOWN | UP | DOWN | |
| Biosynthesis of unsaturated fatty acids | 9 | 2 | 1 | 3 | 1 | 1 |
| Cutin, suberine, and wax biosynthesis | 11 | 5 | 7 | 4 | 12 | 4 |
| Fatty acid biosynthesis | 9 | 3 | 1 | 3 | 2 | 0 |
| Fatty acid degradation | 5 | 2 | 5 | 3 | 5 | 1 |
| Fatty acid elongation | 6 | 5 | 4 | 4 | 4 | 3 |
| Fatty acid metabolism | 14 | 5 | 2 | 5 | 3 | 2 |
| ABC transporters | 16 | 3 | 6 | 3 | 7 | 3 |
| SUM | 48 | 18 | 22 | 16 | 28 | 11 |
| GENE ID | Up/Down-Regulation | Annotation |
|---|---|---|
| Sme2.5_00826.1_g00007.1 | Up | 3-ketoacyl-CoA synthase 3 |
| Sme2.5_00014.1_g00009.1 | Up | 3-ketoacyl-CoA synthase 20 |
| Sme2.5_01347.1_g00005.1 | Up | 3-ketoacyl-CoA synthase 11-like |
| Sme2.5_29857.1_g00001.1 | Down | 3-ketoacyl-CoA synthase 6-like |
| Sme2.5_22772.1_g00001.1 | Down | trans-2-enoyl-CoA reductase |
| Sme2.5_00227.1_g00015.1 | Up | O-acyltransferase WSD1-like |
| Sme2.5_00442.1_g00008.1 | Up | Cytochrome P450 77A1 |
| Sme2.5_00889.1_g00004.1 | Up | cytochrome P450 86A8 |
| Sme2.5_00836.1_g00001.1 | Up | cytochrome P450 86A22 |
| Sme2.5_02292.1_g00008.1 | Up | protein ECERIFERUM 3-like |
| Sme2.5_02154.1_g00006.1 | Up | pleiotropic drug resistance protein 1 |
| Sme2.5_08011.1_g00001.1 | Up | pleiotropic drug resistance protein 1-like |
| Sme2.5_06858.1_g00003.1 | Down | 3-ketoacyl-CoA thiolase 2 |
| Sme2.5_04239.1_g00004.1 | Up | acyl-coenzyme A oxidase 4 |
| Sme2.5_00625.1_g00013.1 | Up | acyl-activating enzyme 16 |
| Sme2.5_03548.1_g00005.1 | Down | fatty acyl-CoA reductase 3-like |
| Sme2.5_00091.1_g00016.1 | Down | putative elongation of fatty acids protein |
| Sme2.5_01164.1_g00001.1 | Up | omega-hydroxypalmitate O-feruloyl transferase |
| Sme2.5_02990.1_g00002.1 | Down | omega-hydroxypalmitate O-feruloyl transferase |
| Sme2.5_01024.1_g00003.1 | Up | ABC transporter B family member 2 |
| Sme2.5_01024.1_g00004.1 | Up | ABC transporter B family member 2 |
| Sme2.5_02844.1_g00004.1 | Up | ABC transporter G family member 32 |
| Sme2.5_29635.1_g00001.1 | Down | ABC transporter G family member 4 |
| Sme2.5_01812.1_g00005.1 | Down | ABC transporter C family member 15 |
| Gene ID | MW (kda) | pI | Protein Length (aa) |
|---|---|---|---|
| Sme2.5_19583.1_g00001.1 | 29,582.9 | 10 | 265 |
| Sme2.5_00826.1_g00007.1 | 47,212.4 | 8.45 | 419 |
| Sme2.5_27871.1_g00001.1 | 37,796.3 | 9.19 | 334 |
| Sme2.5_00871.1_g00008.1 | 57,046.7 | 9.12 | 509 |
| Sme2.5_03969.1_g00002.1 | 56,934.8 | 9.45 | 508 |
| Sme2.5_25073.1_g00001.1 | 57,046.7 | 9.12 | 509 |
| Sme2.5_00014.1_g00009.1 | 45,569.2 | 8.37 | 407 |
| Sme2.5_01347.1_g00005.1 | 58,512.9 | 9.37 | 519 |
| Sme2.5_01521.1_g00005.1 | 57,632.5 | 9.69 | 516 |
| Sme2.5_00064.1_g00011.1 | 55,893.1 | 9.25 | 496 |
| Sme2.5_08668.1_g00001.1 | 59,843.1 | 9.24 | 527 |
| Sme2.5_03767.1_g00003.1 | 39,626.2 | 6.31 | 354 |
| Sme2.5_03248.1_g00007.1 | 52,167.6 | 9.06 | 462 |
| Sme2.5_00238.1_g00006.1 | 43,558.2 | 8.89 | 390 |
| Sme2.5_00238.1_g00007.1 | 32,572.5 | 7.81 | 294 |
| Sme2.5_00238.1_g00005.1 | 55,707.6 | 9.42 | 493 |
| Sme2.5_03511.1_g00001.1 | 59,793.8 | 9.27 | 530 |
| Sme2.5_06196.1_g00003.1 | 55,317.6 | 9.58 | 493 |
| Sme2.5_10876.1_g00001.1 | 56,023.5 | 9.44 | 496 |
| Sme2.5_01170.1_g00008.1 | 22,573.9 | 8.49 | 204 |
| Sme2.5_29857.1_g00001.1 | 25,430.2 | 9.56 | 225 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, A.; Huang, Q.; Li, J.; Zhu, W.; Liu, X.; Wu, X.; Zha, D. Comparative Transcriptome Analysis Reveals Gene Expression Differences in Eggplant (Solanum melongena L.) Fruits with Different Brightness. Foods 2022, 11, 2506. https://doi.org/10.3390/foods11162506
Zhang A, Huang Q, Li J, Zhu W, Liu X, Wu X, Zha D. Comparative Transcriptome Analysis Reveals Gene Expression Differences in Eggplant (Solanum melongena L.) Fruits with Different Brightness. Foods. 2022; 11(16):2506. https://doi.org/10.3390/foods11162506
Chicago/Turabian StyleZhang, Aidong, Qianru Huang, Jianyong Li, Weimin Zhu, Xiaohui Liu, Xuexia Wu, and Dingshi Zha. 2022. "Comparative Transcriptome Analysis Reveals Gene Expression Differences in Eggplant (Solanum melongena L.) Fruits with Different Brightness" Foods 11, no. 16: 2506. https://doi.org/10.3390/foods11162506