
Bacterial Communities Related to Aroma Formation during Spontaneous Fermentation of ‘Cabernet Sauvignon’ Wine in Ningxia, China

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Grapes
2.2. Spontaneous Fermentation and Sample Collection
2.3. DNA Extraction and PCR Reaction
2.4. HTS Analysis
2.5. VOCs Analysis
2.6. Sensory Analysis of Aroma Profile
2.7. Bioinformatics Analysis and Statistical Analysis
3. Results
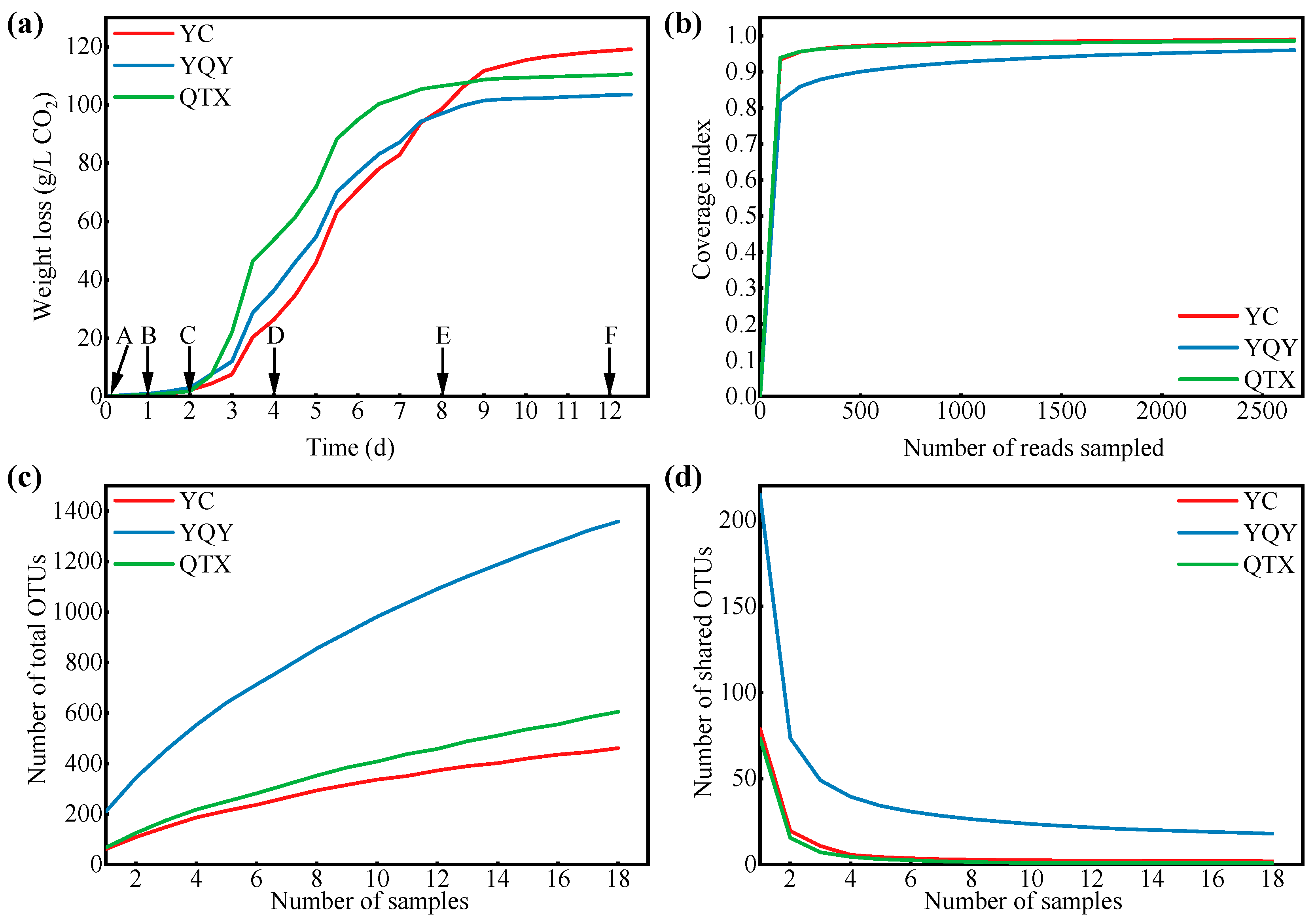
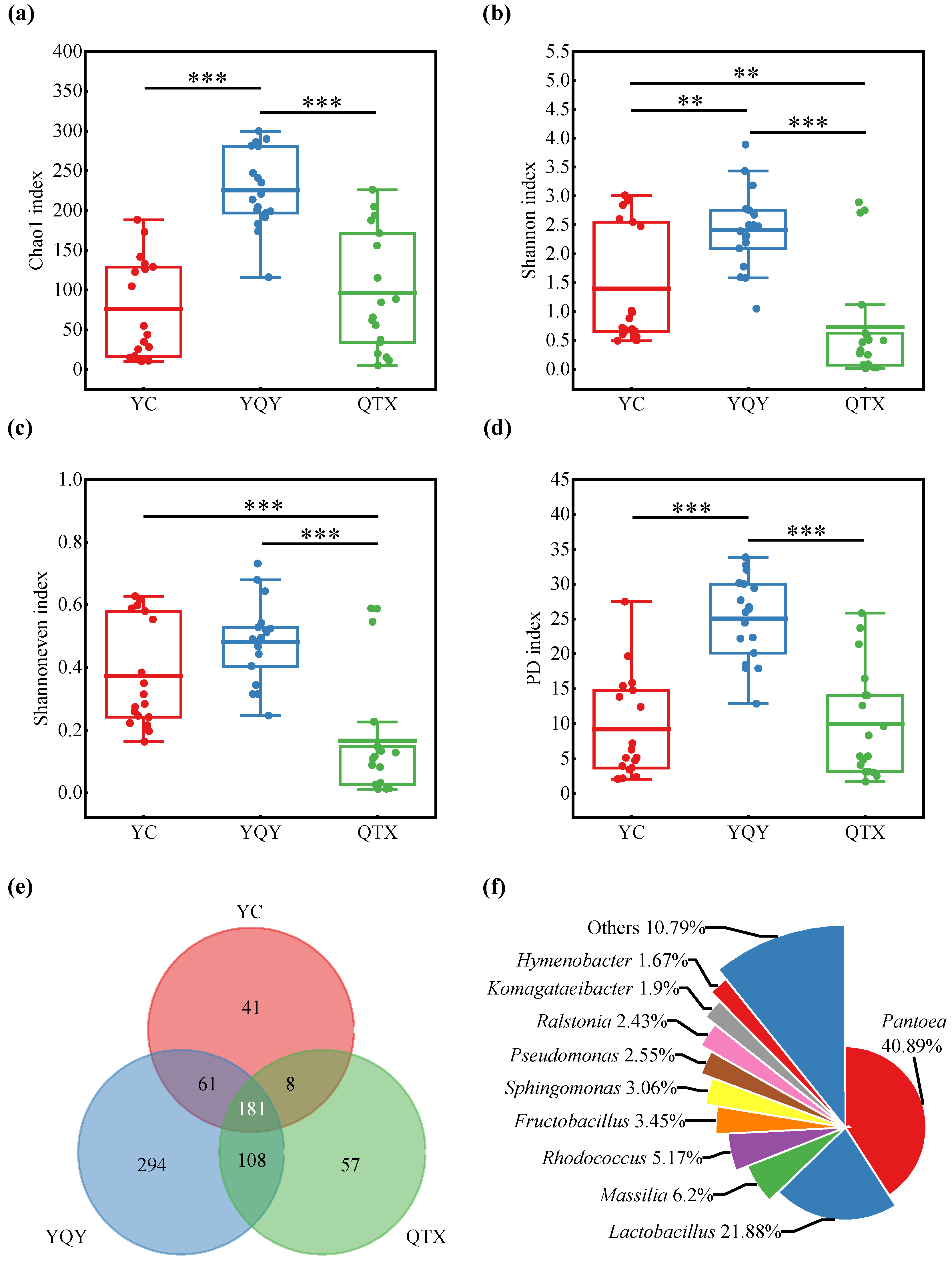
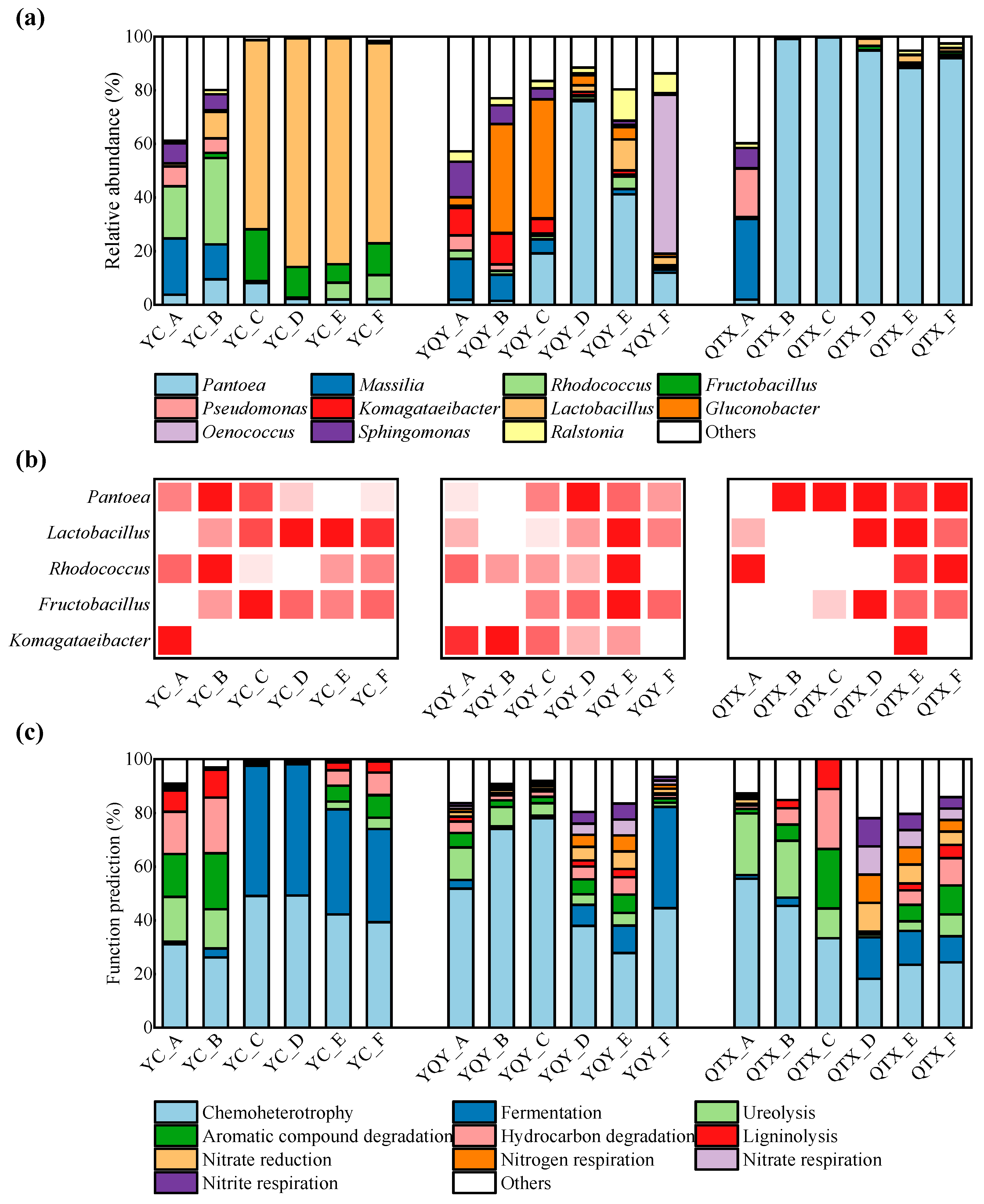
3.1. Bacterial Composition of Different Sub-Regions
3.2. Bacterial Composition of Different Fermentation Stages
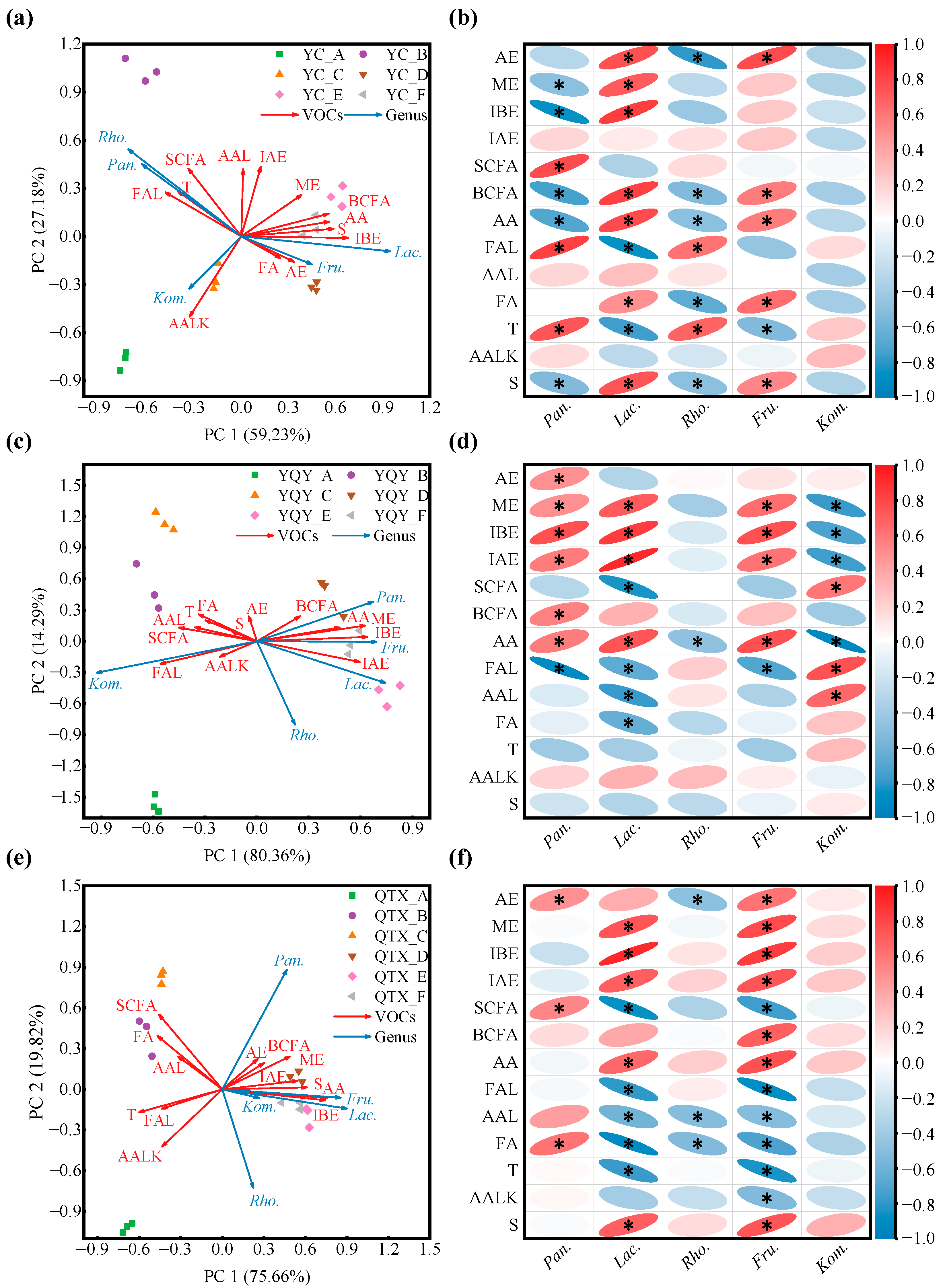
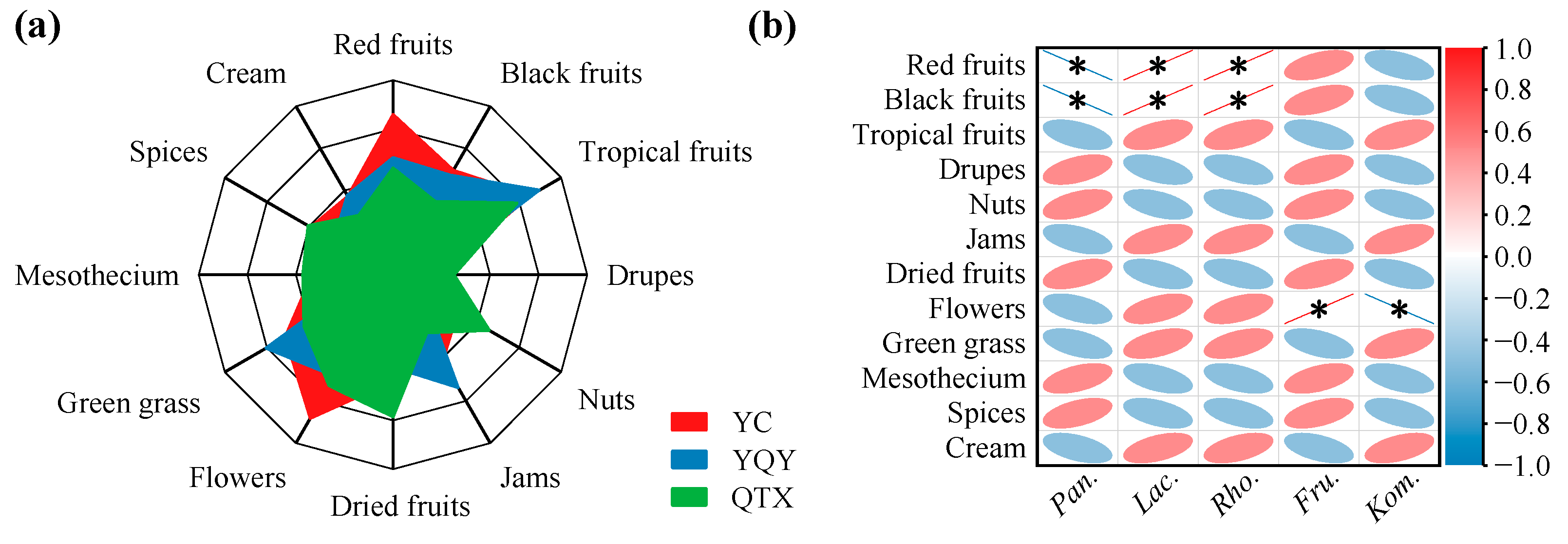
3.3. Correlation Analysis between Core Bacteria and Wine Aroma/Ethanol
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lappa, I.K.; Kachrimanidou, V.; Pateraki, C.; Koulougliotis, D.; Eriotou, E.; Kopsahelis, N. Indigenous yeasts: Emerging trends and challenges in winemaking. Curr. Opin. Food Sci. 2020, 32, 133–143. [Google Scholar] [CrossRef]
- Van Leeuwen, C.; Seguin, G. The concept of terroir in viticulture. J. Wine Res. 2006, 17, 1–10. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Thorngate, J.H.; Richardson, P.M.; Mills, D.A. Microbial biogeography of wine grapes is conditioned by cultivar, vintage, and climate. Proc. Natl. Acad. Sci. USA 2014, 111, E139–E148. [Google Scholar] [CrossRef]
- Gilbert, J.; Van Der Lelie, D.; Zarraonaindia, I. Microbial terroir for wine grapes. Proc. Natl. Acad. Sci. USA 2014, 111, 5–6. [Google Scholar] [CrossRef]
- Griggs, R.G.; Steenwerth, K.L.; Mills, D.A.; Cantu, D.; Bokulich, N.A. Sources and assembly of microbial communities in vineyards as a functional component of winegrowing. Front. Microbiol. 2021, 12, 836. [Google Scholar] [CrossRef]
- González, R.; Morales, P. Truth in wine yeast. Microb. Biotechnol. 2022, 15, 1339–1356. [Google Scholar] [CrossRef] [PubMed]
- Belda, I.; Zarraonaindia, I.; Perisin, M.; Palacios, A.; Acedo, A. From vineyard soil to wine fermentation: Microbiome approximations to explain the “terroir” concept. Front. Microbiol. 2017, 8, 821. [Google Scholar] [CrossRef]
- Pinto, C.; Pinho, D.; Cardoso, R.; Custódio, V.; Fernandes, J.; Sousa, S.; Pinheiro, M.; Egas, C.; Gomes, A.C. Wine fermentation microbiome: A landscape from different Portuguese wine appellations. Front. Microbiol. 2015, 6, 905. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, P.; Chen, D.; Howell, K. From the vineyard to the winery: How microbial ecology drives regional distinctiveness of wine. Front. Microbiol. 2019, 10, 2679. [Google Scholar] [CrossRef]
- Rivas, G.A.; La Hens, D.V.; Delfederico, L.; Olguin, N.; Bravo-Ferrada, B.M.; Tymczyszyn, E.E.; Semorile, L.; Brizuela, N.S. Molecular tools for the analysis of the microbiota involved in malolactic fermentation: From microbial diversity to selection of lactic acid bacteria of enological interest. World. J. Microb. Biot. 2022, 38, 19. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Collins, T.S.; Masarweh, C.; Allen, G.; Heymann, H.; Ebeler, S.E.; Mills, D.A. Associations among wine grape microbiome, metabolome, and fermentation behavior suggest microbial contribution to regional wine characteristics. MBio 2016, 7, e00631-16. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Portillo, M.; Franquès, J.; Araque, I.; Reguant, C.; Bordons, A. Bacterial diversity of Grenache and Carignan grape surface from different vineyards at Priorat wine region (Catalonia, Spain). Int. J. Food. Microbiol. 2016, 219, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Kecskeméti, E.; Berkelmann-Löhnertz, B.; Reineke, A. Are epiphytic microbial communities in the carposphere of ripening grape clusters (Vitis vinifera L.) different between conventional, organic, and biodynamic grapes. PLoS ONE 2016, 11, e0160852. [Google Scholar] [CrossRef]
- Morgan, H.H.; Du Toit, M.; Setati, M.E. The grapevine and wine microbiome: Insights from high-throughput amplicon sequencing. Front. Microbiol. 2017, 8, 820–834. [Google Scholar] [CrossRef]
- Bubeck, A.M.; Preiss, L.; Jung, A.; Dörner, E.; Podlesny, D.; Kulis, M.; Maddox, C.; Arze, C.; Zörb, C.; Merkt, N.; et al. Bacterial microbiota diversity and composition in red and white wines correlate with plant-derived DNA contributions and botrytis infection. Sci. Rep. 2020, 10, 13828. [Google Scholar] [CrossRef]
- Carpena, M.; Fraga-Corral, M.; Otero, P.; Nogueira, R.A.; Garcia-Oliveira, P.; Prieto, M.A.; Simal-Gandara, J. Secondary aroma: Influence of wine microorganisms in their aroma profile. Foods 2021, 10, 51. [Google Scholar] [CrossRef]
- Fowell, R.R. The identification of wild yeast colonies on lysine agar. J. Bacteriol. 1965, 28, 373–383. [Google Scholar] [CrossRef]
- Pallmann, C.L.; Brown, J.A.; Olineka, T.L.; Cocolin, L.; Mills, D.A.; Bisson, L.F. Use of WL medium to profile native flora fermentations. Am. J. Enol. Viticult. 2001, 52, 198–203. [Google Scholar]
- Battistelli, N.; Perpetuini, G.; Piva, A.; Pepe, A.; Sidari, R.; Wache, Y.; Tofalo, R. Cultivable microbial ecology and aromatic profile of “mothers” for Vino cotto wine production. Food Res. Int. 2021, 143, 110311. [Google Scholar] [CrossRef]
- Ghosh, S.; Bagheri, B.; Morgan, H.H.; Divol, B.; Setati, M.E. Assessment of wine microbial diversity using ARISA and cultivation-based methods. Ann. Microbiol. 2015, 65, 1833–1840. [Google Scholar] [CrossRef]
- González-Arenzana, L.; Santamaría, P.; Gutiérrez, A.R.; López, R.; López-Alfaro, I. Lactic acid bacteria communities in must, alcoholic and malolactic Tempranillo wine fermentations, by culture-dependent and culture-independent methods. Eur. Food Res. Technol. 2017, 243, 41–48. [Google Scholar] [CrossRef]
- Wang, C.; Wu, C.; Qiu, S. Yeast diversity investigation of Vitis davidii Föex during spontaneous fermentations using culture-dependent and high-throughput sequencing approaches. Food Res. Int. 2019, 126, 108582. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, T.; Xu, X.; Ji, Y.; Jiang, X.; Shi, X.; Wang, B. Investigation of volatile compounds, microbial succession, and their relation during spontaneous fermentation of Petit Manseng. Front. Microbiol. 2021, 12, 717387. [Google Scholar] [CrossRef] [PubMed]
- Rivas, G.A.; Guillade, A.C.; Semorile, L.C.; Delfederico, L. Influence of climate on soil and wine bacterial diversity on a vineyard in a non-traditional wine region in Argentina. Front. Microbiol. 2021, 12, 726384. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Ding, Y.; Gao, F.; Zhang, L.; Wang, L.; Li, H.; Wang, H. Community succession of the grape epidermis microbes of Cabernet Sauvignon (Vitis vinifera L.) from different regions in China during fruit development. Int. J. Food. Microbiol. 2022, 362, 109475. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Feng, C.; Li, R.; Chen, Q. Isolation and identification of Saccharomyces cerevisiae strain of East Helan Mountain Area in Ningxia. Food Res. Dev. 2017, 38, 176–180. (In Chinese) [Google Scholar] [CrossRef]
- Gao, H.; Yin, X.; Jiang, X.; Shi, H.; Yang, Y.; Wang, C.; Dai, X.; Chen, Y.; Wu, X. Diversity and spoilage potential of microbial communities associated with grape sour rot in eastern coastal areas of China. PeerJ 2020, 8, e9376. [Google Scholar] [CrossRef]
- Zhang, W.; Tian, Y.; Sun, Y.; Zhang, X. Diversity of yeasts in spontaneous fermentation must of Eastern Helan Mountain Areas. China Brew. 2020, 39, 30–35. (In Chinese) [Google Scholar] [CrossRef]
- Shen, T.; Liu, R.; Huang, X.; Xu, Z.; Chen, W. Effects of irrigation amount on rhizosphere soil microbial diversity of ‘Cabernet Sauvignon’. Non-Wood For. Res. 2021, 39, 116–125. (In Chinese) [Google Scholar] [CrossRef]
- Zhao, Y.; Li, J.; Xie, W.; Zhao, H. Microbial diversity of wine grape skins from four different regions in China. Food Ferment. Ind. 2021, 47, 53–60. (In Chinese) [Google Scholar] [CrossRef]
- Committee, C.N.S.M. General Analysis Methods for Wine and Fruit Wine: GB/T 15038-2006; China Standard Press: Beijing, China, 2006; pp. 5–8. [Google Scholar]
- Gump, B.H.; Zoecklein, B.W.; Fugelsang, K.C. Prediction of prefermentation nutritional status of grape juice. In Methods in Biotechnology, 1st ed.; Spencer, J.F.T., Ragout de Spencer, A.L., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 14, pp. 283–296. [Google Scholar]
- Lu, Y.; Sun, F.; Wang, W.; Liu, Y.; Wang, J.; Sun, J.; Mu, J.; Gao, Z. Effects of spontaneous fermentation on the microorganisms diversity and volatile compounds during ‘Marselan’ from grape to wine. LWT-Food Sci. Technol. 2020, 134, 110193. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, K.; Gu, X.; Sun, X.; Jin, G.; Zhang, J.; Ma, W. Flavor chemical profiles of Cabernet Sauvignon wines: Six vintages from 2013 to 2018 from the Eastern Foothills of the Ningxia Helan Mountains in China. Foods 2022, 11, 22. [Google Scholar] [CrossRef]
- Högnadóttir, Á.; Rouseff, R.L. Identification of aroma active compounds in orange essence oil using gas chromatography–olfactometry and gas chromatography–mass spectrometry. J. Chromatogr. A. 2003, 998, 201–211. [Google Scholar] [CrossRef]
- Kong, C.; Li, A.; Jin, G.; Zhu, X.; Tao, Y. Evolution of volatile compounds treated with selected non-Saccharomyces extracellular extract during Pinot Noir winemaking in monsoon climate. Food Res. Int. 2019, 119, 177–186. [Google Scholar] [CrossRef]
- Sun, D.; Qu, J.; Huang, Y.; Lu, J.; Yin, L. Analysis of microbial community diversity of muscadine grape skins. Food Res. Int. 2021, 145, 110417. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Cavagnaro, T.R.; De Bei, R.; Nelson, T.M.; Stephen, J.R.; Metcalfe, A.; Gilliham, M.; Breen, J.; Collins, C.; López, C.M.R. Wine terroir and the soil bacteria: An amplicon sequencing-based assessment of the Barossa valley and its sub-regions. Front. Microbiol. 2021, 11, 597944. [Google Scholar] [CrossRef]
- Orlofsky, E.; Zabari, L.; Bonito, G.; Masaphy, S. Changes in soil bacteria functional ecology associated with Morchella rufobrunnea fruiting in a natural habitat. Environ. Microbiol. 2021, 23, 6651–6662. [Google Scholar] [CrossRef]
- Malensek, M.; Pallickara, S.; Pallickara, S. Analytic queries over geospatial time-series data using distributed hash tables. IEEE T. Knowl. Data Eng. 2016, 28, 1408–1422. [Google Scholar] [CrossRef]
- Emam, K.E.; Benlarbi, S.; Goel, N.; Rai, S.N. The confounding effect of class size on the validity of object-oriented metrics. IEEE T. Knowl. Data Eng. 2001, 27, 630–650. [Google Scholar] [CrossRef]
- Mamet, S.D.; Lamb, E.G.; Piper, C.L.; Winsley, T.; Siciliano, S.D. Archaea and bacteria mediate the effects of native species root loss on fungi during plant invasion. ISME J. 2017, 11, 1261–1275. [Google Scholar] [CrossRef]
- Etschmann, M.; Bluemke, W.; Sell, D.; Schrader, J. Biotechnological production of 2-phenylethanol. Appl. Microbiol. Biot. 2002, 59, 1–8. [Google Scholar] [CrossRef]
- Ingram, L.O. Ethanol tolerance in bacteria. Crit. Rev. Biotechnol. 1990, 9, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.; Madeira, S.C.; Mendes-Ferreira, A.; Sá-Correia, I. Impact of assimilable nitrogen availability in glucose uptake kinetics in Saccharomyces cerevisiae during alcoholic fermentation. Microb. Cell Fact. 2012, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Bartle, L.; Sumby, K.; Sundstrom, J.; Jiranek, V. The microbial challenge of winemaking: Yeast-bacteria compatibility. FEMS Yeast Res. 2019, 19, foz040. [Google Scholar] [CrossRef] [PubMed]
- Bučková, M.; Puškárová, A.; Ženišová, K.; Kraková, L.; Piknová, L.; Kuchta, T.; Pangallo, D. Novel insights into microbial community dynamics during the fermentation of Central European ice wine. Int. J. Food Microbiol. 2018, 266, 42–51. [Google Scholar] [CrossRef]
- Wang, H.; Hopfer, H.; Cockburn, D.W.; Wee, J. Characterization of microbial dynamics and volatile metabolome changes during fermentation of Chambourcin hybrid grapes from two Pennsylvania Regions. Front. Microbiol. 2020, 11, 614278. [Google Scholar] [CrossRef]
- Oyuela Aguilar, M.; Gobbi, A.; Browne, P.D.; Ellegaard-Jensen, L.; Hansen, L.H.; Semorile, L.; Pistorio, M. Influence of vintage, geographic location and cultivar on the structure of microbial communities associated with the grapevine rhizosphere in vineyards of San Juan Province, Argentina. PLoS ONE 2020, 15, e0243848. [Google Scholar] [CrossRef]
- Chen, L.; Ren, L.; Li, D.; Ma, X. Analysis of microbiomes in three traditional starters and volatile components of the Chinese rice wines. Food Sci. Biotechnol. 2021, 30, 87–96. [Google Scholar] [CrossRef]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.; Harris, H.M.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microb. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Q.; Zou, H.; Liu, Y.; Wang, J.; Gan, K.; Xiang, J. Glucose metabolic flux distribution of Lactobacillus amylophilus during lactic acid production using kitchen waste saccharified solution. Microb. Biotechnol. 2013, 6, 685–693. [Google Scholar] [CrossRef]
- Cappello, M.S.; Zapparoli, G.; Logrieco, A.; Bartowsky, E.J. Linking wine lactic acid bacteria diversity with wine aroma and flavour. Int. J. Food. Microbiol. 2017, 243, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bartowsky, E.J.; Costello, P.J.; Chambers, P.J. Emerging trends in the application of malolactic fermentation. Aust. J. Grape Wine R. 2015, 21, 663–669. [Google Scholar] [CrossRef]
- Lombardi, S.J.; Pannella, G.; Iorizzo, M.; Testa, B.; Succi, M.; Tremonte, P.; Sorrentino, E.; Di Renzo, M.; Strollo, D.; Coppola, R. Inoculum strategies and performances of malolactic starter Lactobacillus plantarum M10: Impact on chemical and sensorial characteristics of Fiano Wine. Microorganisms 2020, 8, 516. [Google Scholar] [CrossRef] [PubMed]
- Devi, A.; Anu-Appaiah, K.A. Mixed malolactic co-culture (Lactobacillus plantarum and Oenococcus oeni) with compatible Saccharomyces influences the polyphenolic, volatile and sensory profile of Shiraz wine. LWT-Food Sci. Technol. 2021, 135, 110246. [Google Scholar] [CrossRef]
- Franquès, J.; Araque, I.; Palahí, E.; Del Carmen Portillo, M.; Reguant, C.; Bordons, A. Presence of Oenococcus oeni and other lactic acid bacteria in grapes and wines from Priorat (Catalonia, Spain). LWT-Food Sci. Technol. 2017, 81, 326–334. [Google Scholar] [CrossRef]
- Endo, A.; Tanizawa, Y.; Tanaka, N.; Maeno, S.; Kumar, H.; Shiwa, Y.; Okada, S.; Yoshikawa, H.; Dicks, L.; Nakagawa, J.; et al. Comparative genomics of Fructobacillus spp. and Leuconostoc spp. reveals niche-specific evolution of Fructobacillus spp. BMC Genom. 2015, 16, 1117. [Google Scholar] [CrossRef]
- Milanović, V.; Osimani, A.; Garofalo, C.; De Filippis, F.; Ercolini, D.; Cardinali, F.; Taccari, M.; Aquilanti, L.; Clementi, F. Profiling white wine seed vinegar bacterial diversity through viable counting, metagenomic sequencing and PCR-DGGE. Int. J. Food. Microbiol. 2018, 286, 66–74. [Google Scholar] [CrossRef]
- Román-Camacho, J.J.; Santos-Dueñas, I.M.; García-García, I.; Moreno-García, J.; García-Martínez, T.; Mauricio, J.C. Metaproteomics of microbiota involved in submerged culture production of alcohol wine vinegar: A first approach. Int. J. Food. Microbiol. 2020, 333, 108797. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Regions | Indices | Fermentation Stages | Variation | |||||
|---|---|---|---|---|---|---|---|---|
| A (0 d) | B (1 d) | C (2 d) | D (4 d) | E (8 d) | F (12 d) | |||
| YC | Chao 1 | 240.36 ± 53.42 | 178.97 ± 34.34 | 22.22 ± 11.42 | 15.50 ± 4.92 | 63.27 ± 40.92 | 178.16 ± 149.53 | * |
| Shannon | 3.48 ± 0.06 | 2.93 ± 0.07 | 0.87 ± 0.14 | 0.54 ± 0.06 | 0.67 ± 0.07 | 0.92 ± 0.17 | ** | |
| Shannoneven | 0.68 ± 0.02 | 0.61 ± 0.02 | 0.31 ± 0.06 | 0.23 ± 0.03 | 0.22 ± 0.02 | 0.25 ± 0.08 | * | |
| PD | 23.09 ± 7.52 | 14.92 ± 1.56 | 3.70 ± 0.26 | 2.26 ± 0.15 | 5.17 ± 0.21 | 10.02 ± 5.48 | ** | |
| YQY | Chao 1 | 355.23 ± 59.47 | 321.27 ± 49.35 | 312.66 ± 35.94 | 292.81 ± 148.86 | 423.41 ± 82.85 | 468.38 ± 34.11 | |
| Shannon | 3.93 ± 0.39 | 2.81 ± 0.14 | 2.43 ± 0.11 | 1.55 ± 0.41 | 2.87 ± 0.16 | 2.11 ± 0.39 | ** | |
| Shannoneven | 0.72 ± 0.04 | 0.54 ± 0.01 | 0.48 ± 0.01 | 0.30 ± 0.05 | 0.52 ± 0.02 | 0.39 ± 0.07 | ** | |
| PD | 30.47 ± 8.38 | 23.73 ± 5.71 | 21.52 ± 2.46 | 26.04 ± 11.29 | 35.80 ± 7.12 | 33.62 ± 2.91 | ||
| QTX | Chao 1 | 291.35 ± 53.49 | 34.48 ± 17.27 | 14.28 ± 2.56 | 92.28 ± 34.46 | 221.93 ± 96.94 | 209.38 ± 67.09 | * |
| Shannon | 3.46 ± 0.06 | 0.07 ± 0.02 | 0.02 ± 0.01 | 0.30 ± 0.04 | 0.81 ± 0.36 | 0.53 ± 0.06 | ** | |
| Shannoneven | 0.66 ± 0.02 | 0.03 ± 0.01 | 0.01 ± 0.00 | 0.09 ± 0.02 | 0.17 ± 0.05 | 0.13 ± 0.01 | ** | |
| PD | 22.65 ± 4.23 | 3.60 ± 0.97 | 2.37 ± 0.62 | 5.03 ± 0.82 | 18.91 ± 12.79 | 12.59 ± 2.39 | ** | |
| VOCs | VIF Value | |
|---|---|---|
| Before Filtration | After Filtration | |
| Acetic esters (AE) | 2091.5 | 3.1 |
| Methyl esters (ME) | 11.7 | 7.3 |
| Ethyl esters (EE) | 9311.0 | >10 |
| Isobutyl esters (IBE) | 12.5 | 3.5 |
| Isoamyl esters (IAE) | 16.7 | 5.8 |
| Straight-chain fatty alcohols (SCFA) | 137.4 | 5.3 |
| Branched-chain fatty alcohols (BCFA) | 561.8 | 4.6 |
| C6 alcohols (C6A) | 24,228.7 | >10 |
| Aromatic alcohols (AA) | 8441.4 | 8.4 |
| Ketones (K) | 24.8 | >10 |
| Fatty aldehydes (FAL) | 43.9 | 8.0 |
| Aromatic aldehydes (AAL) | 8.5 | 6.9 |
| Fatty acids (FA) | 50.8 | 6.6 |
| Terpenes (T) | 7.5 | 6.2 |
| Aromatic alkenes (AALK) | 2579.8 | 6.9 |
| Sulfides (S) | 7.1 | 6.4 |
| Volatile phenols (VP) | NA | >10 |
| Euclidean Distance | Manhattan Distance | |||
|---|---|---|---|---|
| Mantel Coefficient | p-Value | Mantel Coefficient | p-Value | |
| Before filtration | 0.148 | 0.002 | 0.120 | 0.003 |
| After filtration | 0.166 | <0.001 | 0.137 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, Q.; Yang, H.; Sun, L.; Xia, H.; Sun, W.; Wang, Z.; Zhang, J. Bacterial Communities Related to Aroma Formation during Spontaneous Fermentation of ‘Cabernet Sauvignon’ Wine in Ningxia, China. Foods 2022, 11, 2775. https://doi.org/10.3390/foods11182775
Zhang Z, Zhang Q, Yang H, Sun L, Xia H, Sun W, Wang Z, Zhang J. Bacterial Communities Related to Aroma Formation during Spontaneous Fermentation of ‘Cabernet Sauvignon’ Wine in Ningxia, China. Foods. 2022; 11(18):2775. https://doi.org/10.3390/foods11182775
Chicago/Turabian StyleZhang, Zhong, Qingchen Zhang, Hui Yang, Lijun Sun, Hongchuan Xia, Wenjing Sun, Zheng Wang, and Junxiang Zhang. 2022. "Bacterial Communities Related to Aroma Formation during Spontaneous Fermentation of ‘Cabernet Sauvignon’ Wine in Ningxia, China" Foods 11, no. 18: 2775. https://doi.org/10.3390/foods11182775
APA StyleZhang, Z., Zhang, Q., Yang, H., Sun, L., Xia, H., Sun, W., Wang, Z., & Zhang, J. (2022). Bacterial Communities Related to Aroma Formation during Spontaneous Fermentation of ‘Cabernet Sauvignon’ Wine in Ningxia, China. Foods, 11(18), 2775. https://doi.org/10.3390/foods11182775